

Abstract
Objective
To evaluate the prevalence of mutations in the CDKN2A gene encoding p16INK4a and p14ARF in familial pancreatic cancer (FPC).
Summary Background Data
The genetic basis of FPC is still widely unknown. Recently, it has been shown that germline mutations in the p16INK4a tumor suppressor gene can predispose to pancreatic cancer. The presence of p14ARF germline mutations has yet not been determined in this setting.
Methods
Eighteen families with at least two first-degree relatives with histologically confirmed pancreatic cancer and five families with at least one patient with pancreatic cancer and another first-degree relative with malignant melanoma of the German National Case Collection for Familial Pancreatic Cancer were analyzed for CDKN2A germline mutations including p16INK4a and p14ARF by direct DNA sequencing. All participating family members were genetically counseled and evaluated by a three-generation pedigree.
Results
None of 18 FPC families without malignant melanoma revealed p16INK4a mutations, compared to 2 of 5 families with pancreatic cancer and melanoma. Truncating p16INK4a germline mutations Q50X and E119X were identified in the affected patients of pancreatic cancer plus melanoma families. None of the 23 families revealed p14ARF germline mutations.
Conclusions
CDKN2A germline mutations are rare in FPC families. However, these data provide further evidence for a pancreatic cancer–melanoma syndrome associated with CDKN2A germline mutations affecting p16INK4a. Thus, all members of families with combined occurrence of pancreatic cancer and melanoma should be counseled and offered screening for p16INK4a mutations to identify high-risk family members who should be enrolled in a clinical screening program.
In Germany about 11,000 patients develop ductal pancreatic adenocarcinoma each year, making it the fifth most common cause of cancer death. 1 The prognosis is dismal, with an overall 5-year survival rate of less than 5%; only 20% of patients have potentially curative resectable tumors at diagnosis. However, in patients with early-stage tumors, 5-year survival rates of up to 40% can be achieved. 2 Thus, the identification of patients at risk for this disease is an important goal to reduce mortality. It has been estimated that about 3% to 10% of patients with pancreatic cancer have an inherited predisposition for this tumor, although conclusive epidemiologic data are still lacking. 3–5 Based on clinical criteria, patients with inherited pancreatic cancer can be separated in two groups. 6,7 One group includes families with an accumulation of PC only (familial pancreatic cancer [FPC]). The underlying gene defect in these families is unknown. The other group consists of families with inherited tumor syndromes or diseases predisposing them to various degrees to pancreatic cancer. These syndromes include hereditary pancreatitis, 8 Peutz-Jeghers syndrome, 9 ataxia-teleangiectasia, 10 hereditary nonpolyposis colon cancer (HNPCC or Lynch II-syndrome 11), Li-Fraumeni syndrome, 12 Gardner syndrome, 13 hereditary breast and ovarian cancer, 14 and familial atypical multiple mole melanoma (FAMMM). 15 The cumulative risk for the development of pancreatic cancer until the age of 70 years in these syndromes varies between 3% and 40%.
The CDKNA gene localized at chromosome 9p21 encodes the cyclin-dependent kinase inhibitor p16INK4a (MTS1) and the p53 activator p14ARF. Both gene products have an independent first exon (exon 1-alpha and exon 1-beta, respectively) but share exons 2 and 3 and are translated in different reading frames. The genes are involved in the negative control of cell proliferation. p16INK4a produces a G1 cell-cycle arrest by inhibiting phosphorylation of the retinoblastoma protein, and p14ARF acts both at G1/S and G2/M phases in a p53-dependent manner via binding and inhibition of the protein MDM2. 16,17
p16INK4 is inactivated in 95% of sporadic pancreatic cancers, 18 indicating its important role in the tumorigenesis of this disease. It has also been shown that p16INK4a germline mutations contribute to the familial accumulation of pancreatic cancer and melanoma. In 1995 Whelan et al 19 identified a p16INK4a germline mutation in a family with an excess of pancreatic cancer and malignant melanoma. FAMMM is an autosomal dominant inherited disorder associated with p16INK4a germline mutations; it predisposes mutation carriers to multiple atypical nevi and multiple malignant melanomas. In some FAMMM kindreds a high prevalence of pancreatic cancer has been observed. Goldstein et al 20 suggested dividing FAMMM kindreds in two groups with respect to their mutation status of the p16INK4a tumor suppressor gene: p16INK4a mutation negative kindreds, without the occurrence of pancreatic cancer, and p16INK4a mutation positive kindreds, with a significant excess of pancreatic cancer. It was estimated that p16INK4a mutation-associated FAMMM family members have an 22-fold risk for the development of pancreatic cancer. 20 Moreover, Moskaluk et al recently identified a p16INK4a germline mutation in a family having two first-degree relatives with pancreatic cancer, not showing the FAMMM phenotype. 21 Recently, a germline CDKN2a mutation involving p14ARF was identified in an individual with multiple primary melanomas, 22 but the incidence of p14ARF germline mutations in FPC is not known.
Due to the few available data, the role of p16INK4a and p14ARF mutations in the setting of FPC is still not well defined. Therefore, we analyzed FPC families and families with an accumulation of pancreatic cancer and malignant melanoma from the German National Case Collection for Familial Pancreatic Cancer of the Deutsche Krebshilfe (FaPaCa) for the presence of CDKN2A germline mutations including p16INK4a and p14ARF.
METHODS
Eighteen FPC families from the FaPaCa with at least two first-degree relatives with histologically confirmed ductal adenocarcinoma of the pancreas were included in the study. In addition, five families with at least one patient with histologically confirmed pancreatic cancer and at least one first-degree relative with histologically confirmed malignant melanoma were also analyzed, since they might represent families with a pancreatic cancer–melanoma syndrome (PCMS). Family members younger than 18 years of age were not included. Data were collected during genetic counseling after informed consent of the index person and family members. For genetic counseling, most index persons and relatives were visited at home by an experienced clinical geneticist (M.S.-F.). For each family, a complete three-generation pedigree was prepared. All tumors and other diseases, including age of onset, were documented. Clinical diagnoses reported by patients and family members were verified by consulting the original medical records. In addition, each participating family member was evaluated by a standardized 115-item questionnaire that was focused especially on sociodemographics, family and medical history, eating and drinking habits, and risk factors. A blood sample was drawn from all participating family members for DNA extraction, and cancer tissues were collected from affected family members whenever available. All patient materials and records were assessed following a study protocol approved by the Ethics Committee of the Philipps-University Marburg.
CDKN2A Mutation Analysis
Constitutional genomic DNA of participating family members was isolated from peripheral blood leukocytes and frozen tumors using the QIAamp DNA kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. DNA of paraffin-embedded carcinomas was extracted via a modified protocol of Wright and Manos. 23 The CDKN2A mutation analysis was performed as described previously. 24 In brief, primers encompassing the entire coding region of p16INK4a were used in five separate polymerase chain reaction (PCR) amplifications, 25 with the following oligonucleotides: exon 1-alpha forward X1.31F GGGAGCAGCATGGAGCCG; reverse X1.26R AGTCGCCCGCCATCCCCT, exon 2A forward X2.62F AGCTTCCTTTCCGTCATGC, reverse 286R GCAGCACCACCAGCGTG; exon 2B forward 200F AGCCCAACTGCGCCGAC, reverse 346R CCAGGTCCACGGGCAGA, exon 2C forward 305F TGGACGTGCGCGATGC, reverse X2.42R GGAAGCTCTCTCAGGGTACAAATTC; exon 3 forward X3.90F CCGGTAGGGACGGCAAGAGA, reverse 530R CTGTAGGACCCTCG-GTGACTGATGA. Primers for exon 1-beta of p14ARF were forward TCCCAGTCTGCAGTTAAGG and reverse GTCTAAGTCGTTGTAACCCG. Amplified PCR products were directly sequenced by Taq cycle sequencing (ABI Prism BigDye Terminator kit/ABI Applied Biosystems, Foster City, CA). Data were analyzed using Sequencing Analysis 3.0 and Sequence Navigator 3.01 software (ABI Applied Biosystems). All sequencing results were confirmed by repeated PCR amplification and sequencing on both strands. In case of identified missense mutations, the germline DNA of 50 healthy control persons was also sequenced to evaluate the presence of a polymorphism.
RESULTS
The 18 FPC families had a total of 4 affected patients with pancreatic cancer. Three families had four affected members with pancreatic cancer, 2 families had three affected members, and 13 families had two affected members. In 1 family three generations were affected; in 11 families two generations were affected; in 6 families one generation was affected. Of the 44 patients with pancreatic cancer, 24 were men and 20 were women. The median age of diagnosis of pancreatic cancer was 63 years (range 41–81); six patients were younger than 50 years at diagnosis. Other associated tumor types were breast cancer in four families, prostate cancer in two families, and cancers of the lung, esophagus, stomach, and colon in one family each.
The mutation analysis of affected index patients with FPC revealed p16INK4a sequence alterations in 2 of 18 families. In one family with two affected patients, a T-to-A base change at nucleotide position 324 leading to a conserved amino acid change from valine to glutamate at codon 95 (V95E) was identified in the germline of the 58-year-old index patient with pancreatic cancer. In another family with two pancreatic cancer patients, a G-to-A transversion at nucleotide position 482 leading to an amino acid change from alanine to threonine at codon 148 (A148T) was detected in both affected family members. Both mutations were also observed at least twice in the germline of 50 healthy control persons. Thus, both alterations are most likely sequence polymorphisms not contributing to the development of pancreatic cancer. No sequence alterations were detected in exon 1-beta coding for the p14ARF transcript.
The five potential PCMS families consisted at least of one affected patient each with pancreatic cancer or cutaneous malignant melanoma. Three families might represent the FAMMM phenotype, since several family members, including the patient with melanoma, had multiple dysplastic nevi. In one family, three family members were affected with malignant melanoma according to the family history. However, with the exception of the index patient, the remaining family members declined to participate in the study, precluding confirmation of the medical data. The median age of onset of pancreatic cancer was 55 years (range 45–67). The median age of onset of malignant melanoma was 45 years (range 19–61). The characteristics of the five PCMS families are summarized in Table 1 and Figures 1 through 4. In two of the five families with pancreatic cancer and melanoma, p16INK4a germline mutations were identified in the affected patients. Neither family was phenotypically recognized to have the FAMMM syndrome. In one family a 19-year-old man developed a solitary melanoma on his back; his father had died of pancreatic cancer at the age of 52 years. In this family both affected patients carried the germline frameshift mutation c.323-324insG, E119X (see Figs. 1 and 2). In the second family, the female index patient who developed pancreatic cancer at age 55 carried the germline nonsense mutation c.188 C>T, Q50X. The same mutation could be detected in the germline DNA of her father, who developed metastatic melanoma at age 52 (see Figs. 3 and 4). Both patients with pancreatic cancer and a p16INK4a germline mutation were nonsmokers. Sequence alterations in exon 1-beta of p14ARF were not identified in the five PCMS families.
Table 1. CHARACTERISTICS OF PCMS FAMILIES

ID, identification code; PC, pancreatic cancer; MM, malignant melanoma.
*, 2 MM are not verified by histology, since patients did not participate in the study; * includes p16INK4a and p14ARF.

Figure 1. Pedigree of PCMS family 25-2-4 with CDKN2A germline mutation E119X. dg., age at diagnosis (years); d., age at death, *, p16INK4a mutation carrier.
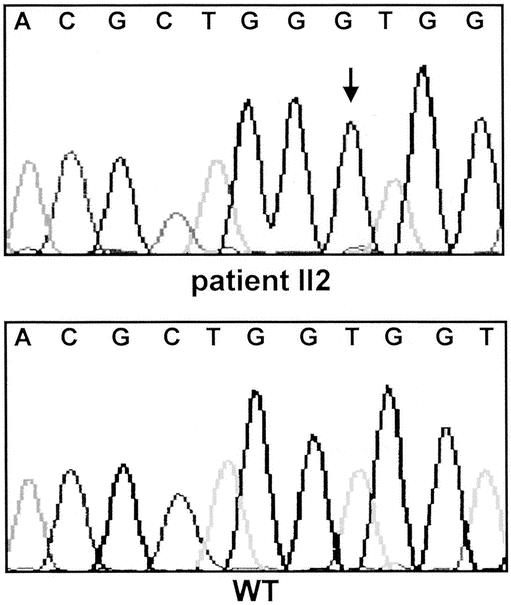
Figure 2. CDKN2A/p16INK4a germline frameshift mutation c.323-324insG, E119X in affected patients of PCMS family 25-2-4. The insertion of guanine is indicated by an arrow. WT, wild-type; II2, index patient with pancreatic cancer.
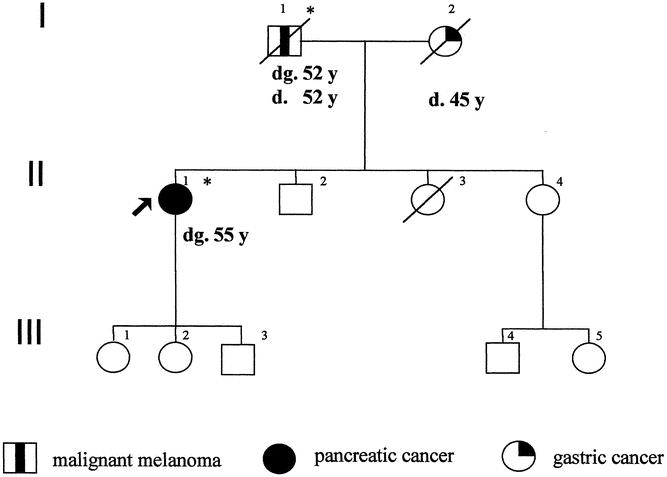
Figure 3. Pedigree of PCMS family 22-5-0256 with CDKN2A germline mutation Q50X. dg., age at diagnosis (years); d., age at death, *, p16INK4a mutation carrier.
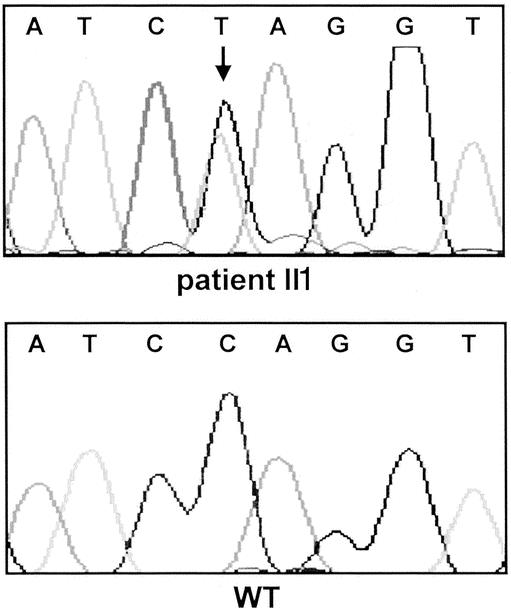
Figure 4. CDKN2A/p16INK4a germline nonsense mutation c.188 C>T, Q50X in affected patients of PCMS family 27-5-0256. The base change from cytosine to thymidine is indicated by an arrow. WT, wild-type sequence.
DISCUSSION
It has long been suggested that there is a familial form of pancreatic cancer, defined as the occurrence of pancreatic cancer in at least two first-degree relatives. 3–5,7,26,27 In recent years several FPC registries have been established to explore the genetics underlying this hereditary tumor syndrome. 3–5,7,28 These registries are invaluable resources in the quest for pathogenic germline mutations in candidate cancer genes. Furthermore, they allow us to study the pattern of aggregation of pancreatic cancer and other cancer types in such families. Previous data of the National Familial Pancreatic Tumor Registry (NFPTR) at the Johns Hopkins University revealed a 18-fold increased relative risk of pancreatic cancer among first-degree relatives of FPC kindreds, reaching a maximum relative risk of 57-fold when three or more family members were affected. 29 The vertical pattern of cancer observed in FPC families to date, including our families, is consistent with an autosomal dominant trait. 3,5,7,30,31 This interpretation, however, must be taken with caution, given the limited number of families and affected individuals available as well as the inability to rigorously exclude common exposures and environmental risk factors such as smoking.
Our results suggest that the predisposition to the development of pancreatic cancer in some families is associated with germline mutations in the CDKN2A tumor suppressor gene. However, this seems to be a rare cause in FPC families without the occurrence of malignant melanomas, since we could not identify relevant CDKN2A germline mutation in 18 FPC families. The p16INK4a alteration A148T identified in our cohort of FPC families is a previously reported variant that neither impairs p16 protein function nor segregates with the disease in melanoma kindreds. 32,33 Similarly, the p16INK4a variant V95E, although not previously described, was also found in two normal controls. Thus, both sequence alterations must be designated as non-disease-associated polymorphisms. Two recent studies also failed to detect p16INK4a mutations in 16 FPC families each, but less stringent criteria were used to define FPC. 21,34 Lal et al 34 included families with pancreatic cancer in second- and third-degree relatives; neither study necessarily verified the diagnosis of pancreatic cancer by histology. This might be an important drawback, since it is known that up to 30% of positive family histories in FPC patients are either questionable or wrong. 7 In addition, we analyzed for the first time CDKN2A exon 1-beta of p14ARF in FPC families and could not identify mutations. This substantiates that germline mutations of the CDKN2A gene do not contribute to the predisposition for pancreatic cancer in FPC families.
Historically, pancreatic cancer in concert with malignant melanoma in a family was first reported by Lynch and Krush in 1968. 35 In 1990, Bergmann et al reported clinical evidence of the integral association of pancreatic cancer and cutaneous malignant melanoma in a subset of families with the FAMMM syndrome. 15 Germline p16INK4a mutations have been detected in 10% to 25% of melanoma-prone kindreds, including families with the FAMMM phenotype. 36,37 Several studies have demonstrated an increased risk of pancreatic cancer of up to 22-fold among melanoma-prone kindreds with p16INK4a mutations. 20,38–40 No pancreatic cancer has yet been described in melanoma-prone families without p16INK4a germline mutations. In addition, in some families with p16INK4a germline mutations, tumors other than malignant melanoma, such as pancreatic cancer, are the predominant tumor phenotype. However, all these studies focused the p16INK4a mutation analysis on melanoma-prone kindreds with two or more first-degree relatives with malignant melanoma. We analyzed families with only two affected first-degree relatives with either pancreatic cancer or malignant melanoma and identified CDKN2A germline mutations leading to truncated p16INK4a in affected patients of two of five such families. Both p16INK4a-mutated families did not reveal the FAMMM phenotype, whereas the other three families without detectable mutation in the coding parts of the CDKN2A gene did. This was surprising, since Lynch et al recently reported p16INK4a germline mutations in eight kindreds prone to melanoma and pancreatic cancer, but all exhibited the FAMMM phenotype. 41 Moreover, in a study on patients with pancreatic cancer and additional other primary cancers, a p16INK4a germline mutation was identified in a patient with pancreatic cancer and cancer of the skin, vocal cord, and thyroid. 42 Altogether, our data substantiate a p16INK4a mutation-associated PCMS, as previously suggested. 19,41
It remains speculative why carriers of the same p16INK4a mutation develop either pancreatic cancer or malignant melanoma, but rarely both. There is no clear evidence for a direct genotype–phenotype association between pancreatic cancer and specific p16INK4a mutations, especially since three common founder mutations (V126D, G101W, H66del19) were observed in melanoma-prone families with and without pancreatic cancer. 35,39 This suggests other genetic or environmental factors modifying the risk of pancreatic cancer in p16INK4a mutation carriers. It has been hypothesized that smoking significantly increases the risk of pancreatic cancer among high-risk members of FPC kindreds. In a study of 214 subjects of 19 FPC kindreds, smokers developed pancreatic cancer one decade earlier than nonsmokers. This association was most pronounced in men. 43 However, both pancreatic cancer patients with p16INK4a mutations in our study were nonsmokers. In addition, the phenotypic prevalence of pancreatic cancer and melanoma among p16INK4a mutation carriers might be explained by an early inactivation of p16INK4a together with modifying genes, partly or completely suppressing the pancreatic cancer or melanoma phenotype.
Our data confirm that the age of onset of melanoma in p16INK4a-mutated families (median 45 years in the current study) is significantly earlier compared with the general population (median 55 years). 37 According to a study by Goldstein et al, pancreatic cancer does not occur at an earlier age in families with p16INK4a mutations. 44 However, the two pancreatic cancer patients of our p16INK4a mutation-carrying PCMS families were diagnosed at ages 52 and 55 years. Although one cannot draw conclusions from two patients, these ages of onset are about 15 years younger than the age of onset in the general German population. 1 This observation is supported by Lynch et al, who recently reported an early age of onset of pancreatic cancer in six of eight FAMMM-PC families. 41
We screened for the first time pancreatic cancer-melanoma families for germline mutations of CDKN2A exon 1-beta of p14ARF. Since we could not identify p14ARF mutations in any of the families, one can hypothesize that the alternate transcript of the CDKN2A gene does not contribute to the development of pancreatic cancer in these families.
Our data suggest that both patients and members of potential PCMS families should be screened for CDKN2A/p16INK4a mutations to identify high-risk family members for the development of pancreatic cancer and malignant melanoma. 45 However, there are strong limitations regarding counseling and pancreatic cancer screening. The penetrance of pancreatic cancer in CDKN2A-mutated families remains to be established; more importantly, there are no reliable diagnostic markers or imaging procedures for the early detection of pancreatic cancer or even better for its precursor lesions. In addition, it remains to be proven that the early detection of pancreatic cancer or its precursor lesions will prolong life expectancy. Nevertheless, uncovering a predisposing gene defect will allow a more precise risk assessment for the development of pancreatic cancer, as well as the evaluation of screening modalities for early detection in members of families with potential PCMS. During the counseling procedure, family members can be offered participation in a controlled screening program for both pancreatic cancer and malignant melanoma. Ideally, screening should be performed in a research setting within multidisciplinary institutional review board-approved protocols, allowing a precise evaluation of the screening program with special regard toward outcome.
Screening for cutaneous melanomas is easily done by regular physical examination of the skin. Although the limitations of screening for pancreatic cancer must be emphasized, there are some reports that high-risk family members of pancreatic cancer-prone families might benefit from close surveillance programs. Recently, Rulyak and Brentnall reported their experience with screening 35 high-risk members of 13 FPC families. 46 Twelve of the 35 high-risk individuals who were thought to have manifested dysplasia of the pancreas on the basis of clinical history, coupled with subtle abnormalities on endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography, underwent prophylactic pancreatectomy. Histopathologic examination of all 12 specimens demonstrated pancreatic dysplasias (mostly PanIN2), but no pancreatic cancer. None of the 35 patients under surveillance has developed pancreatic cancer during follow-up of up to 48 months. The authors concluded that thorough screening of family members in FPC families using endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography is an effective method for identifying individuals with pancreatic dysplasia before the onset of invasive pancreatic cancer, although it is too early to arrive at absolute conclusions about the benefits of currently available screening programs for pancreatic cancer. We do not know the actual risk of a PanIN2 and even PanIN3 lesion to progress into pancreatic cancer. Therefore, considering the potentially lethal complications of total pancreatectomy, this therapeutic option might well be an overtreatment. Furthermore, endoscopic retrograde cholangiopancreatography is considered an invasive screening procedure and may not be adequate for long-term screening. However, we have started to enroll our high-risk family members in an annual clinical screening program for the early diagnosis and treatment of pancreatic cancer and malignant melanoma. This screening program includes magnetic resonance imaging with magnetic resonance cholangiopancreatography and endoscopic ultrasonography as first-line imaging procedures for pancreatic cancer. 7,47 The screening program starts 5 years before the youngest age of onset in the family, or no later than age 45 years. Given the lack of data, we would consider pancreatectomy only in individuals with lesions suspicious for pancreatic cancer on imaging.
In summary, this study confirms that CDKN2A germline mutations including p16INK4a and p14ARF are rare in FPC families without the occurrence of melanoma. It provides further evidence for the existence of a pancreatic cancer–melanoma syndrome, which is in some families is associated with CDKN2A germline mutations affecting p16INK4a but not p14ARF and might be characterized by an early onset of malignant melanoma and pancreatic cancer. Thus, all families with co-occurrence of pancreatic cancer and melanoma should be offered genetic counseling and a chance to participate in clinical screening programs in which testing for CDKN2A/p16INK4a mutations may lead to a more precise individual risk assessment. This will provide important information about the risk of pancreatic cancer and malignant melanoma and about the efficacy of imaging procedures for pancreatic cancer screening in mutation carriers. Careful evaluation of such PCMS families, enrolled in surveillance programs, should lead to improved clinical management of these families and ideally longer life expectancy in p16INK4a mutation carriers.
Acknowledgments
The authors thank all families participating in the study. For enrolling families into FaPaCa we are grateful to the following members of the FaPaCa study group: Dr. Michael Lörken, Department of Surgery, University Aachen; Dr. Thomas Eberl, Dr. Soeren Dülsner, Department of Internal Medicine, Hospital Augsburg; Dr. Nikolaus Zügel, Dr. Karl Breitschaft, Department of Surgery, Hospital Augsburg; Dr. Lope-Estevéz Schwarz, Department of Surgery, Robert-Roessle-Klinik Berlin; Dr. Helmut Friess, Dr. Markus Böhnert, Department of Surgery, University Bern; Dr. Karsten Schulmann, Department of Internal Medicine Ruhr-University Bochum; Dr. Jens Rudolph, Department of Surgery, University Bonn; Dr. Joachim Heise, Department of Surgery, University Düsseldorf; Dr. Michael Ghadimi, Dr. Olaf Horstmann, Department of Surgery University Göttingen; Dr. Jan Schmidt, Dr. Sven Eisold, Department of Surgery, University Heidelberg; Dr. Wolfram von Bernstorff, Department of Surgery, University Kiel; Dr. Klaus Prenzel, Department of Surgery, University Köln; Dr. Helmut Witzigmann, Ria Metzner, Department of Surgery, University Leipzig; Dr. Christina Schleicher, Dr. Thomas Mūudel Department of General Surgery, University Münster; Dr. Tobias Grundei, Dr. Holger Vogelsang, Department of Surgery, University Munich; Dr. Michael Ernst, Department of Surgery, Hospital Neubrandenburg; Dr. Helmut Messmann, Dr. Esther Endlicher, Department of Internal Medicine, University Regensburg; Prof. Dr. Thomas Gress, Dr. Wolfgang Böck, Dept. of Internal Medicine, University Ulm. The authors thank Mrs. Elvira Przypadlo and Mrs. Margarete Schneider for their work in the study office.
Discussion
Prof. L. Fernández-Cruz: Dr. Bartsch, you should be congratulated for the number of families you have studied. Your paper merits more questions than comments.
My first question is, you and others describe PC–melanoma families in whom some patients develop PC, some melanomas, some both, and the age of onset varies greatly. Is there any evidence for a genotype–phenotype correlation like in the familial adenomatous polyposis (FAP)?
Another question refers to the fact that you identified alterations in 2 of 18 FPC families. These were determined as non-disease-associated polymorphisms in contrast to the two alterations in the PC–melanoma families. Since this discrimination is very important for the proband, don’t you think that functional assays always should be performed to ensure the quality of the mutation?
The third question is, you identified p16 mutations in the affected patients in two of your PC–melanoma families and in none of your 18 FPC families. Would you, based on your data, recommend to FPC families without melanoma screening for p16 mutations? What is the cumulative risk for the development of PC in mutation carriers, and how would you screen mutation carriers?
The last question is, are there other unknown genes that predispose to familial pancreatic cancer?
Dr. D.K. Bartsch: Regarding the genotype–phenotype correlation, there is so far, due to the small number of PCMS families analyzed worldwide, about 40 to 50 I would guess, no clear evidence for a genotype–phenotype correlation, although some authors suggest so. It seems that most families with the predominance of pancreatic cancer carry their mutations in the second or third ankyrin domain of the p16 gene, which are very important for the binding to CDK4, which is the partner of p16. But this is just an impression. However, in addition to the type of mutation, other factors might influence the development of pancreatic cancer in these families, such as environmental exposures, especially smoking, and the action of so-called modifier genes, so that pancreatic cancer or melanoma may not be expressed in mutation carriers.
Your second question was a technical question referring to whether a functional assay should always be performed for determination of the quality of the p16 alteration identified. I think it is not always necessary. Nonsense or frameshift mutations that lead to a premature stop and a truncated p16 protein always impair the p16 function. This is the case for the two families I have shown here. The problem missense mutations, because you don’t know whether they have relevance or not. If a sequence alteration like our A148T alteration will be identified in 100 healthy control persons, it represents most likely a polymorphism. Nowadays, it is pretty easy to check a large p16 database at the NIH whether the alteration you have found is a mutation or not, since a large number of mutations and polymorphisms are listed there. To be absolutely sure for missense alterations that are yet not specified, one should perform a functional assay.
Regarding your third question, 18 families are definitely not enough to decide not to recommend p16 mutation analysis to FPC families without melanoma. I would still recommend it, since the experimental effort is not extensive. But I would inform the proband that the possibility of a mutation in this gene is rather small. I also believe if more FPC families are analyzed, we will have the data to omit the p16 screening from FPC families without melanoma.
Regarding the cumulative risk, there is one study that evaluated the risk of pancreatic carcinoma in p16 mutation carriers based on the analysis of 19 FAMMM families in the Netherlands. In this study by Vasen et al, p16 mutation carriers had a cumulative risk of 19% for developing pancreatic cancer by the age 75 years. However, this number is based on melanoma families and not only on the analysis of PC–melanoma families, so the risk is probably a little higher. We recommend during our counseling procedure to mutation carriers the participation in a controlled screening program for both PC and malignant melanoma. Screening for cutaneous melanomas will be done by annual physical examination of the skin starting by the age of 18 years. Although the limitations of screening for PC have to be emphasized, there are first reports that high-risk family members of PC-prone families might benefit from close surveillance programs. Therefore, we started to screen mutation carriers annually with physical examination, CA19-9, bilirubin, phosphatase, magnetic resonance imaging (MRI) with magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography (EUS) as first-line imaging procedures. The screening program for PC starts 5 years below the youngest age of onset in the family, at latest by the age of 45 years.
Regarding your last question, we just conducted together with the EUROPAC study group a study on the prevalence of BRCA2 mutations in 26 familial pancreatic cancer families and identified germline mutations in 5 of 26 families. This underscores for the first time the importance of BRCA2 for familial pancreatic cancer. Furthermore, just this month a putative new gene locus for familial pancreatic cancer was reported on chromosome 4q32–34 by Eberle and colleagues. It is now the challenge to identify the potential predisposing gene located there.
Footnotes
Supported by grants no. 70-2363-Ba2 and 70-2828-Ba3 of the Deutsche Krebshilfe.
Correspondence: Detlef K. Bartsch, MD, FaPaCa Study Group, Department of Surgery, Philipps-University Marburg, Baldingerstrasse, 35043 Marburg/Germany.
E-mail: bartsch@mailer.uni-marburg.de
Accepted for publication April 2002.
References
- 1.Arbeitsgemeinschaft Bevölkerungsbezogener Krebsregister in Deutschland. Krebs in Deutschland-Häufigkeiten und Trends. 2. Ausgabe Saarbrücken, 1999.
- 2.Yeo CJ, Cameron JL. Prognostic factors in ductal pancreatic cancer. Langenbecks Arch Surg 1998; 383: 129–133. [DOI] [PubMed] [Google Scholar]
- 3.Lynch HT. Genetics and pancreatic cancer. Arch Surg 1994; 129: 266–268. [DOI] [PubMed] [Google Scholar]
- 4.Crowley KE, Aston CE, McNamara PJ, et al. Familial aggregation of other cancers in families with pancreatic cancer. Am J Hum Genet 1997; 61: A196. [Google Scholar]
- 5.Hruban RH, Petersen GM, Ha PK, et al. Genetics of pancreatic cancer: from genes to families. Surg Oncol Clin North Am 1998; 7: 1–23. [PubMed] [Google Scholar]
- 6.Hruban RH, Petersen GM, Goggins M, et al. Familial pancreatic cancer. Ann Oncol 1999; 10 (suppl 4): 69–73. [PubMed] [Google Scholar]
- 7.Bartsch DK, Sina-Frey M, Ziegler A, et al. Update of familial pancreatic cancer in Germany. Pancreatology 2001; 1: 510–516. [DOI] [PubMed] [Google Scholar]
- 8.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. J Natl Cancer Inst 1997; 89: 442–446. [DOI] [PubMed] [Google Scholar]
- 9.Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med 1987; 316: 1511–1514. [DOI] [PubMed] [Google Scholar]
- 10.McKusick VA. Ataxia teleangiectatica. In: McKusick MA, ed. Mendelian inheritance in man, 9th ed. Baltimore: John Hopkins University Press, 1990: 68–78.
- 11.Lynch HT, Smyrk TC, Watson P, et al. Genetics, natural history, tumor spectrum, and pathology of hereditary non-polyposis colorectal cancer: an updated review. Gastroenterology 1993; 104: 1535–1549. [DOI] [PubMed] [Google Scholar]
- 12.Li FP, Fraumeni JF Jr. Soft tissue sarcomas, breast cancer, and other neoplasms: a familial syndrome? Ann Intern Med 1969; 71: 747–752. [DOI] [PubMed] [Google Scholar]
- 13.Johan G, Offerhaus A, Giardiello FM, et al. The risk of upper gastrointestinal cancer in familial adenomatous polyposis. Gastroenterology 1992; 102: 1980–1982. [DOI] [PubMed] [Google Scholar]
- 14.Phelan CM, Lancaster JM, Tonin P, et al. Mutation analysis of the BRCA2 gene in 49 site-specific breast cancer families. Nature Genet 1996; 13: 120–122. [DOI] [PubMed] [Google Scholar]
- 15.Bergmann W, Watson P, de Jong J, et al. Systemic cancer and the FAMMM syndrome. Br J Cancer 1990; 61: 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993; 366: 704–707. [DOI] [PubMed] [Google Scholar]
- 17.Stott FJ, Bates S, James MC, et al. The alternative product from the human CDKN2a locus, p14 (ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J 1998; 17: 5001–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rozenblum E, Schutte M, Goggins M, et al. Tumor suppressing pathways in pancreatic carcinomas. Cancer Res 1997; 57: 1731–1734. [PubMed] [Google Scholar]
- 19.Whelan AJ, Bartsch D, Goodfellow PJ. Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med 1995; 333: 975–977. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein AM, Fraser MC, Struewing JP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med 1995; 333: 970–974. [DOI] [PubMed] [Google Scholar]
- 21.Moskaluk CA, Hruban H, Lietman A, et al. Novel germline p16(INK4) allele (Asp145Cys) in a family with multiple pancreatic carcinomas. Mutations in brief no. 148. Hum Mutat 1998; 12: 70. [DOI] [PubMed] [Google Scholar]
- 22.Rizos H, Puig S, Badenas C, et al. A melanoma-associated germline mutation in exon 1beta inactivates p14ARF. Oncogene 2001; 20: 5543–5547. [DOI] [PubMed] [Google Scholar]
- 23.Wright DK, Manos MM. Sample preparation from paraffin-embedded tissues. In: Innis MA, Gelfand DH, Sninsky JJ, et al, eds. PCR protocols: a guide to methods and applications. San Diego: Academic Press, 1990: 153–158.
- 24.Gerdes B, Ramaswamy A, Ziegler A, et al. p16INK4a is a prognostic marker in resected ductal pancreatic cancer. Ann Surg 2002; 235: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet 1994; 8: 15–21. [DOI] [PubMed] [Google Scholar]
- 26.Ehrenthal D, Haeger L, Griffin T, et al. Familial pancreatic adenocarcinoma in three generations: a case report and review of the literature. Cancer 1987; 59: 1661–1664. [DOI] [PubMed] [Google Scholar]
- 27.Grajower MM. Familial pancreatic cancer. Ann Intern Med 1983; 98: 111. [DOI] [PubMed] [Google Scholar]
- 28.Finch MD, Howes N, Ellis I, et al. Hereditary pancreatitis and familial pancreatic cancer. Digestion 1997; 58: 564–569. [DOI] [PubMed] [Google Scholar]
- 29.Tersmette AC, Petersen GM, Offerhaus GJ, et al. Increased risk of incident pancreatic cancer among first-degree relatives of patients with familial pancreatic cancer. Clin Cancer Res 2001; 7: 738–744. [PubMed] [Google Scholar]
- 30.Lynch HT, Fitzsimmons ML, Smyrk TC. Familial pancreatic cancer: clinicopathologic study of 18 nuclear families. Am J Gastroenterol 1990; 85: 54–60. [PubMed] [Google Scholar]
- 31.Lynch HT, Fusaro L, Smyrk TC, et al. Medical genetic study of eight pancreatic cancer prone families. Cancer Invest 1995; 13: 141–149. [DOI] [PubMed] [Google Scholar]
- 32.Ranade K, Hussussian CJ, Sikorski RS, et al. Mutations associated with familial melanoma impair p16INK4a function. Nat Genet 1995; 10: 114–116. [DOI] [PubMed] [Google Scholar]
- 33.Pollock PM, Pearson JV, Hayward NK. Compilation of somatic mutations of the CDKN2 gene in human cancers: non-random distribution of base substitutions. Genes Chromosome Cancer 1996; 15: 77–88. [DOI] [PubMed] [Google Scholar]
- 34.Lal G, Liu G, Schmocker B, et al. Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res 2000; 60: 409–416. [PubMed] [Google Scholar]
- 35.Lynch HT, Krush AJ. Heredity and malignant melanoma: implications for early cancer detection. Can Med Assoc J 1968; 99: 17–21. [PMC free article] [PubMed] [Google Scholar]
- 36.Gruis NA, Sandkuijl LA, van der Velden PA, et al. CDKN2 explains part of the clinical phenotype in Dutch familial atypical multiple-mole melanoma (FAMMM) syndrome families. Melanoma Res 1995; 5: 169–177. [DOI] [PubMed] [Google Scholar]
- 37.Goldstein AM, Tucker MA. Screening for CDKN2a mutations in hereditary melanoma. J Natl Cancer Inst 1997; 89: 676–677. [DOI] [PubMed] [Google Scholar]
- 38.Ghiorzo P, Ciotto P, Martelli M, et al. Characterization of lingurian melanoma families and risk of occurrence of other neoplasia. Int J Cancer 1999; 83: 441–448. [DOI] [PubMed] [Google Scholar]
- 39.Ciotti P, Strigini P, Bianchi-Scarra G. Familial melanoma and pancreatic cancer. N Engl J Med 1996; 334: 468–469. [DOI] [PubMed] [Google Scholar]
- 40.Borg A, Sandberg T, Nilson K, et al. High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2a mutation-positive melanoma families. J Natl Cancer Inst 2000; 92: 1260–1266. [DOI] [PubMed] [Google Scholar]
- 41.Lynch HT, Brand RE, Hogg D, et al. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families. The familial atypical multiple mole melanoma-pancreatic carcinoma syndrome. Cancer 2002; 94: 84–96. [DOI] [PubMed] [Google Scholar]
- 42.Gerdes B, Bartsch DK, Ramaswamy A, et al. Multiple primary tumors as an indicator for p16INK4a germline mutations in pancreatic cancer patients? Pancreas 2000; 21: 369–375. [DOI] [PubMed] [Google Scholar]
- 43.Rulyak SJ, Lowenfels AB, Maisonneuve P, et al. Smoking as a risk factor in familial pancreatic cancer kindreds. Pancreatology 2001; 1: 551. [Google Scholar]
- 44.Goldstein AM, Struewing JP, Chidambaram A, et al. Genotype-phenotype relationships in U.S. melanoma-prone families with CDKN2a and CDK4 mutations. J Natl Cancer Inst 2000; 92: 1006–1010. [DOI] [PubMed] [Google Scholar]
- 45.Lynch HAT, Randall EB, Lynch JF, et al. Genetic counseling and testing for germline p16 mutations in two pancreatic cancer-prone families. Gastroenterology 2000; 119: 1756–1760. [DOI] [PubMed] [Google Scholar]
- 46.Rulyak SJ, Brentnall TA. Inherited pancreatic cancer: surveillance and treatment strategies for affected families. Pancreatology 2001; 1: 477–485. [DOI] [PubMed] [Google Scholar]
- 47.Gerdes B, Kress R, Rieder H, et al. Familiäres Pankreaskarzinom: Studienkonzept einer Nationalen Fallsammlung mit Früherkennungsprogramm für Hochrisikopersonen. Zae FQ 2002; 96: 253–257. [PubMed] [Google Scholar]