

Abstract
Objective
To evaluate the in vivo effects of hypertonic saline (HTS) resuscitation on the interactions of endothelial cells (ECs) and polymorphonuclear neutrophils (PMNs) and vascular permeability after hemorrhagic shock.
Summary Background Data
The PMN has been implicated in the pathogenesis of EC damage and organ injury following hemorrhagic shock. Compared to Ringer’s lactate (RL), HTS resuscitation diminishes PMN and EC adhesion molecule expression and organ sequestration of PMNs.
Methods
In a murine model of hemorrhagic shock (50 mmHg for 45 minutes followed by resuscitation) using intravital microscopy on cremaster muscle, the authors studied PMN–EC interactions and vascular leakage (epifluorescence after 50 mg/kg fluorescent albumin) in three resuscitation groups: HTS (shed blood + 4 cc/kg 7.5% HTS, n = 12), RL (shed blood + RL [2× shed blood volume], n = 12), and sham (no hemorrhage or resuscitation, n = 9). EC ICAM-1 expression was evaluated by immunohistochemistry. Data, presented as mean ± SEM, were evaluated by analysis of variance with Bonferroni correction.
Results
There were no differences between groups in flow mechanics. Compared to RL, HTS animals (t = 90 minutes) displayed diminished PMN rolling and PMN adhesion to EC at time intervals beyond t = 0. There were no differences between the sham and HTS groups. Vascular leakage was 45% lower in HTS than in RL-resuscitated animals. Cremaster EC ICAM-1 expression was similar in the two groups.
Conclusions
Using HTS instead of RL to resuscitate hemorrhagic shock diminishes vascular permeability in vivo by altering PMN–EC interactions. HTS could serve as a novel means of immunomodulation in hemorrhagic shock victims, potentially reducing PMN-mediated tissue injury.
Over 30 years ago, the Vietnam War established the current standard use of isotonic crystalloid fluids (normal saline [NS] and Ringer’s lactate [RL]) for the resuscitation of hemorrhagic shock. Subsequent studies have demonstrated that crystalloids represent an effective and inexpensive means to restore intravascular volume and additionally offer a survival advantage over colloids in the resuscitation of traumatic hemorrhagic shock. 1
More recently, “small volume resuscitation”2 with 4 mL of 7.5% NaCl per kilogram of body weight of hypertonic saline (HTS) has been proposed in the treatment of hemorrhagic shock. Whereas isotonic fluid administration requires large volumes, hypertonic resuscitation offers the advantages of ease of transport, speed of administration, and almost instantaneous hemodynamic effect. The intravascular administration of 7.5% NaCl rapidly creates a potent transcapillary osmotic gradient causing intravascular movement of water from the interstitium, endothelial cells (ECs), and red blood cells, a process that could reduce third-space fluid sequestration in the lungs of patients with traumatic pulmonary contusions or in the brain following head trauma. 3 Early animal studies showed that HTS resuscitation rapidly restored mean arterial pressure (MAP), peripheral tissue perfusion, cardiac contractility, and oxygen consumption, mainly through vasodilatation of precapillary resistance vessels and increases in cardiac preload. 4–6 Although large multicentric randomized human clinical trials subsequently confirmed that HTS resuscitation was safe and efficacious, they failed to demonstrate a clear survival advantage over standard isotonic resuscitation. 4,7 Nonetheless, one multicentric randomized control trial has demonstrated fewer postresuscitation complications, such as ARDS, renal failure, and coagulopathies, with the use of HTS. 4
Many of the complications following the resuscitation of hemorrhagic shock may be related to alterations in host immunity. In particular, the polymorphonuclear neutrophil (PMN), one of the principal host immune effector cells, has been implicated in the development of organ dysfunction and death following sepsis, burns, and multiple trauma as well as hemorrhagic shock resuscitation. 8–10 Although the PMN is essential in protecting the host from traumatic and infectious insults, it may also turn its potent defenses inappropriately against the host, activated by and contributing to the severity of injury.
The sequential events in the passage of PMNs from the vasculature to their sites of action have been well characterized. First marginating to the periphery of the vessel, the PMN rolls on the vessel wall, interacting with ECs through surface selectins (L, E, and P). 11 These weak interactions allow PMN β2-integrins (CD18/CD11) to strongly interact with endothelial receptors of the Ig superfamily (ICAM-1, ICAM-2), 12 resulting in firm adhesion of the two cell types and permitting subsequent PMN diapedesis between ECs to reach the injured site. 13
The inappropriate upregulation of these PMN–EC interactions in the ischemia/reperfusion injury of hemorrhagic shock resuscitation is believed to be an important step in the host’s progression to systemic inflammation and subsequent remote organ injury. There are data suggesting that activated PMNs are then sequestered in end organs, where they unleash a cytotoxic arsenal of proteases and oxygen radicals, causing injury to endothelium and resulting in vascular leakage, tissue edema, and eventually organ damage. 8,10,14
Much evidence now exists demonstrating that HTS resuscitation of hemorrhagic shock alters PMN activity. When human PMNs are incubated in hypertonic media, they display decreased cytotoxicity, cellular activation, superoxide production, and elastase release. 15–19 In vivo studies suggest that both neutrophil and endothelial adhesion molecule expression is reduced in HTS-resuscitated animals as compared to those receiving RL. 15,20–22 These alterations in adhesion molecules suggest that HTS may impart functional changes in PMNs and ECs. More importantly, animal models of hemorrhagic shock resuscitated with HTS have shown reductions in lung and liver injury, diminished bronchioalveolar lavage PMNs, and diminished pulmonary myeloperoxidase (MPO - total PMN content) as well as decreases in mortality. 15,21,23
Models of hemorrhagic shock resuscitation studied with intravital microscopy suggest that the beneficial effects of HTS may be due to altered EC–PMN interactions. 24–26 However, the consequences of such altered EC–PMN interactions following HTS resuscitation of hemorrhagic shock remain elusive, and the links to altered adhesion molecule expression and to subsequent tissue edema are unclear. We thus hypothesized that through diminished endothelial ICAM-1 expression, HTS resuscitation of hemorrhagic shock lessens venular EC–PMN interactions, leading to decreased in vivo vascular leakage in remote organs. We tested this hypothesis in a murine model of resuscitated hemorrhagic shock using intravital microscopy of cremaster muscle.
METHODS
Materials and Solutions
Sodium chloride (0.9% NaCl) and RL solutions were purchased from Baxter Corporation (Toronto, Canada), 2-methylbutane (isopentane) and frozen tissue embedding medium (Histo Prep) from Fisher Scientific Ltd. (Montreal, Canada), 5% NMS mouse serum from Dako Diagnostics Canada Inc. (Mississauga, Canada), heparin (10,000 USP units/mL) from Organon Teknika (Toronto, Canada), and bovine fluorescein isothiocyanate (FITC)-labeled albumin and 0.5% BSA from Sigma Chemicals (St. Louis, MO). The anesthetics used were ketamine from Wyeth-Ayerst (Guelph, Canada) and xylazine from Bayer (Etobicoke, Canada). The monoclonal antibodies used were hamster antimouse CD54 3-E2 (primary, 1:50 dilution) and biotin conjugated mouse antihamster IgG cocktail (secondary, 1:50 dilution) from BD Pharmigen Inc. (Mississauga, Canada). Tris buffer (50 mmol/L Tris/HCl, 150 mmol/L NaCl, pH 7.6), 0.03% H2O2 solution, buffered bicarbonate solution (BBS; NaCl 132 mmol/L, KCl 4.7 mmol/L, CaCl2 2 mmol/L, MgCl2 1.2 mmol/L, NaOH 18 mmol/L, pH 7.4), and HTS (7.5% NaCl in dH2O) were prepared in our laboratory.
Intravital Microscopy Model
Approval for these studies was obtained from the McGill University Animal Care Committee. CD1 male mice (Charles River, St. Constant, Canada), 25 to 30 g, were fed ad libitum and housed in standard care facilities for 3 to 5 days before study. Acclimated mice were anesthetized by intraperitoneal injection (xylazine 6.7 mg/kg, ketamine 13.4 mg/kg), and the right internal jugular vein and carotid artery were cannulated with polyethylene (PE)-10 tubing. The internal jugular vein catheter was used for administration of resuscitation fluids, FITC-labeled albumin, and intermittent 50-μL boluses of xylazine/ketamine solution as required to maintain anesthesia. Blood pressure was monitored continuously through a pressure transducer (Living Systems Instrumentation, Burlington, VT) connected to the carotid artery catheter.
After closing the cervical incision, the mouse cremaster was prepared for intravital microscopy as described previously. 27 Briefly, a longitudinal incision was made in the scrotal tissue, and the cremaster muscle was exteriorized and dissected free of the testicle and epididymis. The cremaster was fixed to a Plexiglas stage at five points with 4-0 silk and continuously perfused with thermostat-controlled (37°C) BBS for the remainder of the experiment. The stage was placed on a Nikon TE 300 inverted microscope (Nikon Canada, Montreal, Canada), and the tissue was imaged at 2,120× magnification.
Nonbranching postcapillary venules 20 to 40 μm in diameter were evaluated for the velocity of blood flow at their center (VRBC) using an optical Doppler velocimeter (Microcirculation Research Institute, College Station, TX) and were considered suitable only if VRBC was 2.0 to 4.0 mm/s. Live microscopic images were captured with a high-definition black-and-white video camera (CCD High Performance Camera, COHU, San Diego, CA), transferred to a monitor (Trinitron color monitor, SSM-14NE, Sony, Toronto, Canada), and recorded on videotape with a video recorder (VR564, RCA, Toronto, Canada) for subsequent analysis without knowledge of treatment groups. A video Time-Date Generator (model WJ-810, Panasonic, Toronto, Canada) projected the time, date, and stopwatch function onto the monitor at all times. A temperature probe (Bio Medic Data Systems, Seaford, DE) introduced subcutaneously after cannulation of vessels was used to measure temperature intermittently. Body temperature was maintained at 37°C with a radiant heat lamp throughout the study period. All animals were killed by xylazine/ketamine overdose followed by cervical dislocation at the completion of experiments.
Experimental Protocol and Randomization of Study Groups
Hemorrhagic shock was induced by withdrawing blood from the carotid artery catheter over 10 to 20 minutes with a tuberculin syringe previously flushed with 25 U heparin until MAP reached 50 mm Hg. Hypotension was maintained for 45 minutes, by further blood withdrawals if MAP rose above 50 mmHg, or by reinfusions of 0.05 mL of withdrawn blood if MAP fell below 40 mmHg. The syringe containing the withdrawn blood was placed in a warmed (37°C) shaker (Roto Mix, Barnstead/Thermolyne, Dubuque, IA) until reinfusion. After 45 minutes of hypotension, the animals were resuscitated using the regimen randomly assigned before anesthesia.
The 33 mice were randomized to one of three resuscitation regimens. In the HTS group, 12 mice were resuscitated with 4 cc/kg of 7.5% hypertonic saline immediately followed by the entire volume of withdrawn blood. In the RL group, 12 mice were resuscitated with RL, infusing twice the volume of withdrawn blood, immediately followed by the withdrawn blood itself. Both fluid regimens have been demonstrated to adequately resuscitate mice after such hemorrhagic shock. 23,28 The sham group, composed of nine mice, underwent vascular cannulation and cremaster preparation but was not subjected to hemorrhage or resuscitation. Once resuscitated, both RL and HTS mice were studied for sequential EC–PMN interactions using video recordings and epiluminescence. Post mortem the cremaster was prepared for immunohistochemistry as outlined below.
Quantification of EC–PMN Interactions
Seven-minute video recordings of each animal’s microvasculature were captured at the following time intervals: baseline (before hemorrhage and 30 minutes after a stabilization period following the completion of surgery), during hypotension, immediately after resuscitation (t = 0), every 15 minutes during the subsequent hour (t = 15, 30, 45, 60), and 90 minutes after resuscitation (t = 90). These recordings were subsequently played back off line and EC–PMN interactions assessed without prior knowledge of treatment.
We measured central vessel flow velocity (VRBC) using Doppler velocimetry and calculated mean velocity (VMEAN) from VRBC using the formula: VMEAN = VRBC/1.6. Shear rate (γ), in sec−1, was calculated using the formula: γ = 8 × (VRBC/DV), where DV is the venular diameter measured directly off line using calipers. Shear stress was calculated using the formula: shear stress = 0.25 × shear rate. PMNs were not labeled with fluorescent markers that could affect their adhesive properties and, as available evidence suggests, all leukocytes visualized were considered to be PMNs. 29 Neutrophil rolling was defined as the number of neutrophils crossing a line perpendicular to the long axis of the vessel that were moving at a rate slower than erythrocytes over a period of 2 minutes. Rolling velocity was calculated as the mean transit time of 10 neutrophils over a given 100-μm length of postcapillary venule and was expressed in μm/s. Neutrophil adherence was defined as the number of cells stationary for a minimum of 30 seconds in a 100-μm length of venule during a 5-minute period. Preadherence was defined as the number of immobile neutrophils in the same 100-μm vessel section at the initiation of counting. Total neutrophil adherence was the sum of neutrophil adherence and preadherence for a given sample.
Fluorescent Quantification of Vascular Permeability
Ninety minutes after resuscitation and following the completion of all video recordings, 50 mg/kg fluorescein isothiocyanate (FITC)–labeled bovine serum albumin was injected intravenously through the jugular catheter. Fifteen minutes later, epiluminescence microscopy was performed using a fluorescence source powered by a high-pressure mercury lamp (HB 10103AF, Nikon Canada, Toronto, Canada). Using a high-definition black-and-white video camera (CCD High Performance Camera, COHU, San Diego, CA), moving images were transferred to a computer (Dimension XPS B866r, Dell Canada, Toronto, Canada) and captured by a frame grabber from Scion Image processing software (Scion Image for Windows, Scion Corp., Frederick, MD). Using a frozen digital image of the fluorescent postcapillary venule, gray levels (0 [black] to 256 [white]) were measured in three equal areas within the venule (venular intensity, Iv), as well as three separate equal areas in the perivenular space (perivenular intensity, Ip) (Fig. 1). Using the means of Ip and Iv measurements, vascular leakage (permeability index [PI]) was calculated with the formula Ip/Iv =PI.
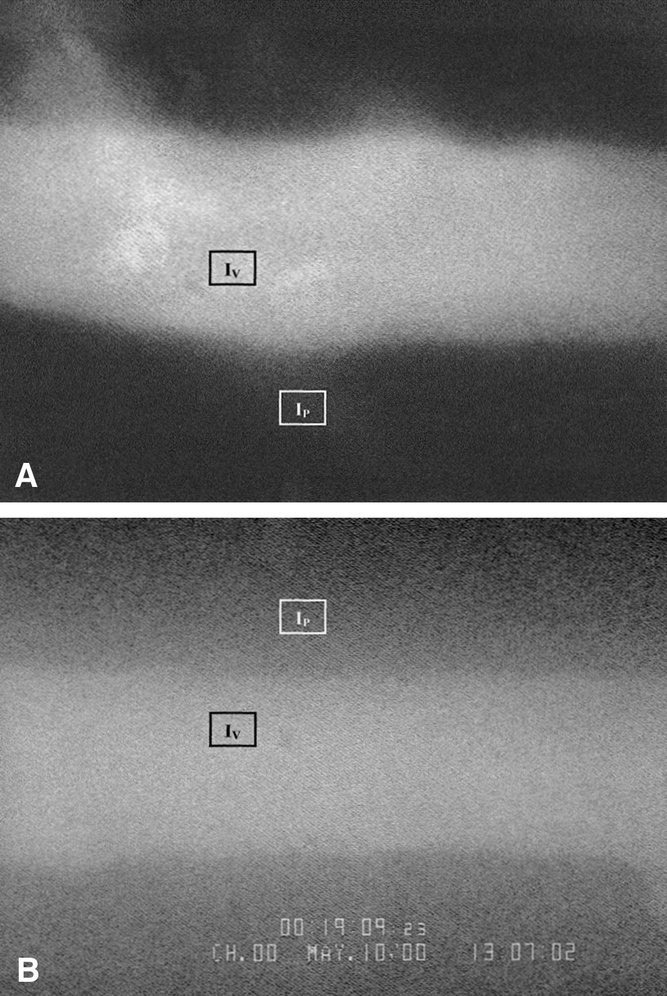
Figure 1. Representative examples of fluorescent intravital microscopic images contrasting the low leakage of FITC-labeled albumin from postcapillary venules in HTS (A) compared with RL (B) animals. IP (perivenular light intensity) and IV (venular light intensity) represent regions evaluated for fluorescence.
Cremaster EC Expression of ICAM-1
Following all recordings and fluorescent evaluations and before sacrifice, the cremaster was delicately resected, placed in embedding medium, snap-frozen in isopentane submerged in liquid nitrogen, and stored at −80°C. Five-micrometer-thick frozen sections were cut on a cryostat, stained with hamster antimouse ICAM-1 mAb 3E2, washed, and stained with biotinylated mouse antihamster IgG cocktail. After further rinses with Tris-buffer, the slides were developed using a standard DAB detection kit (Ventana Basic DAB Detection Kit, Ventana Medical Systems Inc., Tucson, AZ) and counterstained with hematoxylin for 4 minutes. After mounting, the slides were examined at a magnification of 250× and graded semiquantitatively for intensity of vascular endothelial cell ICAM-1 expression by one of us (R.P.M.) without prior knowledge of treatment group, using the following scale: no staining = 0, weak staining = 1, moderate staining = 2, and strong staining = 3. Ten vessels per slide were graded and a mean grade was calculated for each animal. 30
Systemic Total White Blood Cell and PMN Counts
Four hundred microliters of blood removed by cardiac puncture before animal sacrifice was used for hemoglobin, white blood cell count, and differential determination. The Royal Victoria Hospital hematology laboratories performed these analyses using an Advia Hematology System (model 120, Bayer Systems, Hialeah, FL).
Statistical Analysis
All data are presented as mean ± standard error of the mean unless otherwise stated. Differences between groups were compared using analysis of variance with Bonferroni correction with Systat 8 data analysis software (SPSS, Chicago, IL). P < .05 was deemed statistically significant.
RESULTS
Vascular and Hemodynamic Mechanics
Measurements of VRBC, VMEAN, shear rates, and shear velocities in the three groups and at all time intervals were not statistically different (P > .05) (Table 1, to be considered for deposition at the National Auxiliary Publications Service). All groups displayed similar PMN rolling, PMN rolling velocity, and PMN adherence at baseline as well as during hypotension (Figs. 2, 3, and 4). Postcapillary venular diameter (range 20.0–39.0 μm, mean 27.11 ± 0.23 μm), duration of hypotension (mean 45.1 ± 3.2 minutes), withdrawn blood volume (mean 485.3 ± 81.56 μL), and postmortem hemoglobin values (mean 111.6 ± 34.1 g/L) were similar in all groups (P > .05 for all comparisons). RL-resuscitated animals took longer to resuscitate than HTS animals (20.3 ± 7.5 vs. 12.6 ± 4.2 minutes, P = .01), but no differences between groups were found in the time it took to hemorrhage animals (mean 7.93 ± 3.9 minutes). HTS and RL groups displayed similar blood pressures (P > .05) at all time points, but the mice in the sham group had significantly higher blood pressures than both HTS and RL animals during the hypotension time point and onwards from 30 minutes following resuscitation (Fig. 5).
Table 1. MEASUREMENTS OF VASCULAR AND HEMODYNAMIC MECHANICS

Except for the hypotensive period, central flow velocity (VRBC), mean flow velocity (VMEAN), vessel shear rates, and shear stress showed no statistical differences between groups. All results are shown as mean ± SD, and unless otherwise stated, P > .05 for all comparisons between groups.
** Sham vs either RL or HTS:P < .01.
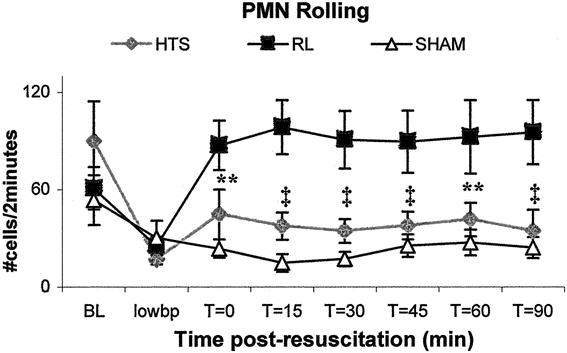
Figure 2. RL animals displayed more rolling neutrophils than either HTS or sham groups. Values are expressed as mean number of rolling neutrophils per 2-minute period ± SEM. **RL versus sham only, P < .05. ‡RL versus either HTS or sham, P < .05. There were no significant differences between HTS and sham at any time point.
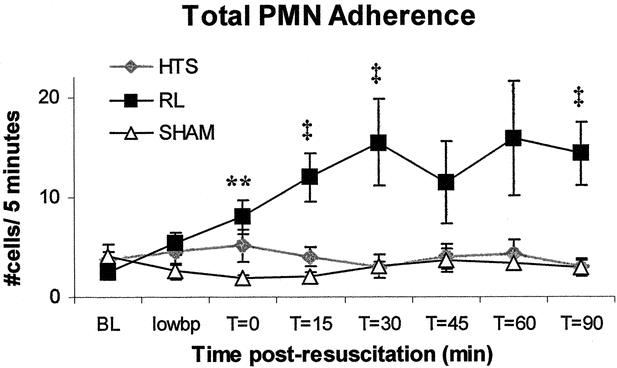
Figure 3. As compared to HTS and sham, RL animals had higher numbers of neutrophils adhering to endothelial cells. Values are expressed as mean number of stationary PMNs on a given 100-μm vessel length ± SEM. **RL versus sham only, P < .05. ‡RL versus either HTS or sham, P < .05. There were no significant differences between HTS and sham at any time point.
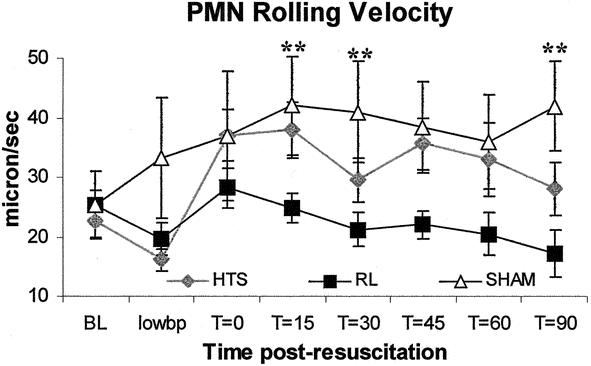
Figure 4. HTS animals tended to have higher PMN rolling velocities than RL aniimals. Values are expressed as the mean velocity (μm/s) of 10 PMNs crossing a given vessel section ± SEM. **RL versus sham only, P < .05. There were no significant differences between HTS and sham at any time point.
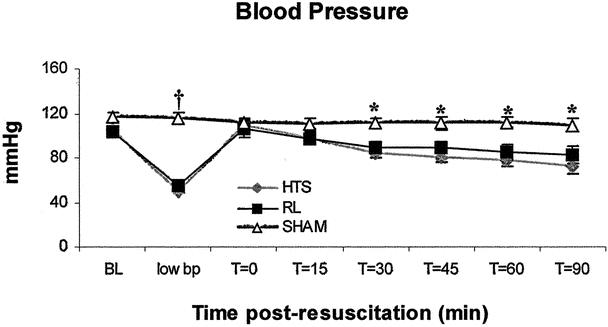
Figure 5. Compared to the two resuscitation groups, sham animals had a significantly higher blood pressure 30 minutes after resuscitation and thereafter. Values are expressed as mean (mmHg) ± SEM. *Sham versus either HTS or RL, P < .05. †Sham versus either HTS or RL, P < .001. There were no significant differences between HTS and RL at any time point.
Effect of Hypertonic Saline on PMN Rolling and Adhesion to EC
HTS-resuscitated animals displayed less than half the number of rolling neutrophils seen in RL-resuscitated animals at all time points following resuscitation. RL-resuscitated animals had a threefold greater number of rolling neutrophils than animals in the sham group (Figs. 2 and 6). PMN rolling was similar in the sham and HTS groups (P > .05 at all time points). Total neutrophil adherence to endothelium was similar in the HTS and sham groups throughout the experiments, but after 15 minutes adherence levels in RL-resuscitated animals increased significantly to almost four times those observed in the sham and HTS groups at t = 60 (Figs. 3 and 6). PMN rolling velocity tended to be greater in both HTS and sham groups compared to RL at all time points following resuscitation (Fig. 6). Although during hypotension both resuscitated groups had neutrophil rolling velocities that were half those of sham animals, these did not differ significantly between HTS and sham animals in the remaining time points.
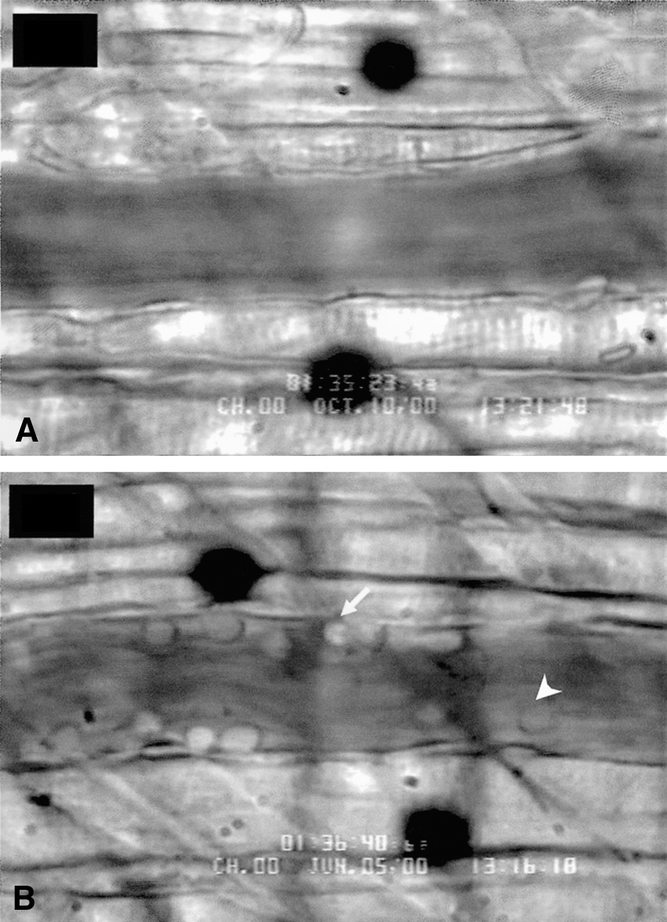
Figure 6. Representative examples of intravital microscopic images contrasting the lack of PMN rolling (arrowhead) and PMN adherence (arrow) in HTS (A) as compared to RL (B) vessels 90 minutes after resuscitation.
Macromolecular Leakage in HTS-Resuscitated Animals
Vascular permeability (PI) was slightly higher in HTS than in sham mice (36% ± 0.02 and 27% ± 0.02, P = .03), but both groups displayed significantly less leakage than RL mice (66% ± 0.2, P < .01 vs. either HTS or sham) (Figs. 1 and 7).
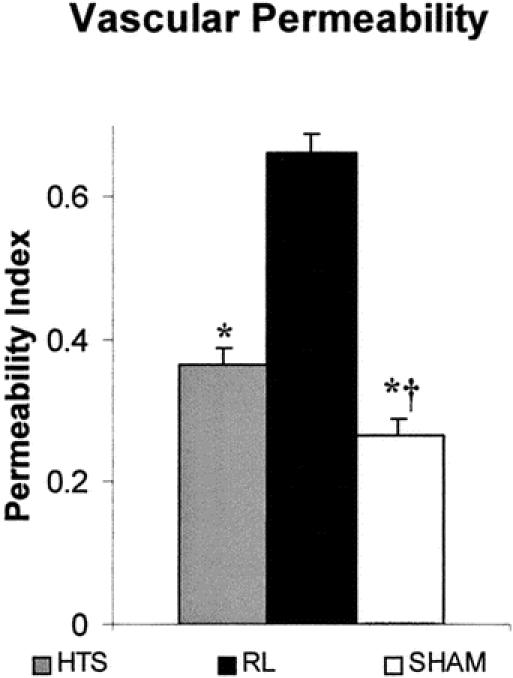
Figure 7. Vascular permeability index (PI) (where IP/IV = PI) is an estimation of vessel leakage and was higher in HTS as compared to sham animals, while RL animals had the highest PI of all. Values are expressed as % permeability index ± SEM. †Versus HTS, P < .05. *Versus RL, P < .01.
Cremaster ICAM-1 Expression in Resuscitation Groups
Cremasteric muscle vascular EC expression of ICAM-1 was similar in both HTS and RL animals (mean grade 1.69 ± 0.3 and 1.82 ± 0.2, respectively, P > .05).
Total White Blood Cell and PMN Counts
Both circulating leukocyte (P < .05) and neutrophil counts (P = .06) tended to be higher in HTS animals (72.1 ± 31.4 and 1.98 ± 0.86, respectively) than in both sham (3.16 ± 0.36 and 0.14 ± 0.08, respectively) and RL animals (14.9 ± 10.14 and 0.28 ± 0.12, respectively). The proportion of circulating leukocytes that were PMNs was similar in all groups (data not shown).
DISCUSSION
For the last two decades HTS has become an increasingly popular resuscitation fluid, showing a trend to increased posttraumatic survival both in the early phase (<12 hours after trauma) and in the late phase (days to weeks after trauma). 4,6,7,31–33 The early phase effect is believed to be due to a functional increase in cardiac preload, primarily by inducing an osmotic fluid shift from the cells and interstitium into the vasculature and leading to increases in systemic blood pressure and cardiac output as well as decreases in small vessel capacitance. 4,31 In contrast, the late beneficial effects of HTS resuscitation are unlikely to be due to simple physiologic alterations. The late morbidity and mortality of traumatic hemorrhage have been attributed to the “second hit”; that is, a relatively less important inflammatory event (pneumonia, aspiration, minor surgery) that occurs days to weeks after the traumatic injury and initiates progression to the systemic inflammatory response syndrome (SIRS), organ failure, and eventual death. 34 Clinical and animal studies conducted in the past decade have provided strong evidence suggesting that this late effect of HTS resuscitation is due to an immune modulation of leukocytes, in particular neutrophils. 21,35–37
We believe that the present study provides novel evidence demonstrating that HTS alters interactions between neutrophils and ECs. In the present study, using a “single-hit” murine model of hemorrhagic shock, resuscitation with HTS specifically diminished the number of rolling and adherent PMNs while tending to increase PMN rolling velocities; there were no differences in these parameters between HTS-treated mice and the sham controls.
The effect of hypertonic saline on PMN–EC interactions has been studied previously. In vitro studies of cultured endothelial monolayers demonstrate diminished PMN adherence, 15 whereas in vivo models of reperfused local pressure-induced ischemia display fewer rolling and “sticking” venular PMNs after pretreatment with HTS. 38 Using intravital microscopy of pial windows, HTS administration after cerebral percussion injury or systemic thermal injury also reduced EC–PMN interactions. 39,40 Liver intravital microscopy has probably yielded the most exciting findings to date about EC–PMN interactions following the differential fluid resuscitation of hemorrhagic shock. Indeed, several studies report decreases in capillary and sinusoidal luminal narrowing and improved flow parameters, 25,41–43 as well as diminished EC–PMN interactions (sinusoids and postsinusoidal venules) in animals resuscitated with hypertonic fluids. 24–26 Because none of these studies evaluated the effects of HTS alone, but rather used HTS-sugar resuscitation regimens, it is difficult to reach a conclusion on the individual effect of HTS. Dextrans and other sugars have been shown to exert by themselves powerful antiadhesive influences on PMNs. 44–46 Furthermore, these studies in hepatic tissue employ fluorescent labeling of PMNs (rhodamine, acridine orange), even though such fluorescent markers are also known to affect the adhesive capacities of PMNs. 47 Moreover, as liver sinusoids are not lined by typical endothelium, it may be inappropriate to apply findings from the hepatic microvasculature to the rest of the systemic vasculature. An additional difference between the hepatic and systemic microvasculatures is that in the former, the possible “trapping” of swollen and stiff PMNs may occur due to the narrowing of sinusoidal passages following crystalloid resuscitation. 48–50 Thus, the unique liver microvascular architecture may not be representative of other organs, as PMNs may arrest and be sequestered by mechanisms other than standard transmigration.
Although knowledge about PMN–EC interactions is extensive, there is less evidence demonstrating an associated HTS-mediated decrease in vascular permeability following hemorrhagic shock resuscitation. HTS given after local pressure-induced ischemia reduced in vivo fluorescent dextran leakage, which was correlated with diminished EC–PMN interactions. 38 A rodent cheek pouch intravital microscopic study evaluating vascular leakage in systemic endotoxic shock reported diminished EC–PMN interactions and leakage after HTS resuscitation. 51 Following the resuscitation of hemorrhagic shock, postmortem studies revealed less radioactive albumin leakage into pulmonary tissue if HTS was used for resuscitation instead of RL. 15,52 The present study represents the first attempt at characterizing in vivo postcapillary venous permeability following resuscitation of systemic hemorrhagic shock. Leakage of labeled albumin was evaluated with epifluorescence microscopy and revealed almost 50% less leakage if HTS was used for resuscitation instead of RL. This enhanced vascular leakage in RL animals rapidly reached a plateau after 15 to 30 minutes, whereas HTS animals never showed a significant increase in vascular permeability. Although we cannot establish a direct causal relationship between HTS-mediated decreases in EC–PMN interactions and vascular leakage, we believe that we have demonstrated at least a strong association.
Much evidence exists supporting HTS-mediated modulation of PMN and EC adhesion molecule expression. The in vitro activation of PMNs from HTS-resuscitated animals and humans results in diminished L-selectin 15,20 and CD11b 15,18,19,37,53 expression on the PMN surface. Diminished in vitro endothelial ICAM-1 protein and mRNA expression has been reported when LPS activation was followed by incubation in HTS. 54 In vivo studies evaluating pulmonary and hepatic ICAM-1 mRNA expression in HTS-resuscitated animals have also demonstrated diminished receptor levels as compared to RL controls. 15,54 Moreover, in another in vivo study, Sun et al. 22 found diminished splenic ICAM-1 protein expression and diminished pulmonary and splenic ICAM-1 gene expression in HTS-treated as compared to RL-treated rats. Our experiments, however, did not demonstrate differences in ICAM-1 expression between the different resuscitation groups. This could indicate that this adhesion receptor may not be the principal target of HTS-mediated effects, or that our model did not provide a sufficiently strong activating stimulus to produce such effects in the cremaster muscle. A “two-hit” model of hemorrhage followed by an infectious insult, 15,23 for example, may have allowed for greater PMN activation and enabled us to demonstrate differential adhesion molecule expression. Animal ICAM-1 knockout models and adhesion molecule blockade may be more definite methods of establishing the true effects of HTS on ICAM-1 or other adhesion molecules.
Resuscitation of hemorrhagic shock, particularly when using RL, has also been associated with reductions in circulating leukocytes. 20 Certain authors suggest that this may be the result of increased interactions of PMNs with the vessel wall. 21 Consequently, it could be argued that fewer EC–PMN interactions secondary to HTS resuscitation explain higher circulating neutrophil levels. The present study confirmed a tendency to neutrophilia in HTS-resuscitated animals and a relative neutropenia in RL-resuscitated animals.
There are a few potential sources of error in our resuscitated hemorrhagic shock model. Since we did not label the circulating PMNs, and since other leukocytes 55 are known to interact with endothelium, we cannot be certain that all interacting leukocytes were neutrophils; nevertheless, studies of interacting leukocytes in similar models indicate that the vast majority of such visible leukocytes are indeed PMNs. 29 Animals in the RL group were resuscitated with volumes 10-fold greater than the HTS animals in addition to reinfused blood (mean 1.12 ± 0.12 vs. 0.13 ± 0.003 mL, respectively, P < .001). This large intravascular fluid challenge may have effectively fluid-overloaded the animals resuscitated with RL, at least initially, so that an additional detrimental mechanical effect on the vessel wall of these animals cannot be entirely discounted. Moreover, related to the different resuscitation volumes, RL animals took longer to resuscitate than HTS animals, effectively prolonging the duration of hypotension and possibly of ischemia. In contrast, HTS and RL but not sham animals displayed a gradually decreasing blood pressure 30 minutes following resuscitation (Fig. 5). We did not evaluate the adequacy of resuscitation in either group, and the question of whether both groups were equally well resuscitated could be raised. Nonetheless, differences in PMN rolling and adhesion were already seen before this time point, beginning shortly after resuscitation and indicating an early differential effect. Furthermore, protocols using resuscitation with shed blood and once/twice that volume of RL or 4 cc/kg 7% HTS, in addition to shed blood, have been used by several other groups on different rodent models. 15,21,23
In summary, compared to RL, HTS resuscitation of a single-hit hemorrhagic shock murine model results in faster-moving postcapillary venular neutrophils, with diminished rolling and adherence to ECs. Furthermore, RL-resuscitated animals displayed an early and sustained elevation in vascular leakage in vivo that was not observed in HTS-resuscitated animals. These novel findings further contribute to the understanding of the natural history of hemorrhagic shock and the sequential steps that can lead its victims to organ failure and subsequent death. The future use of HTS resuscitation and its immunomodulation following hemorrhagic shock may potentially reduce PMN-mediated tissue injury leading to organ failure in trauma victims and severely ill patients.
Acknowledgments
The authors thank Ms. Carmen Loiselle and Mr. David Hori for help in conducting experiments and Ms. Mary Bouldadakis and Mrs. Dominique Charette in the preparation of the manuscript.
Footnotes
Supported in part by Medical Research Council, research grant # MT-12937 from the Medical Council of Canada. J.L.P. is the recipient of the 2000 Surgical Infection Society/Merck & Co. Resident Research Fellowship.
Correspondence: Nicolas V. Christou, MD, PhD, FRCS(C), FACS, FCCM, McGill University Health Center, Royal Victoria Hospital, Room C5.53, 687 Pine Avenue West, Montreal, Quebec H3A 1A1, Canada.
E-mail: nicolas.christou@muhc.mcgill.ca
Accepted for publication February 18, 2002.
References
- 1.Velanovich V. Crystalloid versus colloid fluid resuscitation: a meta-analysis of mortality. Surgery 1989; 105: 65–71. [PubMed] [Google Scholar]
- 2.Nakayama S, Sibley L, Gunther RA, et al. Small-volume resuscitation with hypertonic saline (2,400 mOsm/liter) during hemorrhagic shock. Circ Shock 1984; 13: 149–159. [PubMed] [Google Scholar]
- 3.Shackford SR, Bourguignon PR, Wald SL, et al. Hypertonic saline resuscitation of patients with head injury: a prospective, randomized clinical trial. J Trauma 1998; 44: 50–58. [DOI] [PubMed] [Google Scholar]
- 4.Mattox KL, Maningas PA, Moore EE, et al. Prehospital hypertonic saline/dextran infusion for post-traumatic hypotension. The U.S.A. Multicenter Trial. Ann Surg 1991; 213: 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velasco IT, Pontieri V, Rocha e Silva M Jr, et al. Hyperosmotic NaCl and severe hemorrhagic shock. Am J Physiol 1980; 239: H664–673. [DOI] [PubMed] [Google Scholar]
- 6.Kreimeier U, Thiel M, Peter K, et al. Small-volume hyperosmolar resuscitation. Acta Anaesthesiol Scand Suppl 1997; 111: 302–306. [PubMed] [Google Scholar]
- 7.Wade CE, Kramer GC, Grady JJ, et al. Efficacy of hypertonic 7.5% saline and 6% dextran-70 in treating trauma: a meta-analysis of controlled clinical studies. Surgery 1997; 122: 609–616. [DOI] [PubMed] [Google Scholar]
- 8.Botha AJ, Moore FA, Moore EE, et al. Early neutrophil sequestration after injury: a pathogenic mechanism for multiple organ failure. J Trauma 1995; 39: 411–417. [DOI] [PubMed] [Google Scholar]
- 9.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med 1989; 320: 365–376. [DOI] [PubMed] [Google Scholar]
- 10.Botha AJ, Moore FA, Moore EE, et al. Base deficit after major trauma directly relates to neutrophil CD11b expression: a proposed mechanism of shock-induced organ injury. Intens Care Med 1997; 23: 504–509. [DOI] [PubMed] [Google Scholar]
- 11.Bevilacqua MP, Nelson RM. Selectins. J Clin Invest 1993; 91: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Etzioni A. Integrins–the glue of life. Lancet 1999; 353: 341–343. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed N, Christou N. Systemic inflammatory response syndrome: interactions between immune cells and the endothelium. Shock 1996; 6 (Suppl 1): S39–42. [PubMed] [Google Scholar]
- 14.Cochrane CG, Aikin BS. Polymorphonuclear leukocytes in immunologic reactions. The destruction of vascular basement membrane in vivo and in vitro. J Exp Med 1966; 124: 733–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizoli SB, Kapus A, Fan J, et al. Immunomodulatory effects of hypertonic resuscitation on the development of lung inflammation following hemorrhagic shock. J Immunol 1998; 161: 6288–6296. [PubMed] [Google Scholar]
- 16.Hampton MB, Chambers ST, Vissers MC, et al. Bacterial killing by neutrophils in hypertonic environments. J Infect Dis 1994; 169: 839–846. [DOI] [PubMed] [Google Scholar]
- 17.Ciesla DJ ME, Zallen G, Biffl WL, et al. Hypertonic saline attenuation of neutrophil cytotoxic function is reversed upon return to normotonicity. Surg Forum 1999;:189–191.
- 18.Ciesla DJ, Moore EE, Zallen G, et al. Hypertonic saline attenuation of polymorphonuclear neutrophil cytotoxicity: timing is everything. J Trauma 2000; 48: 388–395. [DOI] [PubMed] [Google Scholar]
- 19.Rhee P, Wang D, Ruff P, et al. Human neutrophil activation and increased adhesion by various resuscitation fluids. Crit Care Med 2000; 28: 74–78. [DOI] [PubMed] [Google Scholar]
- 20.Angle N, Hoyt DB, Cabello-Passini R, et al. Hypertonic saline resuscitation reduces neutrophil margination by suppressing neutrophil L selectin expression. J Trauma 1998; 45: 7–12. [DOI] [PubMed] [Google Scholar]
- 21.Angle N, Hoyt DB, Coimbra R, et al. Hypertonic saline resuscitation diminishes lung injury by suppressing neutrophil activation after hemorrhagic shock. Shock 1998; 9: 164–170. [DOI] [PubMed] [Google Scholar]
- 22.Sun LL, Ruff P, Austin B, et al. Early up-regulation of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression in rats with hemorrhagic shock and resuscitation. Shock 1999; 11: 416–422. [PubMed] [Google Scholar]
- 23.Coimbra R, Hoyt DB, Junger WG, et al. Hypertonic saline resuscitation decreases susceptibility to sepsis after hemorrhagic shock. J Trauma 1997; 42: 602–606. [DOI] [PubMed] [Google Scholar]
- 24.Bauer M, Marzi I, Ziegenfuss T, et al. Comparative effects of crystalloid and small volume hypertonic hyperoncotic fluid resuscitation on hepatic microcirculation after hemorrhagic shock. Circ Shock 1993; 40: 187–193. [PubMed] [Google Scholar]
- 25.Vollmar B, Lang G, Menger MD, et al. Hypertonic hydroxyethyl starch restores hepatic microvascular perfusion in hemorrhagic shock. Am J Physiol 1994; 266 (5 Pt 2):H1927–1934. [DOI] [PubMed] [Google Scholar]
- 26.Corso CO, Okamoto S, Ruttinger D, et al. Hypertonic saline dextran attenuates leukocyte accumulation in the liver after hemorrhagic shock and resuscitation. J Trauma 1999; 46: 417–423. [DOI] [PubMed] [Google Scholar]
- 27.Baez S. An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res 1973; 5: 384–394. [DOI] [PubMed] [Google Scholar]
- 28.Coimbra R, Junger WG, Hoyt DB, et al. Hypertonic saline resuscitation restores hemorrhage-induced immunosuppression by decreasing prostaglandin E2 and interleukin-4 production. J Surg Res 1996; 64: 203–209. [DOI] [PubMed] [Google Scholar]
- 29.Tangelder GJ, Janssens CJ, Slaaf DW, et al. In vivo differentiation of leukocytes rolling in mesenteric postcapillary venules. Am J Physiol 1995; 268: H909–915. [DOI] [PubMed] [Google Scholar]
- 30.Giaid A, Michel RP, Stewart DJ, et al. Expression of endothelin-1 in lungs of patients with cryptogenic fibrosing alveolitis. Lancet 1993; 341: 1550–1554. [DOI] [PubMed] [Google Scholar]
- 31.Vassar MJ, Fischer RP, O’Brien PE, et al. A multicenter trial for resuscitation of injured patients with 7.5% sodium chloride. The effect of added dextran 70. The Multicenter Group for the Study of Hypertonic Saline in Trauma Patients. Arch Surg 1993; 128: 1003–1013. [DOI] [PubMed] [Google Scholar]
- 32.Holcroft JW, Vassar MJ, Perry CA, et al. Perspectives on clinical trials for hypertonic saline/dextran solutions for the treatment of traumatic shock. Braz J Med Biol Res 1989; 22: 291–293. [PubMed] [Google Scholar]
- 33.Vassar MJ, Perry CA, Gannaway WL, Holcroft JW. 7.5% sodium chloride/dextran for resuscitation of trauma patients undergoing helicopter transport. Arch Surg 1991; 126: 1065–1072. [DOI] [PubMed] [Google Scholar]
- 34.Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am 1995; 75: 257–277. [DOI] [PubMed] [Google Scholar]
- 35.Smith RM, Giannoudis PV. Trauma and the immune response. J R Soc Med 1998; 91: 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Junger WG, Hoyt DB, Davis RE, et al. Hypertonicity regulates the function of human neutrophils by modulating chemoattractant receptor signaling and activating mitogen-activated protein kinase p38. J Clin Invest 1998; 101: 2768–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angle N, Cabello-Passini R, Hoyt DB, et al. Hypertonic saline infusion: can it regulate human neutrophil function? Shock 2000; 14: 503–508. [PubMed] [Google Scholar]
- 38.Nolte D, Bayer M, Lehr HA, et al. Attenuation of postischemic microvascular disturbances in striated muscle by hyperosmolar saline dextran. Am J Physiol 1992; 263: H1411–1416. [DOI] [PubMed] [Google Scholar]
- 39.Hartl R, Medary MB, Ruge M, et al. Hypertonic/hyperoncotic saline attenuates microcirculatory disturbances after traumatic brain injury. J Trauma 1997; 42: S41–47. [DOI] [PubMed] [Google Scholar]
- 40.Barone M, Jimenez F, Huxley VH, et al. Morphologic analysis of the cerebral microcirculation after thermal injury and the response to fluid resuscitation. Acta Neurochir Suppl 1997; 70: 267–268. [DOI] [PubMed] [Google Scholar]
- 41.Mazzoni MC, Borgstrom P, Arfors KE, et al. Dynamic fluid redistribution in hyperosmotic resuscitation of hypovolemic hemorrhage. Am J Physiol 1988; 255: H629–637. [DOI] [PubMed] [Google Scholar]
- 42.Mazzoni MC, Borgstrom P, Warnke KC, et al. Mechanisms and implications of capillary endothelial swelling and luminal narrowing in low-flow ischemias. Int J Microcirc Clin Exp 1995; 15: 265–270. [DOI] [PubMed] [Google Scholar]
- 43.Corso CO, Okamoto S, Leiderer R, et al. Resuscitation with hypertonic saline dextran reduces endothelial cell swelling and improves hepatic microvascular perfusion and function after hemorrhagic shock. J Surg Res 1998; 80: 210–220. [DOI] [PubMed] [Google Scholar]
- 44.Messmer K. Capillary Functions and White Cell Interaction. Basel, New York: Karger, 1991.
- 45.Steinbauer M, Harris AG, Messmer K. Effects of dextran on microvascular ischemia-reperfusion injury in striated muscle. Am J Physiol 1997; 272: H1710–1716. [DOI] [PubMed] [Google Scholar]
- 46.Matsumiya A, Yamaguchi M, Nakano H, et al. Dextran sulfate inhibits E-selectin-mediated neutrophil adhesion to endotoxin-activated vascular endothelial cells. Life Sci 1999; 64: L9–17. [DOI] [PubMed] [Google Scholar]
- 47.Saetzler RK, Jallo J, Lehr HA, et al. Intravital fluorescence microscopy: impact of light-induced phototoxicity on adhesion of fluorescently labeled leukocytes. J Histochem Cytochem 1997; 45: 505–513. [DOI] [PubMed] [Google Scholar]
- 48.McCuskey RS, Reilly FD. Hepatic microvasculature: dynamic structure and its regulation. Semin Liver Dis 1993; 13: 1–12. [DOI] [PubMed] [Google Scholar]
- 49.Bauer M, Zhang JX, Bauer I, et al. ET-1 induced alterations of hepatic microcirculation: sinusoidal and extrasinusoidal sites of action. Am J Physiol 1994; 267: G143–149. [DOI] [PubMed] [Google Scholar]
- 50.Nishida J, McCuskey RS, McDonnell D, et al. Protective role of NO in hepatic microcirculatory dysfunction during endotoxemia. Am J Physiol 1994; 267: G1135–1141. [DOI] [PubMed] [Google Scholar]
- 51.de Carvalho H, Matos JA, Bouskela E, et al. Vascular permeability increase and plasma volume loss induced by endotoxin was attenuated by hypertonic saline with or without dextran. Shock 1999; 12: 75–80. [DOI] [PubMed] [Google Scholar]
- 52.Moon PF, Hollyfield-Gilbert MA, Myers TL, et al. Fluid compartments in hemorrhaged rats after hyperosmotic crystalloid and hyperoncotic colloid resuscitation. Am J Physiol 1996; 270: F1–8. [DOI] [PubMed] [Google Scholar]
- 53.Rizoli SB, Kapus A, Parodo J, et al. Hypertonic immunomodulation is reversible and accompanied by changes in CD11b expression. J Surg Res 1999; 83: 130–135. [DOI] [PubMed] [Google Scholar]
- 54.Oreopoulos GD, Hamilton J, Rizoli SB, et al. In vivo and in vitro modulation of intercellular adhesion molecule (ICAM)-1 expression by hypertonicity. Shock 2000; 14: 409–415. [DOI] [PubMed] [Google Scholar]
- 55.Broide DH, Stachnick G, Castaneda D, et al. Inhibition of eosinophilic inflammation in allergen-challenged TNF receptor p55/p75- and TNF receptor p55-deficient mice. Am J Respir Cell Mol Biol 2001; 24: 304–311. [DOI] [PubMed] [Google Scholar]