

Abstract
Objective:
Because histologic features can be unreliable in determining the malignant potential of pancreatic endocrine neoplasms (PENs), we characterized the methylation patterns of PENs to develop a molecular marker system useful for clinical prognosis.
Summary Background Data:
Aberrant promoter methylation of tumor suppressor genes is associated with a loss of gene function that can afford selective growth advantages to neoplastic cells. Gene hypermethylation, coupled with sporadic genetic mutations, defines the heterogeneous biology of human neoplasms.
Methods:
Forty-eight well-differentiated PENs were subjected to methylation-specific polymerase chain reaction to detect aberrant methylation associated with 11 candidate tumor suppressor genes (INK4a/p16, APC, O6-MGMT, hMLH1, p73, E-cadherin, RAR-β, p14ARF, GST-π, TIMP3, and RASSF1A). Methylation differences among PENs, subdivided according to tumor size, lymph node status, or liver metastasis, were analyzed, and the association of gene methylation with tumor recurrence and patient survival was evaluated.
Results:
Aberrant hypermethylation of any of the 11 tumor suppressor genes was detected in 87% of the PENs. In decreasing order of frequency, the 5 most commonly methylated genes were: RASSF1A (75%), INK4a/p16 (40%), O6-MGMT (40%), RAR-β (25%), and hMLH1 (23%). In general, tumors larger than 5 cm and those associated with lymph node or hepatic metastases exhibited a higher frequency of methylation at each promoter site compared with PENs without malignant histologic features. The methylation of specific tumor suppressor genes was an independent predictor of early PEN recurrence and decreased 5-year survival following surgical resection. The accumulation of methylation of multiple tumor suppressor genes was associated with early tumor recurrence and reduced survival among a subpopulation of patients with lymph node-negative PENs.
Conclusions:
Aberrant methylation of multiple tumor suppressor genes is associated with advanced tumor stage and identifies molecularly distinct PENs with identical histologic characteristics. The methylation status of specific tumor suppressor genes is predictive of PEN behavior.
Aberrant promoter methylation of tumor suppressor genes, especially those responsible for cell cycle control, is associated with advanced endocrine tumors of the pancreas. The methylation status of specific tumor suppressor genes is a useful molecular marker system for predicting clinical behavior of pancreatic endocrine neoplasms.
Pancreatic endocrine neoplasms (PENs), commonly referred to as islet cell tumors, are a unique group of malignancies often characterized by a clinical neuroendocrine syndrome attributable to the selective overproduction and humoral circulation of pancreas-specific hormones. Approximately 2000 new cases of PENs are diagnosed each year in the United States; of these cases, 60-70% are associated with a clinical syndrome resulting from the secretion of a single functional hormone.1 The remaining one third of PENs secrete no clinically detectable biologically active hormones and most often present as space occupying lesions causing obstructive jaundice, upper gastrointestinal luminal obstruction, bleeding, or abdominal pain. While many functional PENs follow an indolent course, a substantial proportion of functional and nonfunctional tumors are defined by aggressive biology, resulting in early locoregional invasion of lymph node basins and adjacent organs, as well as metastases to the liver and beyond.2,3 Although histologic characteristics, including tumor size, pleomorphism, neurovascular invasion, and metastases to the regional lymph nodes or liver, have been used for predicting long-term survival, these factors have not proven to be entirely reliable prognostic markers for PENs after surgical resection.3-6
In recent years, several studies have reported a number of genes with important implications in the tumorigenesis of the pancreatic islets. These have included deletional mutations in the MEN1 gene on chromosome 11q, activation of the HER-2/neu proto-oncogene, overexpression of cyclin D1, point mutations in the DPC4/Smad4 gene, and deletion of a putative tumor suppressor gene on chromosome 3p.7-11 In addition to the genetic mutations of MEN1, molecular alterations of the cell cycle control gene, INK4a/p16, appear to be particularly important contributors to the pathogenesis of PENs.12-15 While homozygous deletions of INK4a/p16 have been found to variable degrees in PENs, the presence of epigenetic alterations, namely aberrant hypermethylation involving the promoter region of this locus, has been reported in PENs regardless of functional status.12-14
The combination of genetic and epigenetic alterations involving tumor suppressor genes contributes to the biology of several solid human tumors. Of the several epigenetic mechanisms that play a role in tumorigenesis (including chromosomal histone deacetylation and loss of genomic imprinting), DNA methylation of tumor suppressor gene promoter sites has been studied most extensively for its potential to predict tumor behavior.16-18 Silencing of tumor suppressor gene expression by promoter hypermethylation at CpG-rich islands is common among several human malignancies.19 Several reports have validated that the hypermethylation of promoter regions for such genes as INK4a/p16, E-cadherin, and hMLH1 correlates directly with the loss of transcription of these tumor suppressor genes in a variety of tumors.19-22
The purpose of this investigation was to determine the methylation status of several tumor suppressor genes to gain insight into the heterogeneous biology affiliated with nonfunctional PENs. The unique methylation pattern associated with each PEN in our study was analyzed for its ability to predict clinical outcomes after surgical resection.
METHODS
Human Tissues
Tumor samples were obtained from 48 retrospectively identified patients who underwent resection of a pancreatic endocrine neoplasm (PEN) at The Johns Hopkins Hospital between 1991 and 2001 (inclusive). Permission for cataloging and processing all samples for this study was obtained in accordance with the guidelines set forth by the institutional review board (IRB), termed the Joint Committee on Clinical Investigation. All tissue specimens, including primary PENs, and when applicable, associated metastases, along with disease-free tumor margins, were paraffin-embedded and sectioned sequentially at a thickness of 5-10 μm each. Characterization of the PEN samples, including tumor size, pleomorphism/differentiation, immunohistochemical staining, neurovascular invasion, and lymph node/extrapancreatic spread was carried out by 2 gastrointestinal pathologists examining the tissue sections. Tissue margins, associated with the primary tumors, were found free of disease in 41 (85%) of 48 of the collected specimens. To establish the definitional pathologic diagnosis of a PEN, immunohistochemical analyses for markers associated with neuroendocrine differentiation (ie, synaptophysin and chromogranin) were performed for each of the tumors. Specific immunostaining for pancreatic islet secretagogues (insulin, gastrin, glucagon, vasoactive intestinal peptide, somatostatin, and serotinin) was applied selectively to cases associated with a clinical neuroendocrine syndrome or at the surgeon’s request. Table 1 indicates the tumor characteristics and associated patient demographics for the samples included in this study. Overall, 52% of the tumors were derived from male patients, and 90% of the patients were Caucasian. Only 3 neoplasms were associated with a specific functional neuroendocrine syndrome, and these included 2 gastrinomas and 1 glucagonoma.
TABLE 1. PEN Characteristics

DNA Preparation
Two sequential 5- to 10-μm sections from each PEN (primary or metastatic) and associated tumor-free margin were deparaffinized with xylene and digested overnight at 50°C with proteinase K buffered in 1% SDS (pH 8). DNA was isolated by phenol-chloroform extraction and ethanol precipitation.23 Approximately 10 μg of DNA was partially purified from the 2 10-μm tissue sections.
Methylation-Specific PCR (MSP)
The methylation status of the promoter regions for 11 candidate tumor suppressor genes was determined by the method of MSP. The classic MSP technique was further modified as a nested two-step approach to increase the sensitivity of detecting allelic hypermethylation at targeted sequences and to facilitate the examination of multiple gene loci.23-25 Initially, 1 μg of tissue DNA was bisulfite treated according to previously described protocols to render unmethylated cytosines to uracil.25 The bisulfite-treated DNA was column-purified over Wizard clean-up resin (Promega, Madison, WI) and ethanol precipitated. Step 1 of the nested MSP was carried out with primer sets for 4-6 individual genes in each reaction. PCR products of step 1 were diluted 1:1000 and subjected to the second step of MSP, which incorporated 1 set of primers for each gene (labeled as unmethylated or methylated) that were designed to recognize bisulfite-induced modifications of unmethylated cytosines. All of the primer sequences and PCR conditions for this nested-MSP approach have been published previously.26 Both steps of the nested MSP used a 25-μL reaction volume, 0.5 μL of Jump Start Red TaqDNA polymerase (Sigma, St. Louis, MO), and 1 μL of DNA template. DNA isolated from normal peripheral lymphocytes from healthy individuals served as a negative methylation control. Human placental DNA was treated in vitro with SssI methyltransferase (NEB, Beverly, MA) to create completely methylated DNA at all CpG-rich regions. In vitro methylated DNA (IVD) served as the positive methylation control. MSP products were analyzed on 6% polyacrylamide gel electrophoresis.
Follow-Up Measurements
Clinical follow-up data were ascertained retrospectively through the Johns Hopkins Hospital patient record system after permission from the IRB. The dates of tumor recurrence and/or death after surgical resection were recorded. Complete follow-up data for recurrence and disease-free survival were obtained in 87% of all patients. The mean follow-up was 28 months, with the longest postoperative follow-up interval being 105 months. Table 1 contains the salient survival and tumor recurrence data for the patients with adequate follow-up. Adjuvant chemotherapy, administered at Johns Hopkins Hospital or at outside institutions, was received by 8 patients.
Statistical Analysis
The Fisher exact test was used to analyze the differences in methylation status of tumor suppressor genes among PENs subdivided according to malignant histologic characteristics, including tumor size, lymph node or hepatic metastasis. A two-tailed P < 0.05 was considered statistically significant (Stata Release 6, College Station, TX). A univariate Cox regression analysis was applied to the differences among individual histologic variables relating to survival or early tumor recurrence. Recurrence-free survival was measured from the time of diagnosis to the time of first recurrence or last follow-up. Kaplan-Meier curves were plotted to graphically display the prognostic value of methylation findings and histologic features on recurrence and survival.27 The log-rank test was used to compare the differences between survival groups. To identify independent predictors of recurrence-free survival, a multivariate Cox regression analysis, adjusting for tumor histologic characteristics, including size, differentiation, margin status, and lymph node/hepatic metastases, was used to compare the survival distributions of groups categorized according to the methylation status of specific tumor suppressor genes.28
RESULTS
Hypermethylation of PENs
We examined the methylation status of 11 candidate tumor suppressor genes, INK4a/p16, APC, O6-methyl-guanine methyltransferase (O6-MGMT), hMLH1, p73, E-cadherin, retinoic acid receptor beta 2 (RAR-β), ras association domain family protein 1 isoform A (RASSF1A), tissue inhibitor of metalloproteinases 3 (TIMP3), glutathione S-transferase pi (GST-π), and p14ARF, established to have roles in human cancer formation and progression (Table 2). 19 We afforded particular attention to genes involved directly with cell cycle control: INK4a/p16, APC, p73, and p14ARF.19, 29 Figure 1 depicts the promoter methylation findings for the INK4a/p16 gene responsible for inhibition of cyclin-dependent kinase-4 phosphorylation of the retinoblastoma gene product.30 While methylation of INK4a/p16 was present in 40% of all PENs, no promoter methylation was detected within the tumor-free tissue margins adjacent to the primary neoplasms (Fig. 2). In this study, none of the tumor-free margins demonstrated methylation of the tumor suppressor genes among our candidate group of 11. Even RASSF1A, which was frequently methylated in the PENs, was not methylated within the normal pancreas margins adjacent to the primary neoplasms. The presence of methylation within the neoplasms and not within the adjacent histologically normal pancreatic parenchyma supports the paradigm that aberrant promoter hypermethylation of specific genes and subsequent loss of gene function is a neoplastic-specific process.18,31 Moreover, unlike neoplasms arising in the setting of chronic exposure to carcinogens or inflammation, such as squamous carcinomas of the upper aerodigestive tract in heavy smokers or colorectal adenocarcinomas in patients with ulcerative colitis, PENs do not appear to evolve from broad methylation field defects of the pancreas.21,32
TABLE 2. Tumor Suppressor Gene Location and Function
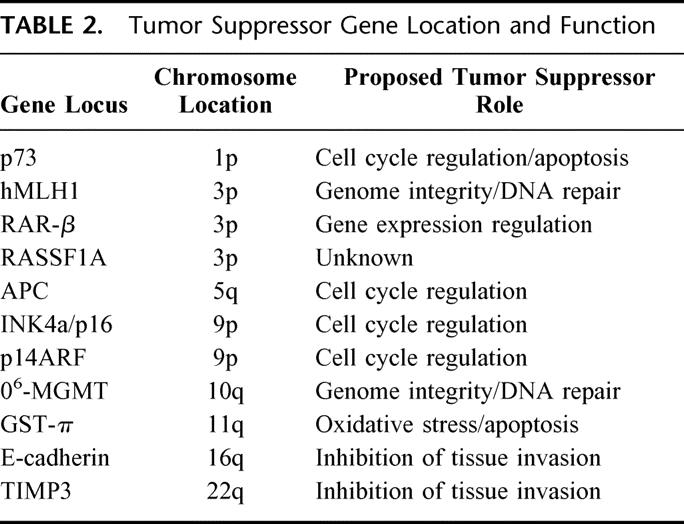
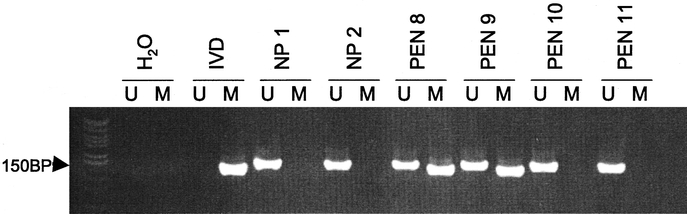
FIGURE 1. Amplified products after the second stage of nested methylation-specific PCR (MSP) for the INK4A/p16 gene. Lanes marked (U) signify unmethylated promoter CpG islands, and (M) bands denote the presence of gene promoter hypermethylation. In vitro methylated DNA (IVD) served as the positive control. Water (H2O) served as the system control. The MSP results for pancreatic tissue samples, including normal pancreas (NP) and representative endocrine tumors (PEN), are shown. PEN samples #8 and #9 contain INK4a/p16 gene methylation; whereas, samples #10 and #11 do not. The presence of unmethylated promoter regions among the methylated PEN samples (#8 and #9) represents inclusion of normal surrounding pancreas in addition to the neoplastic specimens.
FIGURE 2. Methylation of INK4A/p16 was not found in the tumor-free pancreatic margin (N) of surgical specimens containing a PEN (T). The presence of unmethylated (U) and methylated (M) promoter regions for the p16 gene within the normal pancreas (N) adjacent to a pancreatic neuroendocrine tumor (T) are shown for 4 representative patients.
Aberrant promoter methylation associated with each of the tumor suppressor genes for all PENs is summarized in Figure 3. Overall, we found aberrant hypermethylation of at least 1 gene in 41 of 48 neoplasms (85%). Methylation of at least 2 genes was present in 78% of all PENs, and multigene methylation, involving ≥ 3 genes, was documented in 52% of cases. In decreasing order of frequency, the 5 most frequently methylated genes were: RASSF1A (75%), INK4a/p16 (40%), O6-MGMT (40%), RAR-β (25%), and hMLH1 (23%).

FIGURE 3. A methylation profile of 11 tumor suppressor genes in 48 PENs. Shaded boxes represent the presence of aberrant gene promoter methylation. The total number of methylated gene markers for each tumor is indicated in the right column. The frequency of methylation for each gene within the entire group of PENs is noted.
Synchronous hepatic metastases were present in 14 of the 48 PENs (29%) that were studied. From these cases, we were able to compare the methylation patterns associated with hepatic metastases and the corresponding primary pancreatic neoplasm in 4 patients. Figure 4 demonstrates that the methylation status of each primary PEN matched that of its secondary hepatic metastasis, except for patient 28, whose neoplasm acquired methylation of an additional locus (INK4a/p16) during metastatic spread. Thus, for the most part, heritable traits of gene promoter methylation appear to be preserved during metastatic spread of pancreatic islet cell tumors.

FIGURE 4. A comparison of the methylation findings for primary endocrine neoplasms (P) associated with secondary hepatic metastases (M) in 4 individuals who underwent simultaneous pancreatic resection and liver metastasectomy. Methylated genes are represented as black blocks for the primary tumors and as gray blocks for the synchronous hepatic metastases.
Previous studies have demonstrated that the acquisition of methylation at multiple tumor suppressor genes is associated with advanced forms of human cancer.26,33,34 Since 52% of the PENs in this study were found to have methylation involving at least 3 loci, we studied differences in multigene methylation among neoplastic phenotypes classically associated with malignant islet cell tumor behavior, specifically large tumor burden and concomitant locoregional metastases.6 Differences in multigene methylation between PENs categorized according to malignant pathohistological features did not reach statistical significance, but methylation for multiple loci was found more frequently in neoplasms ≥ 5 cm and for those with lymph node metastases (Fig. 5). PENs with hepatic metastases or neurovascular invasion did not demonstrate any differences in multigene methylation compared with neoplasms without these histologic findings. With the exception of methylation at RAR-β, which was significantly more common for lymph node-positive PENs (P = 0.012), no significant differences in individual gene methylation were apparent for neoplasms with benign or malignant histopathologic characteristics.
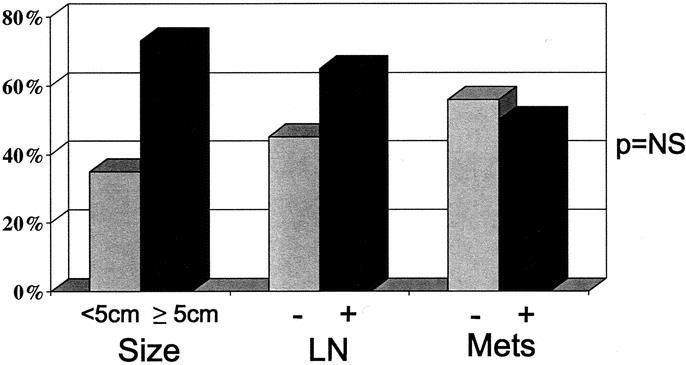
FIGURE 5. The frequency of multigene methylation, involving ≥ 3 genes, within PENs categorized according to established prognostic histologic features, namely size ≥ 5 cm, presence (+) of lymph node involvement (LN), or hepatic metastasis (Mets).
Molecular Markers for PEN Behavior
To verify the reliable histologic features predictive of malignant PEN behavior, we performed a univariate analysis of several factors with regard to clinical outcome. We achieved adequate follow-up in 42 (87%) of 48 patients included for study. Of these patients, 31% experienced the first recurrence of their PEN within 24 months from the time of surgery. Despite this high rate of early tumor recurrence, the median overall survival for our patients was just over 6 years, and the 5-year disease-free survival was 63%. Table 3 presents the survival risks associated with several histopathologic factors. Using a univariate Cox regression model, we found that poor tumor differentiation, lymph node or hepatic metastases, neurovascular invasion, and a positive surgical margin were significant predictors of poor clinical outcome. We then used a multivariate regression model, adjusting for primary tumor size and the malignant pathologic markers identified above, to analyze the relationship between methylation of the 11 tumor suppressor genes and survival (Table 3). The methylation status of 4 of the 11 candidate genes correlated with patient survival (shown in Table 3), but only methylation at the INK4A/p16 locus reached statistical significance. In addition, the presence of multigene methylation, involving 3 or more genes, was associated with significantly decreased patient survival.
TABLE 3. Predictors of Patient Mortality
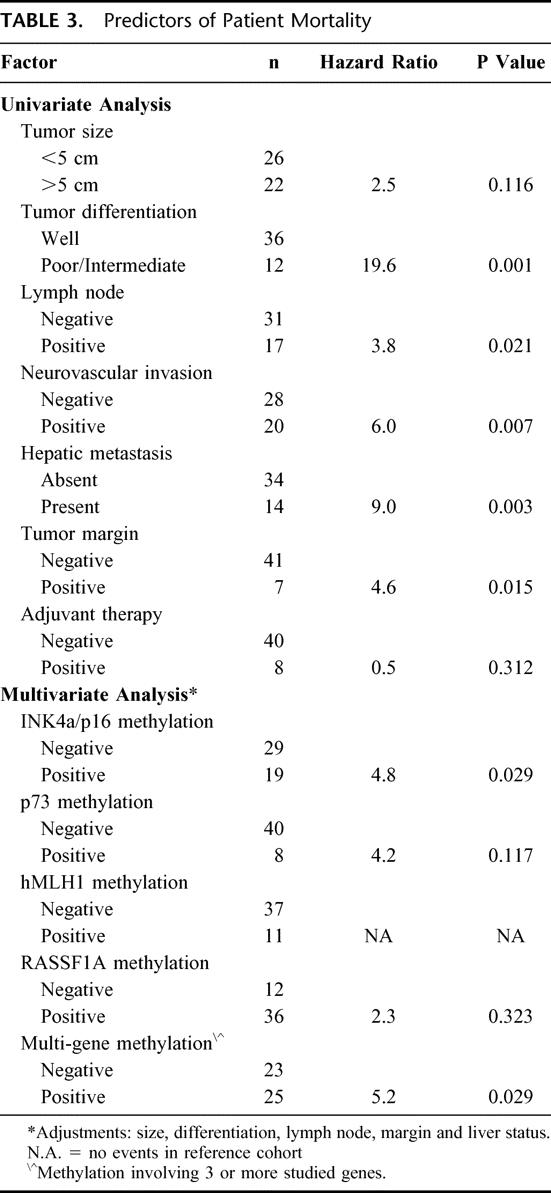
Based on multivariate analysis, the presence of methylation at INK4a/p16 and at 3 or more loci was an independent predictor of decreased patient survival after surgical resection for a PEN. This apparent survival disadvantage associated with gene hypermethylation was further studied with Kaplan-Meier analysis. With the exception of hMLH1 gene methylation, which demonstrated an inverse survival relationship due to its association with microsatellite instability, the presence of methylation at each of the other 10 loci was associated with decreased 5-year survival.20 The 2 molecular markers, holding the strongest association with poor clinical outcome after surgical resection, were INK4a/p16 and p73 gene methylation. At 5 years, patients with PENs harboring INK4a/p16 promoter methylation experienced a 44% tumor-free survival, compared with 72% for neoplasms without INK4a/p16 methylation (P = 0.15). The 5-year survival for patients with p73-methylated tumors was 57% versus 67% for PENs without methylation at this locus (P = 0.021). Patient survival according to multigene methylation status is displayed in Figure 6. The presence of methylation at 3 or more tumor suppressor genes is associated with decreased tumor-free survival, 45% versus 73% at 5 years (P = 0.09).
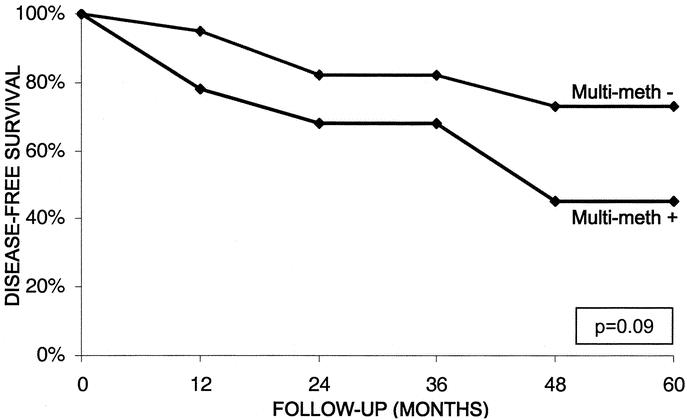
FIGURE 6. Disease-free survival at 5 years for patients with PENs characterized by the presence (+) or absence (-) of multigene methylation involving ≥ 3 gene markers. Statistical significance between the 2 survival groups was analyzed with the log-rank test (P = 0.09) and a multivariate regression model (P = 0.029).
Clearly, PENs associated with lymph node metastases carry an unfavorable prognosis. Five-year survival was significantly reduced in patients with lymph node-positive tumors (34%) compared with lymph node-negative tumors (79%; P = 0.013). Among PENs without lymph node involvement, multigene methylation was able to stratify patients into subpopulations with different survival functions. Figure 7 demonstrates that lymph node-negative PENs with multigene methylation are associated with decreased survival compared with lymph node-negative PENs without multigene methylation, 57% versus 90%, respectively (P = 0.07). Comparing gene methylation differences within tumor subgroups categorized according to malignant pathologic features, such as the presence of hepatic metastases, neurovascular invasion, or high tumor grade, we did not identify any further methylation markers capable of stratifying clinical outcomes for PENs with identical pathologic features.
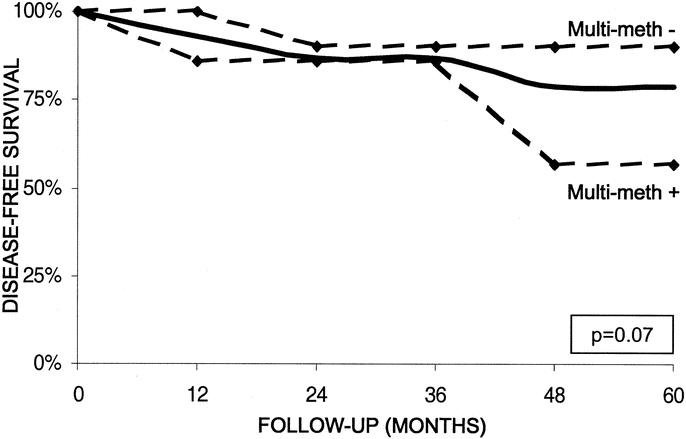
FIGURE 7. Five-year disease-free survival for patients following surgical resection of a LN-negative PEN. The overall survival for patients with a LN-negative PEN is shown by the solid black line. Survival for patients with LN-negative PENs is shown according to the presence (+) or absence (-) of multigene methylation (dashed lines). The log-rank test was used to analyze the differences in survival function (P = 0.07).
DISCUSSION
In contrast to neoplasms arising from the exocrine pancreas, the molecular events associated with PEN evolution and progression remain largely unknown. The genetic mutations of common tumor suppressor genes (p53, DPC4) and oncogenes (K-ras) that characterize exocrine neoplasms of the pancreas are not typically found in PENs.35,36 One exception is the frequent inactivation of the INK4A/p16 gene on chromosome 9p, a common finding in both endocrine and nonendocrine tumors of the pancreas.13,14,19,31,33 While both genetic and epigenetic alterations of INK4A/p16 have been reported, aberrant hypermethylation at this locus has been found in over 50% of PENs regardless of functional status.13,14 Our study was performed to characterize the epigenetic alterations, particularly hypermethylation of several candidate tumor suppressor genes, including INK4A/p16, associated with the pathogenesis of PENs.
Most (45 of 48) of the PENs in this study were not associated with a functional neuroendocrine syndrome or positive immunostaining for a specific pancreas-related peptide hormone. For these nonfunctional PENs, in contrast to functional islet tumors arising in patients with or without multiple endocrine neoplasia type 1, there is little knowledge regarding tumor biology and neoplastic progression. Similar to previous studies, we discovered frequent promoter hypermethylation at the INK4A/p16 gene (40%).14 Methylation at this promoter region was second in frequency only to methylation at the ras-associated domain family gene (RASSF1A) on chromosome 3p21 (75% methylated), a putative tumor suppressor gene that is frequently methylated in several human cancers.37 The exact sequence of inactivation of these genes during the initiation and later promotion of PEN growth remains to elucidated.
The methylation status of each tumor suppressor gene in our candidate group was compared with established histologic characteristics that classically define PEN malignant behavior, namely lymph node or neurovascular invasion and hepatic metastases.4-6 Hypermethylation at each gene was more common in PENs with a primary tumor diameter ≥ 5 cm or neoplasms associated with lymph node metastases. Similarly, Serrano et al reported that INK4A/p16 gene methylation correlated only with malignant gastrinomas associated with lymph node and not liver metastases, thus failing to demonstrate an indisputable effect of INK4a/p16 gene methylation on tumor aggressiveness.13 Since INK4a/p16 gene inactivation is a relatively early event during carcinogenesis, this finding is not surprising. Cumulatively, methylation at multiple genes, defined as 3 or more genes in our study, was associated with large tumor size and lymph node involvement. However, PENs with synchronous hepatic metastases did not demonstrate increased methylation at individual or multiple loci compared with neoplasms without liver disease. When we redefined multigene methylation to include ≥ 4 genes, notable, albeit nonsignificant, differences became more apparent for each of these 3 histologic categories, although at reduced frequencies. Methylation involving at least 4 genes was more common in PENs with either lymph node or liver metastases (30% and 21%, respectively) compared with PENs without these malignant features (19% and 16%). To a certain degree, the methylation of multiple tumor suppressor genes appears to be associated with more aggressive forms of PENs.
We examined whether patterns of multigene methylation were simply biologic signatures of aggressive PENs or reflected sequential accumulation of methylation at multiple loci during tumor progression. Unfortunately, from the 14 PENs with synchronous hepatic metastases, we were only able to study metastatic tumors correlating to primary lesions in 4 cases. Although 1 of these neoplasms (sample 28-M) acquired methylation at 1 additional gene during metastasis, the other 3 cases demonstrated complete matching of the methylation profiles for the primary and metastatic tumors. From these data, we cannot conclude that gene methylation accumulates during tumor progression or even during metastatic spread. However, this consistency of methylation between primary and secondary neoplasms may have clinical application. The methylation status of individual or multiple genes could be used to follow tumor aggressiveness in response to surgical and/or medical therapy or to prove which primary tumor (if more than one) is responsible for any synchronous or metachronous metastasis. Also, specific methylation patterns of histopathologically normal tissues within the surgical specimen, including adjacent organs, lymph nodes, or distant sites, may be indicative of occult regional or systemic tumor spread.
Despite numerous studies establishing the prognostic validity of pathohistological characteristics, it remains challenging to determine which PENs will pursue a malignant course.4,6,38 Identification of some of the epigenetic changes contributing to the molecular biology of nonfunctional PENs may lead to a reliable marker system for predicting the clinical outcomes of these tumors before and after surgical resection. The presence of methylation at specific genomic sites is not only predictive of survival, but forecasts early tumor recurrence after complete surgical extirpation of PENs. As a result, gene methylation may be able to subcategorize patients with histologically identical tumors into groups with unique molecular biology amenable to different treatment strategies.
Using a multivariate regression model for survival analysis adjusting for histologic features predictive of malignant behavior, we were able to identify 2 molecular markers with potential value as reliable predictors of patient outcome after surgical resection. Methylation at 3 or more genes or at the INK4a/p16 locus predicted both decreased patient survival at 5 years and tumor recurrence within 24 months of the time of surgery. Postoperatively, 37% of INK4a/p16 methylated PENs recurred, compared with 26% of tumors without INK4a/p16 methylation (P = 0.13). Although INK4a/p16 methylation was an independent prognostic factor for 5-year survival (see Table 3), it failed to reach statistical significance as a predictor of tumor recurrence within 2 years of operation. On the contrary, multigene methylation predicted early tumor recurrence, 40% versus 19% (P = 0.017), in addition to decreased patient survival. The propensity for tumor recurrence and cancer-related death in the presence of these 2 markers likely reflects the contribution of these epigenetic events to PEN biology.
The determination of methylation status at multiple genes serves as an important, and perhaps independent, predictor of PEN behavior following surgical resection. More importantly, methylation markers can identify molecularly distinct tumors with histologically indistinguishable features. When we examined the group of patients with lymph node-negative PENs who were expected to have a favorable prognosis overall, we were able to identify 2 survivor subgroups on the basis of the methylation status of multiple genes. Patients with lymph node-negative PENs harboring multigene methylation experienced a poorer outcome compared with those with lymph node-negative PENs without multigene methylation. As a translational application, tumor methylation status could assign patients into different surgical and adjuvant treatment arms according to their calculated risk for disease progression.
The frequent methylation of several tumor suppressor genes within PENs contributes to the underlying biology of these peculiar tumors. Greater understanding of the sequence of genetic and epigenetic events that lead to neoplastic transformation of the endocrine pancreas will afford more opportunities for developing a reliable molecular marker system for the diagnosis and prognosis of PENs. Furthermore, for the future, therapeutic strategies that reactivate tumor suppressor gene function via active demethylation or inhibition of de novo methylation can perhaps alter neoplastic aggressiveness and affect clinical outcome.
Discussions
Dr. Timothy J. Eberlein (St. Louis, Missouri): DNA methylation of tumor suppressor gene promoter sites is associated with a loss of gene function which will result in selective growth advantages to neoplastic cells. In the present study the authors hypothesize that hypermethylation to tumor suppressor genes coupled with sporadic genetic mutations will better define this relatively rare and heterogeneous group of neoplasms. This technology is relatively new. It has been successfully used to predict tumor behavior in other tumor models. My first question is a technical one. Why did you choose to use DNA from normal T-cells as a negative control instead of DNA from other normal tissue?
You have nicely shown that methylation of specific tumor suppressor genes was an independent predictor of early pancreatic endocrine neoplasm recurrence and decreased five-year survival following surgical resection even in the subpopulation of patients with lymph node negative pancreatic endocrine neoplasms. Yet this does not seem to correlate with hepatic metastasis or neurovascular invasion. Why is this?
You propose a potential therapeutic strategy in your discussion of the manuscript that might reactivate tumor gene function by active de-methylaytion or inhibition of de novo methylation. However, would that still have as great an impact on the systemic disease, especially liver metastasis, as much as local and possible nodal spread?
This is a rare group of tumors even at your tertiary referral center. There are probably not enough patients to really classify subcategories by this methodology, although you have demonstrated a significant way of predicting prognosis. Where will you plan to go from here in terms of using this methodology in these types of rare tumors?
I very much enjoyed reviewing the manuscript and congratulate the authors, especially the presenter, for an excellent presentation in helping us understand the biologic behavior of these tumors. I would also like to thank the Association for the privilege of discussing this manuscript.
Dr. Michael G. House (Baltimore, Maryland): Thank you, Dr. Eberlein, for your comments, and I will try to address the issues that you have raised in sequence.
First, you questioned our selection of a negative control for studies involving methylation-specific PCR. Typically, peripheral lymphocytes from healthy volunteers serve as an excellent control for unmethylated genomic DNA. While we employed normal lymphocytes as a negative control for each of the tumor suppressor genes that we studied, we like to ensure that positive methylation is associated with a neoplastic and not a patient-specific phenomenon. Because our group and others have discovered both partially and fully methylated tumor suppressor genes in older cancer-free individuals, and especially those who smoke chronically, we always validate the presence of methylated alleles in the primary neoplasm and not in the adjacent stroma or nondysplastic epithelium from each patient.
Your second question addresses the failure of this study to demonstrate an increased frequency of multigene methylation among the metastatic islet tumors compared with the nonmetastatic neoplasms. Although a greater frequency of both individual and multiple gene methylation was observed for islet tumors with malignant characteristics, including those associated with synchronous metastases, these differences did not reach statistical significance. While we hope that this simply reflects a population size issue, we cannot exclude the possibility that this is indeed this neoplasm’s biology. In addition to understanding the role of methylation-induced silencing of tumor suppressor genes in endocrine tumor biology, we wanted to correlate an objective marker, here gene methylation, with clinical outcomes, namely recurrence-free survival after surgical resection.
In terms of future studies on islet cell tumors, our goal is to establish a profile of gene methylation that is specific for pancreatic endocrine neoplasms. Although we have studies 11 tumor suppressor genes in this study, we will need to explore hundreds, if not thousands, of genes to accomplish this goal. Not only can we apply additional methylation markers for prognosis, as we did in this study, we would also like to use this PCR based technology as a diagnostic marker system as well. And even though DNA methylation is a covalent biochemical modification, that is essentially irreversible, there are several current drug strategies that are capable of preventing de novo genomic methylation of later generation clonal progeny. Inhibition of DNA methyltransferases may be 1 way to reactivate silenced tumor suppressor genes, and consequently prevent neoplastic progression in established primary tumors.
Dr. Henry A. Pitt (Milwaukee, Wisconsin): I would also like to congratulate the authors on an excellent presentation. I know that you were focusing on methylation, but I am curious about some of the other tumor suppressor genes. An obvious 1 would be p53. Do you have any data on p53?
In Milwaukee in studying our patients with adenocarcinoma of the pancreas we found that p21 was a predictor of outcome and, interestingly, that it was a predictor of response to chemoradiation and/or chemotherapy. More recently, we have looked at thymadilate synthase and have shown that this enzyme is also a predictor of response to chemotherapy.
I know that your islet cell tumors are more rare, that their survival is better, and that they are not very likely to be treated with chemotherapy or with octreotide, but I am curious whether you have looked at whether any of these methylation markers predict way response to hormonal therapy, chemotherapy or chemoradiation.
Dr. Michael G. House (Baltimore, Maryland): Thanks, Dr. Pitt, for your astute comments. One thing that has been attractive about methylation studies of tumor biology, is the facility and reliability with which they can be performed. Unlike genetic studies of variable polymorphisms in large tumor suppressor genes, such as the p53 gene, methylation-specific PCR is a relatively simple technique that can be immediately applied to the clinical laboratory.
In terms of gene methylation markers predicting responses to adjuvant chemoratiation, this study involved only 7 patients who received some form of adjuvant therapy (in the form of radiation, streptozocin, or alkylating agents) so we were unable to study any potential relationships. However, we are interested in such prospective studies, given the potential of Fanconi anemia related gene expression to predict adenocarcinoma responses to cross-linking chemotherapy.
Footnotes
Correspondence: Charles J. Yeo, MD, Professor of Surgery and Oncology, The Johns Hopkins Hospital, 600 N. Wolfe Street, Blalock 606, Baltimore, MD 21287-4606. E-mail: cyeo@jhmi.edu
Supported in part by grants from the Stavros S. Niarchos Foundation and NCI Grant CA-84986 of the Early Detection Research Network.
Presented at the 123rd Annual Meeting of the American Surgical Association, Washington, DC, April 24-26, 2003.
REFERENCES
- 1.Phan GQ, Yeo CJ, Hruban RH, et al. Surgical experience with pancreatic and peripancreatic neuroendocrine tumors: review of 125 patients. J Gastrointest Surg. 1998;2:472-482. [PubMed] [Google Scholar]
- 2.Yu F, Venzon DJ, Serrano J, et al. Prospective study of the clinical course, prognostic factors, causes of death, and survival in patients with long-standing Zollinger-Ellison syndrome. J Clin Oncol. 1999;17:615-630. [DOI] [PubMed] [Google Scholar]
- 3.Jensen RT. Pancreatic endocrine tumors: recent advances. Ann Oncol. 1999;10(Suppl 4):170-176. [PubMed] [Google Scholar]
- 4.Madeira I, Terris B, Voss M, et al. Prognostic factors in patients with endocrine tumours of the duodenopancreatic area. Gut. 1998;43:422-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.La Rosa S, Sessa F, Capella C, et al. Prognostic criteria in nonfunctioning pancreatic endocrine tumours. Virchows Arch. 1996;429:323-333. [DOI] [PubMed] [Google Scholar]
- 6.Wick MR, Graeme-Cook FM. Pancreatic neuroendocrine neoplasms: a current summary of diagnostic, prognostic, and differential diagnostic information. Am J Clin Pathol. 2001;115(Suppl):S28-S45. [DOI] [PubMed] [Google Scholar]
- 7.Wang EH, Ebrahimi SA, Wu AY, et al. Mutation of the MENIN gene in sporadic pancreatic endocrine tumors. Cancer Res. 1998;58:4417-4420. [PubMed] [Google Scholar]
- 8.Chung DC, Brown SB, Graeme-Cook F, et al. Overexpression of cyclin D1 occurs frequently in human pancreatic endocrine tumors. J Clin Endocrinol Metab. 2000;85:4373-4378. [DOI] [PubMed] [Google Scholar]
- 9.Bartsch D, Hahn SA, Danichevski KD, et al. Mutations of the DPC4/Smad4 gene in neuroendocrine pancreatic tumors. Oncogene. 1999;18:2367-2371. [DOI] [PubMed] [Google Scholar]
- 10.Evers BM, Rady PL, Sandoval K, et al. Gastrinomas demonstrate amplification of the HER-2/neu proto-oncogene. Ann Surg. 1994;219:596-601; discussion, 602-604. [DOI] [PMC free article] [PubMed]
- 11.Chung DC, Smith AP, Louis DN, et al. A novel pancreatic endocrine tumor suppressor gene locus on chromosome 3p with clinical prognostic implications. J Clin Invest. 1997;100:404-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan AO, Kim SG, Bedeir A, et al. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene. 2003;22:924-934. [DOI] [PubMed] [Google Scholar]
- 13.Serrano J, Goebel SU, Peghini PL, et al. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J Clin Endocrinol Metab. 2000;85:4146-4156. [DOI] [PubMed] [Google Scholar]
- 14.Muscarella P, Melvin WS, Fisher WE, et al. Genetic alterations in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors: an analysis of p16/MTS1 tumor suppressor gene inactivation. Cancer Res. 1998;58:237-240. [PubMed] [Google Scholar]
- 15.Chung DC, Brown SB, Graeme-Cook F, et al. Localization of putative tumor suppressor loci by genome-wide allelotyping in human pancreatic endocrine tumors. Cancer Res. 1998;58:3706-3711. [PubMed] [Google Scholar]
- 16.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163-167. [DOI] [PubMed] [Google Scholar]
- 17.Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187-191. [DOI] [PubMed] [Google Scholar]
- 18.Baylin SB, Herman JG, Graff JR, et al. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141-196. [PubMed] [Google Scholar]
- 19.Esteller M, Corn PG, Baylin SB, et al. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225-3229. [PubMed] [Google Scholar]
- 20.House MG, Herman JG, Guo M, et al. Prognostic value of hMLH1 hypermethylation and microsatellite instability in pancreatic endocrine neoplasms. Surgery. 2003; in press. [DOI] [PubMed]
- 21.Belinsky SA, Nikula KJ, Palmisano WA, et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A. 1998;95:11891-11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870-6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esteller M, Sanchez-Cespedes M, Rosell R, et al. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999;59:67-70. [PubMed] [Google Scholar]
- 24.Palmisano WA, Divine KK, Saccomanno G, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000;60:5954-5958. [PubMed] [Google Scholar]
- 25.Herman JG, Graff JR, Myohanen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821-9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.House MG, Guo M, Iacobuzio-Donahue C, et al. Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis. 2003;24:193-198. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457-481. [Google Scholar]
- 28.Peto R, Peto R. Asymptomatically efficient rank invariant test procedures. J Roy Stat. 1972;34:185-206. [Google Scholar]
- 29.Larsen F, Gundersen G, Lopez R, et al. CpG islands as gene markers in the human genome. Genomics. 1992;13:1095-1107. [DOI] [PubMed] [Google Scholar]
- 30.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672-1677. [DOI] [PubMed] [Google Scholar]
- 31.Ueki T, Toyota M, Sohn T, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835-1839. [PubMed] [Google Scholar]
- 32.Issa JP. Epigenetic variation and human disease. J Nutr. 2002;132(8 Suppl):2388S-2392S. [DOI] [PubMed] [Google Scholar]
- 33.Ueki T, Toyota M, Skinner H, et al. Identification and characterization of differentially methylated CpG islands in pancreatic carcinoma. Cancer Res. 2001;61:8540-8546. [PubMed] [Google Scholar]
- 34.Park TJ, Han SU, Cho YK, et al. Methylation of O(6)-methylguanine-DNA methyltransferase gene is associated significantly with K-ras mutation, lymph node invasion, tumor staging, and disease free survival in patients with gastric carcinoma. Cancer. 2001;92:2760-2768. [DOI] [PubMed] [Google Scholar]
- 35.Jensen RT. Carcinoid and pancreatic endocrine tumors: recent advances in molecular pathogenesis, localization, and treatment. Curr Opin Oncol. 2000;12:368-377. [DOI] [PubMed] [Google Scholar]
- 36.Hruban RH, Iacobuzio-Donahue C, Wilentz RE, et al. Molecular pathology of pancreatic cancer. Cancer J. 2001;7:251-258. [PubMed] [Google Scholar]
- 37.Pfeifer GP, Yoon JH, Liu L, et al. Methylation of the RASSF1A gene in human cancers. Biol Chem. 2002;383:907-914. [DOI] [PubMed] [Google Scholar]
- 38.Modlin IM, Sandor A. An analysis of 8305 cases of carcinoid tumors. Cancer. 1997;79:813-829. [DOI] [PubMed] [Google Scholar]