

Abstract
cADPR (cADP-ribose), a metabolite of NAD+, is known to modulate intracellular calcium levels and to be involved in calcium-dependent processes, including synaptic transmission, plasticity and neuronal excitability. However, the enzyme that is responsible for producing cADPR in the cytoplasm of neural cells, and particularly at the synaptic terminals of neurons, remains unknown. In the present study, we show that endogenous concentrations of cADPR are much higher in embryonic and neonate mouse brain compared with the adult tissue. We also demonstrate, by comparing wild-type and Cd38−/− tissues, that brain cADPR content is independent of the presence of CD38 (the best characterized mammalian ADP-ribosyl cyclase) not only in adult but also in developing tissues. We show that Cd38−/− synaptosome preparations contain high ADP-ribosyl cyclase activities, which are more important in neonates than in adults, in line with the levels of endogenous cyclic nucleotide. By using an HPLC method and adapting the cycling assay developed initially to study endogenous cADPR, we accurately examined the properties of the synaptosomal ADP-ribosyl cyclase. This intracellular enzyme has an estimated Km for NAD+ of 21 μM, a broad optimal pH at 6.0–7.0, and the concentration of free calcium has no major effect on its cADPR production. It binds NGD+ (nicotinamide–guanine dinucleotide), which inhibits its NAD+-metabolizing activities (Ki=24 μM), despite its incapacity to cyclize this analogue. Interestingly, it is fully inhibited by low (micromolar) concentrations of zinc. We propose that this novel mammalian ADP-ribosyl cyclase regulates the production of cADPR and therefore calcium levels within brain synaptic terminals. In addition, this enzyme might be a potential target of neurotoxic Zn2+.
Keywords: brain, cADP-ribose (cADPR), CD38, NAD+, synaptosome, zinc
Abbreviations: ADPR, ADP-ribose; cADPR, cADP-ribose; cGDPR, cGDP-ribose; GTP[S], guanosine 5′-[γ-thio]triphosphate; NADase, NAD+ glycohydrolase; NGD+, nicotinamide–guanine dinucleotide; PSD95, postsynoptic density 95 kDa; TPEN, N,N,N′,N′-tetrakis(2-pyridylmethyl)-ethylenediamine; [Zn2+]i, intracellular free zinc concentration
INTRODUCTION
Calcium is an intracellular messenger that relays information within cells to regulate their activity. Increases in cytosolic calcium concentration involve several calcium channels from both the cell surface and the intracellular membranes, giving access to the two available sources of calcium: external medium and intracellular calcium stores. Several metabolites of NAD+ have been identified as second messengers that contribute to the complex and versatile cell calcium signalling. cADPR (cADP-ribose) was shown to modulate calcium movements from ryanodine-sensitive endoplasmic reticulum stores (for reviews, see [1,2]). NAADP+ (nicotinic acid–adenine dinucleotide phosphate) was shown to act on as yet unidentified intracellular stores of calcium (for reviews, see [3–5]). ADPR (ADP-ribose) was shown to modulate the activity of a plasma membrane calcium channel, TRPM2, a member of the large TRP (transient receptor potential) channel family [6]. Recently, cADPR was also shown to synergize with ADPR in the activation of TRPM2 channels [7]. Therefore enzymes capable of producing these NAD+ metabolites within cells undoubtedly have key roles in the control of fundamental calcium-dependent cellular processes.
In neural cells, as in other cells, calcium controls a large number of functions. cADPR, which is abundant in the brain [8,9], was shown to modulate calcium movements from ryanodine-sensitive stores both in neurons [10–13] and in astrocytes [14]. It is involved in calcium-dependent neural processes, including synaptic transmission, plasticity and neuronal excitability [13–16]. The cellular events that allow cADPR to control ryanodine-sensitive stores or TRPM2 channels remain to be clarified. However, there is growing evidence that cADPR could be produced in response to the activation of some metabotropic receptors [17–23] and play a critical role at synaptic terminals. However, the identity of the cADPR-synthesizing enzyme that is involved in such signalling pathways remains unknown.
cADPR-synthesizing enzymes known to date form a growing family of proteins that are highly conserved at both genetic and structural levels. All of these enzymes produce both cADPR (ADP-ribosyl cyclase activity) and ADPR [NADase (NAD+ glycohydrolase) activity] from NAD+, but in considerably varying proportions [24]. The first identified enzyme of this family, mainly endowed with ADP-ribosyl cyclase activity, was isolated from the mollusc Aplysia californica [25], where it is present in the cytosolic compartment of synaptic terminals [15]. In mammals, two proteins sharing approx. 30% sequence identity with the Aplysia cyclase have been identified, CD38 [26] and CD157 [or BST-1 (bone marrow stromal cell antigen 1)] [27]. Only CD38, which is a membrane-associated protein and which, unlike the Aplysia enzyme, mainly exhibits an NADase activity, is expressed in the nervous system [28,29]. Previously, we have shown that CD38 is mostly ectocellular in the brain tissue, but it also has intracellular location; however, as it is principally located in the soma of neural cells [30], CD38 activity is unlikely to play a significant role in synaptic transmission in relation to ryanodine-sensitive internal stores. The discovery that the endogenous level of cADPR of adult brain tissue is not changed in Cd38−/− mice [9] is consistent with this conclusion. Moreover, we have also revealed the existence of an ADP-ribosyl cyclase in neural cells from Cd38−/− mice [31]. This novel enzyme, which produces intracellular cADPR and ADPR, is present in brain during development and in the adult, and exhibits low basal activity which can be enhanced by GTP[S] (guanosine 5′-[γ-thio]triphosphate) application. This novel ADP-ribosyl cyclase could be a relay between extracellular signals and intracellular calcium-dependent events that occur at synaptic terminals.
In the present study, we show that synaptosomes from Cd38−/− mouse brains have high intracellular ADP-ribosyl cyclase activity, and that the highest activities are detected in synaptosomes purified from neonate compared with synaptosomes purified from adult animals. This result is consistent with the observation that endogenous brain cADPR content, which is definitely not related to the presence of the CD38 protein, is higher in the developing brain and declines in the adult tissue. We then characterized the ADP-ribosyl cyclase from brain synaptosomes. Interestingly, we found that this ADP-ribosyl cyclase is sensitive to micromolar concentrations of Zn2+ ions, which were described as modulators of synaptic activities [32] and whose homoeostasis imbalance was shown to generate brain injury and defects in cognitive development [33,34].
We propose that the novel mammalian ADP-ribosyl cyclase participates in the production of the cADPR messenger, and also of ADPR, within nerve terminals and might therefore be involved in crucial calcium-dependent pathways in brain development and functioning.
EXPERIMENTAL
Materials
ADP-ribosyl cyclase from A. californica, nucleotide pyrophosphatase from Crotalus atrox venom, NADase from Neurospora crassa, alcohol dehydrogenase from baker's yeast (Saccharomyces cerevisiae), diaphorase from Clostridium kluyveri and resazurin were purchased from Sigma. Alkaline phosphatase from calf intestine was purchased from Roche Diagnostics Corporation. [14C]NAD+ (nicotinamide–[U-14C]adenine dinucleotide, 300 mCi/mmol) was from Amersham Biosciences. Monoclonal antibodies [anti-(synapsin IIa) and anti-PSD95 (postsynoptic density 95 kDa)] were purchased from BD Transduction Laboratories.
Preparation and Western-blot analysis of brain synaptosomes
Synaptosomes were prepared from total brain isolated from adult and neonate Cd38−/− mice using a one-step preparation based on the known isopycnic densities of cellular components, as described in [35]. They were subsequently either directly frozen in liquid nitrogen or dialysed against solution D (containing 10 mM Tris/HCl, pH 7.5, 2.2 mM CaCl2, 0.5 mM Na2HPO4, 0.4 mM KH2PO4, 4 mM NaHCO3 and 80 mM NaCl) for 2 h at 4 °C and were then frozen in liquid nitrogen. Using this protocol, we were able to purify from neonate brains (postnatal days 1 or 2) approx. one-third of the quantity of synaptosomes purified from the same mass of adult brains. Therefore approx. 1% (w/w) of the total protein content of adult tissue corresponds to the purified synaptosomes proteins, whereas only approx. 0.3–0.4% (w/w) of the total protein content of neonate tissue represents the synaptosome proteins.
Synaptosome enrichment was tested by Western-blot analysis of the associated proteins using monoclonal antibodies against two synaptic proteins, i.e. synapsin IIa and PSD95 (at a dilution of 1/2000 and 1/250 respectively). Proteins (6 μg/well) were separated on 12% (w/v) polyacrylamide denaturing gels, followed by an electrotransfer for 2 h at 500 mA to Immobilon P sheets (Millipore). After 1 h of incubation with 4% (w/v) non-fat dried milk powder (Bio-Rad), the primary antibody was added and the samples were incubated overnight at 4 °C. The blots were then stained for 3 h at room temperature (20–22 °C) with horseradish-peroxidase-conjugated anti-mouse IgG (Cappel). After washing the blots, the reactive proteins were detected using the Renaissance chemiluminescence reaction (NEN Life Science Products) followed by exposure to Biomax films (Kodak).
Measurement of endogenous cADPR
The measurement of cADPR from brain tissues was performed essentially as described previously [36], except that fresh tissues were used instead of frozen tissues. We found that the use of fresh tissue did not modify the results that were obtained by comparing the cADPR content of half-brain homogenates, one half-brain being frozen and pulverized to powder before the addition of HClO4 and the other half being mixed directly with the acid; the same results were obtained with both types of homogenates (results not shown). For each purification performed, half-brains were used in the case of adult mice, whereas entire brains were used for embryos and neonates.
The fresh brain tissues from wild-type mice (of known mass) were directly homogenized on ice with a Potter–Elvehjem-type homogenizer in 0.6 M HClO4. The fresh brain tissues from Cd38−/− mice were first homogenized on ice with 100 mM sodium phosphate buffer at pH 8.0, 10 μl aliquots (of a total volume of 800 μl) were removed to determine the protein content using the Bradford method (using BSA as standard), and then (4 min after adding the buffer) the HClO4 was added to a final concentration of 0.6 M. This slightly different protocol, allowing the measurement of protein content in homogenate, was used with the brain tissue from Cd38−/− mice, since we have observed that the cADPR level was stable in this tissue during the first 4 min after buffer addition. This was not the case with wild-type tissues, in which the cADPR level decreases quickly (results not shown). The acid was eliminated by extraction with TFE (1,1,2-trichlorotrifluoroethane) and TNO (tri-n-octylamine), and finally 20 mM sodium phosphate buffer at pH 8.0 was added to the samples. Samples were then reacted overnight at 37 °C with a mix of nucleotide pyrophosphatase, alkaline phosphatase and NADase in the presence of magnesium as in [36] in order to remove all of the contaminating nucleotides. Enzymes were eliminated on ultrafiltration units (Nanosep 10 K; Pall Corporation).
cADPR present in samples was then quantified. The conversion of cADPR into NAD+ was performed by incubating the sample for 1 h at room temperature with 0.3 μg/ml Aplysia ADP-ribosyl cyclase and 10 mM nicotinamide in 35 mM sodium phosphate buffer at pH 8.0. The cycling reaction was then allowed to proceed in 60 mM sodium phosphate buffer at pH 8.0 in the presence of 0.8% ethanol, 10 mM nicotinamide, 40 μg/ml BSA, 20 μg/ml di-aphorase (extemporaneously treated with charcoal), 20 μg/ml alcohol dehydrogenase, 10 μM FMN and 20 μM resazurin. The increase in resorufin fluorescence (excitation at 544 nm and emission at 590 nm) was measured every 1 min for 2 or 3 h using a fluorescence plate reader (FluoStar from BMG Labtechnologies) and compared with standard solutions of cADPR and NAD+.
In order to calculate the cADPR content of the samples isolated from wild-type mice, in pmol/mg of protein, we determined the ratio (quantity of protein/quantity of tissue) in both neonate and adult brain by weighing brains and analysing their protein content using the Bradford assay. The ratios obtained were of 36 μg of protein/mg of tissue in neonates (n=4) and 52 μg of protein per mg of tissue in adults (n=3). In the case of Cd38−/− mice brains, cADPR content calculation was made using the brain homogenate protein content also measured using the Bradford assay.
Enzyme assays with [14C]NAD+ and analysis by HPLC
Synaptosomes (1 mg of protein/ml) were incubated at 32 or 37 °C in 20 mM buffer (citrate/HCl at pH 4.0, Mes/NaOH at pH 5.0 and 6.0, and Tris/HCl at pH 7.0 and 8.0) with 60 μM [14C]NAD+ (8 mCi/mmol), 4 mM EDTA (to inhibit the nucleotide pyrophosphatases; see [31]), and with or without 1% (w/v) Triton X-100. Reactions were stopped at given times (see legends to Figures) by adding 10% (v/v) HClO4, followed by neutralization with potassium carbonate.
HPLC analysis and standards preparation ([14C]cADPR, [14C]ADPR and [14C]AMP) were performed essentially as described previously [31]. Briefly, HPLC separations were performed isocratically on a C18 column using 10 mM ammonium phosphate buffer at pH 5.5 and 1% (v/v) acetonitrile. Radio-detection of the products was performed using an online radioactivity monitor after mixing the eluted products with a scintillation liquid. Reaction products of NAD+ (ADPR, cADPR and AMP) were identified on the chromatograms by comparing with standards injected separately.
Enzymatic tests using the cycling assay
Synaptosomes (0.2–0.4 mg of protein/ml) were generally incubated in solution D with 200 μM NAD+ in the absence or presence of 1% (w/v) of the detergent Emulphogen (polyoxyethylene-10-tridecyl ether). However, in the experiments with various calcium concentrations, synaptosomes were first centrifuged at 54000 g for 30 min, rinsed and resuspended in solutions containing 50 mM Tris/HCl at pH 7.5 and free calcium at 1 nM (2 mM EGTA and 52 μM CaCl2), 1 μM (1 mM EGTA and 1.93 mM CaCl2) or 1 mM (2 mM EGTA and 3 mM CaCl2). Suspensions were subsequently incubated (at 0.4 mg of protein/ml) with NAD+ and Emulphogen.
Reactions were stopped at given times (see legends to Figures) by adding 0.6 M HClO4, followed by neutralization with potassium carbonate. The contaminating nucleotides were eliminated by the use of a mixture of the three nucleotide hydrolytic enzymes as detailed above, except that the overnight reaction was performed in solution D. We ensured that the substrate used in our experiments (200 μM NAD+) was completely degraded during this step (results not shown).
Then the quantity of cADPR produced in the samples was determined: the conversion of cADPR into NAD+ and the cycling reaction were allowed to proceed as detailed above, except that the sodium phosphate buffer was replaced by solution D. Results were compared with standard solutions of cADPR and NAD+ analysed under the same experimental conditions, i.e. metabolites were diluted in solution D. The recovery of cADPR content after extraction with HClO4 and neutralization with potassium carbonate was estimated as 68% of control by tests performed with standard solutions of cADPR.
Assays of GDP-ribosyl cyclase activity
The conversion of NGD+ (nicotinamide–guanine dinucleotide) into cGDPR (cGDP-ribose) was followed fluorimetrically as described previously [30]. The enzyme reaction was started by adding 100 μM NGD+ to synaptosomes (500 μg/ml) purified from adult or neonate Cd38−/− mouse brains that were incubated at 37 °C in solution D in the presence of Emulphogen (0.25% w/v). After 2 h of incubation at 37 °C, Aplysia cyclase (3 μg/ml) was added as a control in order to transform all of the remaining NGD+ into cGDPR. Before each measurement, all materials, including the quartz cuvette, injection syringes and mixer were meticulously washed and incubated for 15 min with boiling water in order to totally eliminate contaminating Aplysia cyclase.
Kinetic analysis
The initial rates of substrate conversion were calculated from a polynomial regression curve. Kinetic parameters were determined from the plot of the initial rates as a function of substrate concentrations using a non-linear regression program (Graph Pad Prism).
RESULTS
Brain tissues contain higher cADPR levels during their development
Previous studies have estimated the cADPR content of brain homogenates isolated from adult mice [9,37]; however, the cADPR level of the developing tissue had never been studied before. In the present study, we examined the cADPR content of brain tissue from embryonic (days 15 or 16), neonate (postnatal day 1 or 2), and adult wild-type and Cd38−/− mice (Figure 1). Using the highly sensitive cycling assay developed by Graeff and Lee [36], we first confirmed results of the literature, showing that the cADPR content of adult brain homogenates prepared from Cd38−/− mice is not significantly different from that of wild-type adult brain homogenates [9]. The cADPR content of adult mouse brain that we measured (3.0±0.8 pmol/mg) was similar to the adult mouse brain content found with an RIA (3.86±0.87 pmol/mg [9]) and to the adult rat brain content measured by a calcium-release bioassay (2.75±0.35 pmol/mg [8]). Using the cycling assay, Graeff and Lee [36] estimated the cerebrum and cerebellum cADPR contents to be 2.2±0.3 and 2.1±0.3 pmol/mg respectively, values which are close to those that we have obtained with the entire brain.
Figure 1. Endogenous cADPR content in developing and adult brain from wild-type and Cd38−/− mice.

Extracts were prepared from tissues isolated from embryos (EM, days 15 or 16), from neonates (NN, days 1 or 2), or adults (AD), and were analysed for cADPR content using the cycling assay as described in the Experimental section. Results are means±S.D.
Very interestingly, the cADPR contents measured during the development of the brain, in embryos and in neonates, were found to be notably higher than the contents of the adult tissue. Indeed, the embryonic cADPR content was estimated to be 7.3±1.1 pmol/mg, and the neonate content was 5.9±0.4 pmol/mg. Moreover, neonate Cd38−/− brain tissue showed cADPR content equivalent to neonate wild-type tissue, and higher than the content of adult Cd38−/− brain tissue.
In conclusion, our results indicate the presence of higher cADPR contents in the developing brain than in the adult tissue: the level being 2.5- and 2-fold higher respectively in embryos and in neonates. Besides, as in the adult tissues [9], the cADPR contents of developing brain are independent of the presence of the CD38 protein.
An intracellular ADP-ribosyl cyclase is present in brain synaptosomes isolated from Cd38−/− mice
cADPR production was shown to occur in response to the activation of metabotropic receptors in several neural cell types [17–23], and was shown to be involved in different neural processes occurring at synaptic connections [14–16]. However, the enzyme that is responsible for producing cADPR in the cytoplasm of neural cells, and particularly at the synaptic terminals of neurons, remains unknown. Interestingly, we have demonstrated previously the presence in brain of a new ADP-ribosyl cyclase, different from the known ADP-ribosyl cyclases CD38 and CD157 (see the Introduction) [31].
For these reasons, we wished to determine whether the novel ADP-ribosyl cyclase was present in brain synaptic terminals. We therefore isolated synaptosomes from adult and neonate Cd38−/− mice brains using the method of Phillips et al. [35] and examined the occurrence of an ADP-ribosyl cyclase activity within these subcellular structures. Synaptosome enrichment in the preparation was determined by Western blot using antibodies against two typical synaptic proteins: synapsin IIa, which is a synaptic vesicles-associated protein, and PSD95, which is a protein from the post-synaptic density. As shown in Figure 2(A), fraction 3, which is found at the 1.25 M/1.0 M sucrose interface, is highly enriched in these markers. The ADP-ribosyl cyclase activity was tested in this synaptosome-enriched fraction using two sensitive assays, i.e. a radioactive HPLC method to follow the transformation of [14C]NAD and identify its reaction products [31,38] and a fluorimetric cycling assay which quantifies the production of cADPR from NAD+ [36]. It should be noted that we have adapted this latter assay, initially developed to study the cADPR contents in tissues [36], to accurately determine ADP-ribosyl cyclase activity.
Figure 2. Analysis of the ADP-ribosyl cyclase activity from Cd38−/− mouse brain synaptosomes by the cycling assay: effects of detergent and free calcium concentration.

(A) Synaptosomes preparation by centrifugation of brain homogenate on a discontinuous sucrose gradient and Western-blot analysis of the collected fractions containing proteins (6 μg/well) using anti-(synapsin IIa) and anti-PSD95 antibodies. Synaptosomes were collected at the 1.25 M/1.0 M sucrose interface (fraction 3; arrow). (B) Synaptosomes (0.4 mg/ml for adult and 0.2 mg/ml for neonate) were incubated (2 h for adult and 1 h for neonate) with 200 μM NAD+ in solution D (containing 10 mM Tris/HCl, pH 7.5, 2.2 mM CaCl2, 0.5 mM Na2HPO4, 0.4 mM KH2PO4, 4 mM NaHCO3 and 80 mM NaCl) in the absence (−) or presence (+) of the detergent Emulphogen (1%, w/v). Reactions were stopped by the addition of HClO4 followed by neutralization. The formation of cADPR was detected using the cycling assay as described in the Experimental section. The results obtained in the absence of detergent was expressed as percentages of the result obtained in the presence of the detergent. (C) Adult synaptosomes (0.4 mg/ml) were incubated for 2 h in 50 mM Tris/HCl, pH 7.5, in the presence of 200 μM NAD+ and 1% (w/v) Emulphogen, and with 1 nM (pCa 9), 1 μM (pCa 6) or 1 mM (pCa 3) free calcium. The experiment was reproduced twice with two different synaptosomal preparations (n=4). Results are means±S.D. Analysis of the cADPR production was performed using the cycling assay.
Brain synaptosomes (fraction 3; 0.2–0.4 mg/ml) were tested for their capacity to transform NAD+ (200 μM) specifically into cADPR after incubation at 37 °C in their conservation medium, i.e. at pH 7.5 and in the presence of millimolar concentrations of CaCl2. Figure 2(B) shows the results obtained with synaptosomes isolated from adult and neonate Cd38−/− brain tissues which were either intact or treated with the detergent Emulphogen (an optically neutral detergent, equivalent to Triton X-100). The cADPR production was analysed using the fluorimetric cycling assay. Under the experimental conditions used (1 h of incubation for neonate, 2 h of incubation for adult), with both adult and neonate brain tissues, the cADPR produced by intact synaptosomes was only approx. 10% of the cADPR synthesized by detergent-treated synaptosomes.
Brain synaptosomes (1 mg/ml) were also tested for their ability to transform [14C]NAD (60 μM) into cADPR and ADPR after incubation at 32 °C and pH 6.0 in the presence of 4 mM EDTA (experimental conditions similar to those already used with brain cells and membrane extracts, in which EDTA was added to inhibit the pyrophosphatases cleaving NAD+ metabolites such as ADPR; see [31]). Figure 3 shows representative HPLC radiochromatograms obtained with intact (control) and Triton X-100-treated synaptosomes isolated from adult (Figures 3A and 3B) and neonate (Figures 3C and 3D) Cd38−/− brain tissues after an incubation of 8 h. In the reactions with intact synaptosomes, peaks corresponding to ADPR were observed (4% NAD+ in the adult and 6.7% NAD+ in the neonate), whereas no cADPR peaks could be detected. In sharp contrast, when the same reactions were performed in the presence of detergent, both ADPR and cADPR peaks were observed: 29% of NAD+ was converted into ADPR and 2% into the cyclic compound in adult synaptosomes; 52% of NAD+ was converted into ADPR and 4% into cADPR in neonate synaptosomes.
Figure 3. Analysis of NAD+-metabolizing activities of synaptosomes from adult and neonate Cd38−/− mice brain by HPLC: effects of detergent.

Synaptosomes (1 mg/ml) prepared from brain tissue of adult (AD; A, B) and neonate (NN; C, D) Cd38−/− mice were incubated for 8 h in 20 mM Mes/NaOH at pH 6.0 and 32 °C with [14C]NAD+ (8 mCi/mmol) and NAD+ (final concentration 60 μM) in the presence of 4 mM EDTA, with (B, D) or without (A, C) 1% (w/v) Triton X-100 (TX). Reactions were stopped by the addition of HClO4 followed by neutralization. Products analysis was performed by HPLC using an online radioactivity detector as described in the Experimental section.
We next examined, using the cycling assay, the effect of calcium concentration on the synaptosome cADPR synthesis activity. To that end, adult synaptosomes (0.4 mg/ml) were incubated for 2 h with NAD+ and Emulphogen in solutions containing various free calcium concentrations at pH 7.5 (Figure 2C). We have observed less than 12% difference between the cADPR production measured at 1 nM, 1 μM and 1 mM free calcium.
Thus, by the use of two different methods, we have demonstrated the presence of an ADP-ribosyl cyclase in synaptosomes isolated from both neonate and adult Cd38−/− brain tissues. We detected much larger ADP-ribosyl cyclase and NADase activities in detergent-treated synaptosomes compared with intact structures, a result indicative of an intracellular location of the enzyme catalytic site. The low activities observed with intact synaptosomes are indeed very likely to be due to the presence of leaky synaptosomes in our preparations. Moreover, free calcium appears to have no major effect on the cADPR production by the synaptosomal enzyme. The enzyme detected in synaptosomes certainly corresponds to the novel enzyme which we have detected previously within brain cells and membrane extracts (P540 extracts obtained by high-speed centrifugation and enriched in endoplasmic reticulum and plasma membranes) isolated from Cd38−/− mice [31].
In order to have access to the full synaptosomal activity, the subsequent experiments were all performed with detergent-treated synaptosomes, i.e. in the presence of 1% (w/v) Triton X-100 or Emulphogen.
Reaction-progress curves of the ADP-ribosyl cyclase activity of synaptosomes isolated from neonate and adult Cd38−/− mouse brain
We analysed the kinetics of cADPR synthesis by brain synaptosomes from Cd38−/− neonate and adult mice using a HPLC method (Figure 4A) and the cycling assay (Figure 4B). Experimental protocols used here were the same as those described above. Incubation times were between 1 and 8 h for the HPLC experiments, and between 5 min and 1 h in the cycling assays, which are much more sensitive.
Figure 4. Kinetics of the ADP-ribosyl cyclase activity of synaptosomes from adult and neonate Cd38−/− brains, measured using both HPLC (A) and the cycling assay (B).
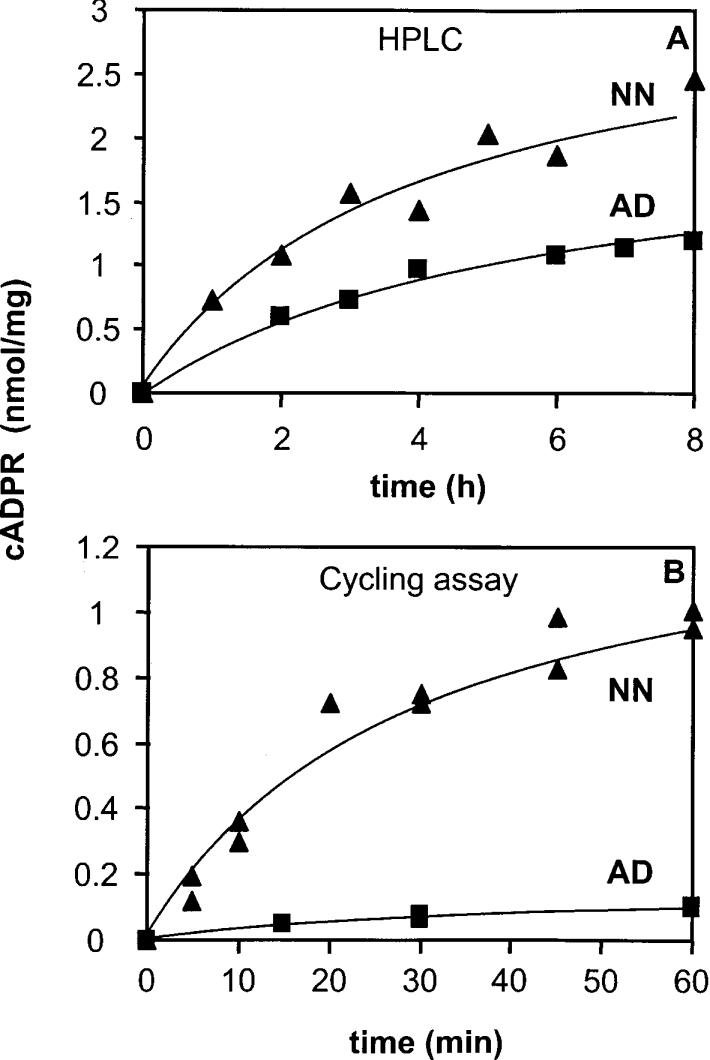
(A) Synaptosomes (1 mg of protein/ml) were incubated at 32 °C for the given times with [14C]NAD+ (8 mCi/mmol) and NAD+ (final concentration 60 μM), 4 mM EDTA, 1% (w/v) Triton X-100 in 20 mM Mes/NaOH at pH 6.0. (B) Synaptosomes [adult (AD) 0.4 mg/ml, neonate (NN) 0.2 mg/ml] were incubated for the given times with 200 μM NAD+ in solution D (see legend to Figure 3) in the presence of 1% (w/v) Emulphogen.
The estimated initial rates of the cADPR production obtained in the presence of 60 μM NAD+ at pH 6.0 and 32 °C (HPLC) are 4.1 pmol/mg per min for the adult brain synaptosomes and 8.4 pmol/mg per min for the neonate brain synaptosomes. The initial rates of the reactions performed with 200 μM NAD+ at pH 7.5 and 37 °C (cycling assay) are 2.3 pmol/mg per min for adult brain synaptosomes and 36 pmol/mg per min for neonate brain synaptosomes.
With both methods, we revealed significantly lower activities in the adult synaptosomes compared with neonate synaptosomes, a result which is consistent with the lower cADPR levels observed in adult tissues compared with the developing tissues (Figure 1). With the cycling assay, which is much more precise than the HPLC method, we found that neonate synaptosomes have approx. 15-fold higher initial rates of cADPR-synthesizing activity than the adult synaptosomes.
Moreover, the initial rate of reaction obtained with neonate synaptosomes using the cycling assay (36 pmol/mg per min) is approx. 10-fold higher than the rate evaluated under similar experimental conditions (200 μM reactive NAD+ at pH 7.5 and 37 °C) with cells isolated from neonate cerebral hemispheres (3.2 pmol/mg per min [31]).
Altogether, these results show a higher cADPR-synthesizing activity in brain synaptosomes from Cd38−/− mouse compared with entire brain cells, and thus an enrichment of the novel ADP-ribosyl cyclase in terminals of brain neural cells. Moreover, higher activities were found in synaptosomes from neonates compared with synaptosomes from adults.
Properties of the adult brain synaptosomal ADP-ribosyl cyclase: Km for NAD+, competition between NAD+ and NGD+, and pH-dependence
We studied further some properties of the synaptosomal ADP-ribosyl cyclase from Cd38−/− mouse brain and determined the Km (app) for the transformation of NAD+ into cADPR using the cycling assay. To that end, adult synaptosomes (0.4 mg/ml) were incubated for 15 or 30 min at pH 7.5 and 37 °C in the presence of increasing concentrations of the substrate in order to determine the initial rates of cADPR synthesis for each NAD+ concentration. The experimental data were fitted assuming a hyperbolic NAD+ concentration-dependence. From the plot of these initial rates (Figure 5A), a Km of 21 μM was estimated for NAD+, i.e. a value within the range of the Michaelis constants determined for the other known mammalian enzymes of the cyclase/NADase family [39,40], and which is slightly higher than the Km estimated for the Aplysia cyclase of 4.6 μM [41]. The Vmax was approx. 6 pmol of cADPR formed/min per mg with adult synaptosomes under these experimental conditions.
Figure 5. Properties of the CD38-independent ADP-ribosyl cyclase: determination of the Km (app) of NAD+ and of the Ki of NGD+.

(A and B) Adult brain synaptosomes (0.4 mg/ml) were incubated for 15 or 30 min at 37 °C in solution D (pH 7.5) with 1% (w/v) Emulphogen and various NAD+ concentrations, in the absence (◇) or in the presence (×) of 100 μM NGD+. Then the cADPR formation was analysed using the cycling assay and reaction rates were estimated. (B) Secondary plot of the Km (app) for NAD+ against NGD+ concentration (n=3 at 0 NGD+; n=1 at 100 μM NGD+; n=2 at 300 μM NGD+). (C and D) Adult brain synaptosomes (1.1 mg of protein/ml) were incubated for 2 or 4 h at 37 °C in the presence of [14C]NAD+ (4 mCi/mmol) and NAD+ (60 μM), 4 mM EDTA, 1% (w/v) Triton X-100 in 20 mM Tris/HCl at pH 7.0 either in the absence or in the presence of 1 mM NGD+. Product analysis was performed by HPLC.
In a previous study [31], we demonstrated that, in contrast with all other known ADP-ribosyl cyclases, including mammalian CD38 and CD157, and Aplysia cyclase, the novel cyclase detected in a membrane extract from Cd38−/− brain was unable to transform NGD+ (a surrogate NAD+ analogue) into cGDPR (a fluorescent analogue of cADPR). In the present study, we tested the ability of NGD+ to compete with the endogenous substrate NAD+ for binding to the active site of the synaptosomal enzyme. Therefore the Km (app) of NAD+ was determined under the same experimental conditions as above, but in the presence of NGD+ (at 100 and 300 μM). NGD+ was shown to inhibit the synthesis of cADPR from NAD+, leading to an increase of the Km (app) for NAD+ (Figure 5A). The Km (app) values for NAD+ were plotted as a function of the NGD+ concentration (Figure 5B). This secondary plot is reasonably linear, indicating that NGD+ is able to displace NAD+ from the active site in a competitive manner. The deduced Ki for NGD+ was estimated to be 24 μM, i.e. in the same range as the Km for NAD+.
We confirmed this inhibitory effect of NGD+ by the use of the HPLC method. Thus NGD+ (1 mM) was shown to inhibit cADPR and ADPR production catalysed by the adult synaptosomal cyclase when using 60 μM substrate. Production of both metabolites was decreased by approx. 50–70% under the experimental conditions used, which include long incubation times in order to have sufficient sensitivity to observe the cADPR production by the HPLC method.
We have then tested the possibility that synaptosomes from Cd38−/− brain could utilize NGD+ as a substrate. To test a possible transformation of NGD+ into cGDPR, fluorimetric measurements with both adult (results not shown) and neonate (see Figure 6) Cd38−/− synaptosomes were performed. Synaptosomes were incubated (at 0.5 mg/ml) for 2 h at 37 °C with Emulphogen in the presence of NGD+ (100 μM). Representative results presented in Figure 6 show that a GDP-ribosyl cyclase activity was barely measurable. The final addition of Aplysia cyclase, which converts the residual substrate into cGDPR, indeed demonstrated that less than 1% (limit of detection) of NGD+ may have been transformed into cGDPR by the synaptosomal enzyme within the time period studied. Additional experiments were performed using an HPLC method (detailed in [42]), which permits the detection of both products, cGDPR and GDPR. Adult and neonate synaptosomes (0.4 mg/ml) were incubated with NGD+ (100 μM) for 2 and 4 h at 37 °C. The HPLC chromatograms show that a very small amount of NGD+ could be transformed into GDPR (approx. 4–5%), while no cGDPR formation could be detected (results not shown). Thus, even if the synaptosomal preparation represents an enriched preparation compared with the membrane fraction studied previously [31], we were unable to observe any cyclization of NGD+ by the use of two different methods. The synaptosomal enzyme from Cd38−/− mouse brain therefore appears to be a very poor GDP-ribosyl cyclase.
Figure 6. Absence of cGDPR formation by the ADP-ribosyl cyclase from Cd38−/− brain synaptosomes.

NGD+ (100 μM) was added to 0.5 mg/ml neonate brain synaptosomes from Cd38−/− mice incubated at 37 °C in solution D. Formation of cGDPR was detected by fluorescence at 420 nm upon excitation at 300 nm. After 2 h of incubation, Aplysia cyclase (0.3 μg/ml) was added to fully transform the residual NGD+ into cGDPR (control).
We then examined the pH-dependence of the synaptosomal enzyme activity using the HPLC method. To that end, the cADPR production by adult synaptosomes was studied after incubations of either 6 h at 37 °C or 7 h at 32 °C in the presence of 60 μM NAD+. Figure 7(A) shows that similar results were observed under both experimental conditions: the ADP-ribosyl cyclase activity, which is optimal at pH between 6.0 and 7.0, is strongly decreased at acidic pH, but is only slightly affected at pH 8.0. Reaction-progress curves of adult synaptosomes were obtained using the cycling assay at pH 6.0 and at pH 7.5 in the presence of 60 μM NAD+ (Figure 7B). We observed that the initial rates of cADPR production are similar at these two pH values. However, the shapes of the kinetics curves were different: at pH 7.5, the cADPR production is clearly slowed down after 2 h of incubation, whereas production is still progressing at pH 6.0 after this time. Such an effect is reminiscent of the paracatalytic inactivation, observed at pH >7.0, for the NADase activity of other members of that enzyme family [39].
Figure 7. pH effects on the CD38-independent synaptosomal ADP-ribosyl cyclase activity.
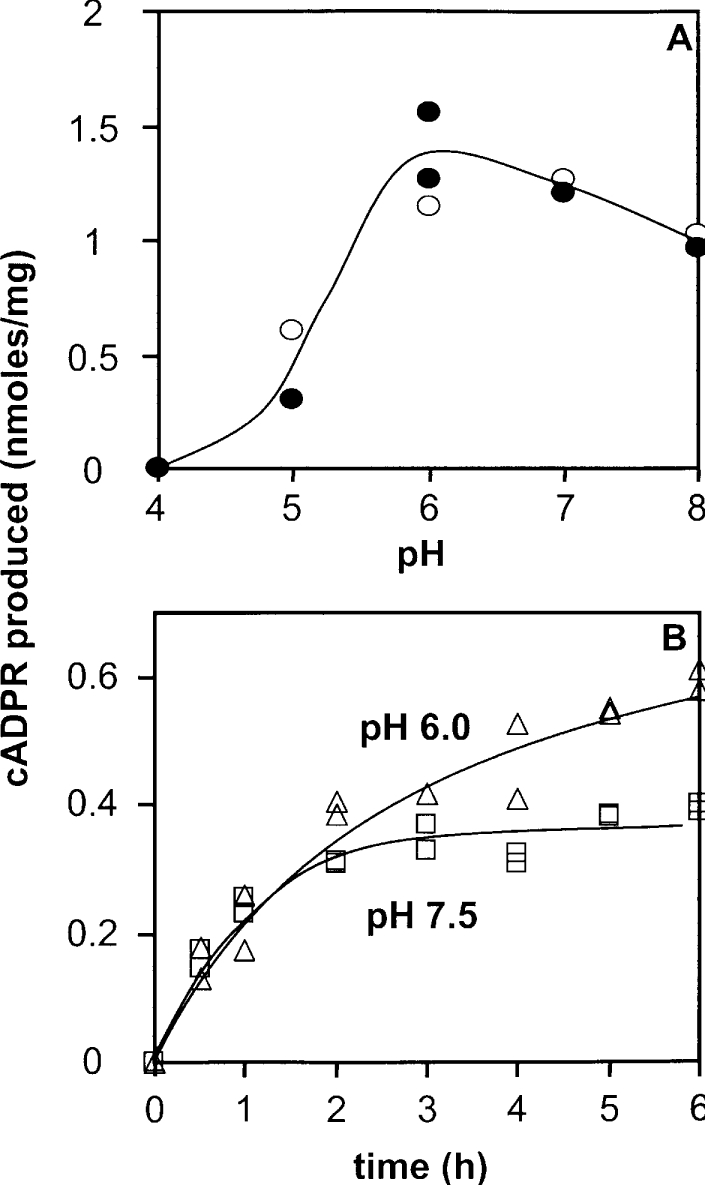
(A) Adult brain synaptosomes (1 mg of protein/ml) were incubated for 6 h at 37 °C or 7 h at 32 °C in the presence of [14C]NAD+ (4 mCi/mmol) and NAD+ (60 μM), 4 mM EDTA, 1% (w/v) Triton X-100 in 20 or 40 mM citrate/HCl (pH 4.0), Mes/NaOH (pH 5.0 and 6.0), or Tris/HCl (pH 7.0 or 8.0). Product analysis was performed using HPLC. (B) Synaptosomes (adult, 0.4 mg/ml) were incubated for the given times with 60 μM NAD+ in solution D (see legend to Figure 3) containing either 30 mM Tris/HCl (pH 7.5) or 80 mM Mes/NaOH (pH 6.0) in the presence of 1% (w/v) Emulphogen. cADPR formation was analysed using the cycling assay.
Altogether, these results show that the brain synaptosomal ADP-ribosyl cyclase has a broad optimal pH at 6.0–7.0. They provide a Km of 21 μM for NAD+. Moreover, they demonstrate that NGD+, despite its inability to be a substrate for the formation of cGDPR, can bind with a good affinity to the active site of the novel enzyme and inhibit the transformation of NAD+ (Ki 24 μM).
Micromolar concentrations of Zn2+ ions inhibit the ADP-ribosyl cyclase detected in Cd38−/− brain synaptosomes
We then studied the effect of Zn2+, which plays major roles in the synaptic activity and during brain development (for reviews, see [32,34,43,44]), on the Cd38−/− synaptosomal ADP-ribosyl cyclase activity.
We have therefore examined the effect of increasing concentrations of zinc acetate on the cADPR production by neonate and adult brain synaptosomes after incubation with 200 μM NAD+. The results presented in Figure 8 show a decrease of the cADPR production, with an IC50 of 1.8 μM with neonate synaptosomes and 0.9 μM with adult synaptosomes under these experimental conditions. To confirm these results, we first verified that zinc does not affect the cADPR determination using the cycling assay. To that end, we have analysed, using the cycling assay, standard solutions of cADPR (10 nM) containing various concentrations of zinc (0, 0.2, 2, 20 and 200 μM). We found that the presence of zinc at these concentrations did not modify the signal (slope of the increase in fluorescence at 590 nm) obtained (results not shown). Next, we also determined that no endogenous zinc from brain tissue remained in our synaptosomes preparations. Thus a zinc-chelating agent, TPEN [N,N,N′,N′-tetrakis(2-pyridylmethyl)-ethylenediamine], was added to brain synaptosomes, which were tested for their ability to synthesize cADPR: the same cADPR production was observed when incubating the adult synaptosomes with 60 μM NAD+ in the presence of 0, 10 or 25 μM TPEN (results not shown). Finally, we also checked that the decrease in the cADPR level was not due to an increase in cADPR hydrolysis. To that end, cADPR (100 nM or 1 μM) was incubated (at 37 °C for 2 h) in the presence of adult synaptosomes (0.4 mg/ml) without or with 10 μM Zn2+, and then worked-up as in the preceding experiments (precipitation with HClO4 and neutralization, treatment with the three hydrolases overnight to eliminate the contaminating nucleotides). The resulting cADPR was then estimated and compared with untreated cADPR. We found that neither the presence of the synaptosomes nor the overnight treatment with the three hydrolases changed the cADPR level (results not shown). Such control experiments demonstrate that the effect of Zn2+ is definitely due to an inhibition of the cADPR synthesis catalysed by the non-CD38 ADP-ribosyl cyclase.
Figure 8. Effect of Zn2+ on the ADP-ribosyl cyclase activity of synaptosomes from neonate and adult Cd38−/− mice.
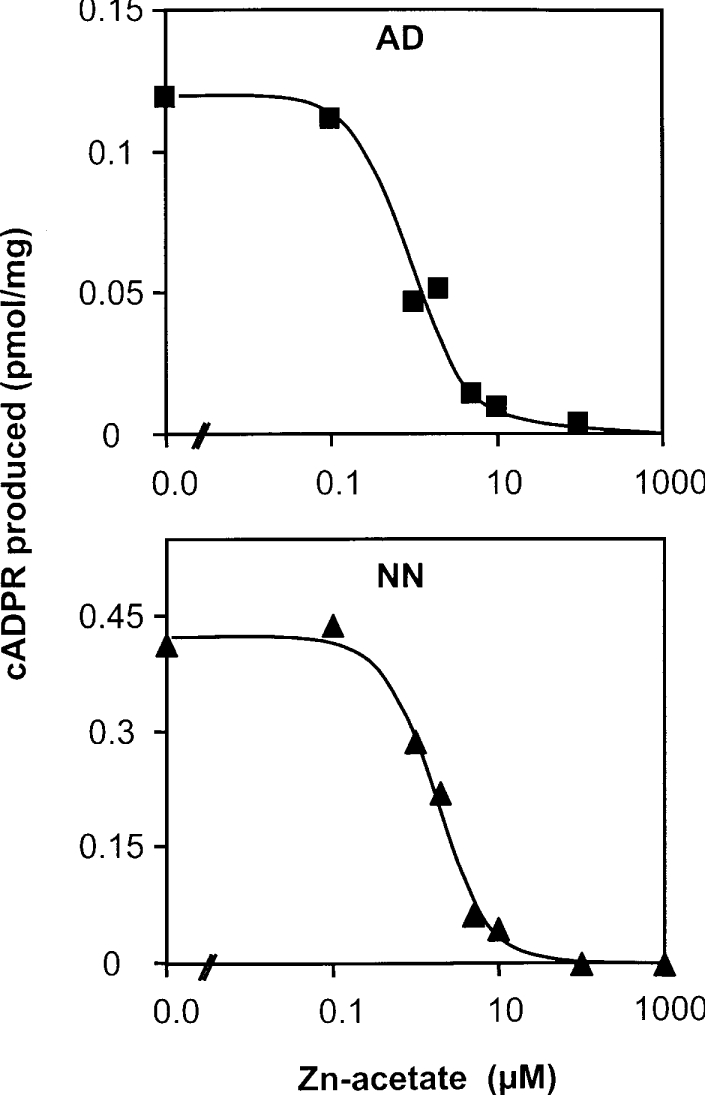
Synaptosomes (0.4 mg/ml for adult and 0.2 mg/ml for neonate) were incubated [2 h for adult (AD) and 1 h for neonate (NN)] with 200 μM NAD+ in solution D (see legend to Figure 2) containing 1% (w/v) Emulphogen and various concentrations of zinc acetate. The cADPR formation was analysed using the cycling assay.
We also have tested the effect of two other divalent ions, Cu2+ and Ni2+, on the synaptosome cyclase activity. Under experimental conditions similar to those used to study the effect of Zn2+ (see the legend to Figure 8), we observed that 10 μM Cu2+ completely inhibited the formation of cADPR by adult brain synaptosomes, whereas 10 μM Ni2+ did not affect this activity (results not shown).
These divalent ions were shown to modify the ADP-ribosyl cyclase activity of known mammalian enzymes at millimolar concentrations: Zn2+, Cu2+ and Ni2+ activate CD38, whereas Zn2+ activates and Cu2+ inhibits CD157. The effects observed on the synaptosomal cyclase are thus clearly different, except the similar inhibitory effect of copper, which was only observed at higher concentrations on CD157 cyclase activity.
Thus our results show, for the first time, a micromolar sensitivity to zinc (and copper) of an intracellular ADP-ribosyl cyclase from brain, an enzyme which we found abundant within synaptosomes.
DISCUSSION
In the present study, we show for the first time that endogenous levels of cADPR are largely higher in developing brain compared with the adult tissue. We also demonstrate that these levels are totally independent of the presence of the CD38 protein, not only in the adult, as already shown by Partida-Sanchez et al. [9], but also during development. Interestingly, we have revealed previously the existence of a novel ADP-ribosyl cyclase in the Cd38−/− mouse brain whose activity was shown to be largely superior in developing brain than in the adult tissue [31]. The non-CD38 enzyme, expressed at all developmental stages, is thus undeniably in charge of the brain cADPR level, which is down-regulated during development. Moreover, the high amounts of cADPR detected in embryonic and neonate brain certainly reflect a key role of this cyclic metabolite during the developmental processes of the central nervous system.
In addition, we show that the novel ADP-ribosyl cyclase detected in Cd38−/− brain is abundant within synaptosomes purified from both adult and neonate mouse brains. This result is remarkable, since cADPR is believed to be involved in processes occurring at synaptic connections, including neurotransmitter release and long-term synaptic depression [15,16,45]. In this context, it must be taken into consideration that, although functional synapses are known to be completely fashioned after birth (around postnatal day 2), the first morphologically identified synapses appear earlier in mouse brain, around embryonic day 16 in the mouse [46]. The amount of synapses then increases continuously, and they are plentiful in the neonate brain. Since we found that the novel ADP-ribosyl cyclase is enriched in terminals of brain neural cells and that its activity is higher in neonate synaptosomes compared with adult synaptosomes, our results raise the question of the role of cADPR and its synthesizing enzyme in morphologically shaped but non-totally efficient synapses. The ADP-ribosyl cyclase and the metabolites it produces (cADPR, but also ADPR) may thus be involved in synaptogenesis, via regulation of calcium signals that are necessary for the appearance of novel constituents of the synapse or to the correct placement of these constituents in the mature synapse.
We have characterized further the ADP-ribosyl cyclase of brain synaptosomes. We showed that it functions best at pH between 6.0 and 7.0 and that the concentration of free calcium has no major effect on its cADPR production. We also showed that it shares some of the properties of other known enzymes of that family (including mammalian CD38 and CD157, and the Aplysia cyclase), such as the ability of synthesizing both cADPR and ADPR, an affinity for NAD+ in the micromolar range (Km 21 μM) and the ability to bind with a similar affinity the NAD+ analogue, NGD+ (Ki 24 μM). This latter result is intriguing, since the novel enzyme happens to be unable to transform NGD+ into the cyclic compound, cGDPR ([31], and the present study), in contrast with all other known enzymes of the family. The inability of the enzyme to cyclize the NGD+ molecule may be due to a different organization of its active site. Thus we describe an ADP-ribosyl cyclase which is competent to transform NAD+ into cADPR, and which is in charge of the endogenous cADPR level found in brain, but which is unable to transform NGD+ into cGDPR. Such results may have an essential impact on the analysis of the physiological role of cADPR and on ‘non-classical’ ADP-ribosyl cyclases which definitely may not be studied only using the NAD+ analogue, NGD+, to establish cyclization reactions.
Given our previous report with brain membrane extracts [31], we have also tested the effect of GTP[S] on the production of NAD+ metabolites by Cd38−/− brain synaptosomes. Results turned out to be more complex to analyse. Thus, in contrast with our previous work, we were unable to detect, under any conditions tested, a significant modification of the cADPR production after addition of GTP[S]. On the other hand, ADPR production was clearly increased (3–7-fold, depending on the medium and time of incubation) after the addition of GTP[S] in the absence of EDTA (results not shown), in agreement with what was observed with the less-purified brain membrane extracts. However, these results deserve further investigation, since new unidentified NAD+ metabolites were also detected on the chromatograms (results not shown).
Another major finding of the present study is that the ADP-ribosyl cyclase from brain synaptosomes is very sensitive to Zn2+, being totally inhibited by micromolar concentrations of that ion. The ADP-ribosyl cyclase activities of CD38 and CD157 were shown, in contrast, to be activated by high (millimolar) concentrations of zinc [47,48]. This opposing effect of zinc on the synaptosomal enzyme is another factor that strongly suggests essential structural differences between the novel enzyme and the other known mammalian ADP-ribosyl cyclases.
Moreover, the sensitivity of the synaptosomal ADP-ribosyl cyclase to zinc is opening new research avenues, because zinc has critical roles in the central nervous system, both on the synaptic activity and during brain development and aging (for reviews, see [32,34,43,44]). The nervous system contains high concentrations of zinc (average total brain zinc 150 μM [49]), which is distributed into three cellular pools: the largest exists in tight association with intracellular proteins (mostly metallothioneins), a second pool is sequestered in pre-synaptic vesicles of certain neurons (principally excitatory glutamatergic neurons of the forebrain), and the third pool is constituted by the free ions in the cytoplasm. Zinc is known to exert a neuromodulatory action at excitatory synapses of ‘gluzinergic neurons’ when released during synaptic transmission. This ion acts not only as an intercellular signalling messenger binding directly to post-synaptic receptors and ionic channels, but also as an intracellular signal by entering into post-synaptic neurons. Fluctuations of the intracellular free zinc concentration, [Zn2+]i, in the cytoplasm of neuronal and glial cells appear to represent important signals for these cells. Intracellular Zn2+ is indeed known to act on several cellular pathways, in order to enhance the antioxidant response, the activity of poly(ADPR) polymerase-1 and of protein kinase C enzymes, etc. Moreover, although [Zn2+]i in resting cells is thought to be maintained at very low levels (in the subnanomolar range), it may be elevated (in the micromolar range) during injurious stimuli [33,50]. Evidence has indicated that the entry of zinc into post-synaptic neurons contributes to the toxic excess occurring during seizure and traumatic brain injury. It has been suggested that high [Zn2+]i promotes neuronal death by inhibiting cellular energy production [43].
We propose that the brain synaptosomal ADP-ribosyl cyclase is a new intracellular target of neurotoxic Zn2+. By acting on this enzyme, Zn2+ may modify neural cell calcium homoeostasis. Altogether, our results strongly suggest a crucial role of the synaptosomal ADP-ribosyl cyclase and of the metabolites it produces, cADPR and ADPR, in calcium-dependent processes that are necessary for the development of the central nervous system and also in the adult brain. Moreover, their action may be linked to Zn2+ cellular signals, particularly during brain injury.
Acknowledgments
We thank Dr Frances Lund for precious discussions and comments on the manuscript. This work was supported by recurrent grants from the French INSERM. We thank the zootechnicians of the CEA-SDV-DRDC Department.
References
- 1.Guse A. H. Cyclic ADP-ribose: a novel Ca2+-mobilising second messenger. Cell. Signalling. 1999;11:309–316. doi: 10.1016/s0898-6568(99)00004-2. [DOI] [PubMed] [Google Scholar]
- 2.Higashida H., Hashii M., Yokoyama S., Hoshi N., Chen X. L., Egorova A., Noda M., Zhang J. S. Cyclic ADP-ribose as a second messenger revisited from a new aspect of signal transduction from receptors to ADP-ribosyl cyclase. Pharmacol. Ther. 2001;90:283–296. doi: 10.1016/s0163-7258(01)00142-5. [DOI] [PubMed] [Google Scholar]
- 3.Rutter G. A. Calcium signalling: NAADP comes out of the shadows. Biochem. J. 2003;373:e3–e4. doi: 10.1042/BJ20030472COM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinnear N. P., Boittin F. X., Thomas J. M., Galione A., Evans A. M. Lysosome–sarcoplasmic reticulum junctions: a trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J. Biol. Chem. 2004;279:54319–54326. doi: 10.1074/jbc.M406132200. [DOI] [PubMed] [Google Scholar]
- 5.Dammermann W., Guse A. H. Functional ryanodine receptor expression is required for NAADP-mediated local Ca2+ signaling in T-lymphocytes. J. Biol. Chem. 2005;280:21394–21399. doi: 10.1074/jbc.M413085200. [DOI] [PubMed] [Google Scholar]
- 6.Perraud A. L., Fleig A., Dunn C. A., Bagley L. A., Launay P., Schmitz C., Stokes A. J., Zhu Q., Bessman M. J., Penner R., et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature (London) 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 7.Kolisek M., Beck A., Fleig A., Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 8.Walseth T. F., Aarhus R., Zeleznikar R. J., Jr, Lee H. C. Determination of endogenous levels of cyclic ADP-ribose in rat tissues. Biochim. Biophys. Acta. 1991;1094:113–120. doi: 10.1016/0167-4889(91)90032-s. [DOI] [PubMed] [Google Scholar]
- 9.Partida-Sanchez S., Cockayne D. A., Monard S., Jacobson E. L., Oppenheimer N., Garvy B., Kusser K., Goodrich S., Howard M., Harmsen A., et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 2001;7:1209–1216. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 10.Hua S. Y., Tokimasa T., Takasawa S., Furuya Y., Nohmi M., Okamoto H., Kuba K. Cyclic ADP-ribose modulates Ca2+ release channels for activation by physiological Ca2+ entry in bullfrog sympathetic neurons. Neuron. 1994;12:1073–1079. doi: 10.1016/0896-6273(94)90315-8. [DOI] [PubMed] [Google Scholar]
- 11.Empson R. M., Galione A. Cyclic ADP-ribose enhances coupling between voltage-gated Ca2+ entry and intracellular Ca2+ release. J. Biol. Chem. 1997;272:20967–20970. doi: 10.1074/jbc.272.34.20967. [DOI] [PubMed] [Google Scholar]
- 12.Hashii M., Minabe Y., Higashida H. cADP-ribose potentiates cytosolic Ca2+ elevation and Ca2+ entry via L-type voltage-activated Ca2+ channels in NG108-NG115 neuronal cells. Biochem. J. 2000;345:207–215. [PMC free article] [PubMed] [Google Scholar]
- 13.Budde T., Sieg F., Braunewell K. H., Gundelfinger E. D., Pape H. C. Ca2+-induced Ca2+ release supports the relay mode of activity in thalamocortical cells. Neuron. 2000;26:483–492. doi: 10.1016/s0896-6273(00)81180-0. [DOI] [PubMed] [Google Scholar]
- 14.Verderio C., Bruzzone S., Zocchi E., Fedele E., Schenk U., De Flora A., Matteoli M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001;78:646–657. doi: 10.1046/j.1471-4159.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- 15.Mothet J. P., Fossier P., Meunier F. M., Stinnakre J., Tauc L., Baux G. Cyclic ADP-ribose and calcium-induced calcium release regulate neurotransmitter release at a cholinergic synapse of Aplysia. J. Physiol. 1998;507:405–414. doi: 10.1111/j.1469-7793.1998.405bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reyes-Harde M., Empson R., Potter B. V., Galione A., Stanton P. K. Evidence of a role for cyclic ADP-ribose in long-term synaptic depression in hippocampus. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4061–4066. doi: 10.1073/pnas.96.7.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higashida H., Yokoyama S., Hashii M., Taketo M., Higashida M., Takayasu T., Ohshima T., Takasawa S., Okamoto H., Noda M. Muscarinic receptor-mediated dual regulation of ADP-ribosyl cyclase in NG108-NG115 neuronal cell membranes. J. Biol. Chem. 1997;272:31272–31277. doi: 10.1074/jbc.272.50.31272. [DOI] [PubMed] [Google Scholar]
- 18.Morita K., Kitayama S., Dohi T. Stimulation of cyclic ADP-ribose synthesis by acetylcholine and its role in catecholamine release in bovine adrenal chromaffin cells. J. Biol. Chem. 1997;272:21002–21009. doi: 10.1074/jbc.272.34.21002. [DOI] [PubMed] [Google Scholar]
- 19.Pollock J., Crawford J. H., Wootton J. F., Seabrook G. R., Scott R. H. Metabotropic glutamate receptor activation and intracellular cyclic ADP-ribose release Ca2+ from the same store in cultured DRG neurones. Cell Calcium. 1999;26:139–148. doi: 10.1054/ceca.1999.0064. [DOI] [PubMed] [Google Scholar]
- 20.Hotta T., Asai K., Fujita K., Kato T., Higashida H. Membrane-bound form of ADP-ribosyl cyclase in rat cortical astrocytes in culture. J. Neurochem. 2000;74:669–675. doi: 10.1046/j.1471-4159.2000.740669.x. [DOI] [PubMed] [Google Scholar]
- 21.Noda M., Yasuda S., Okada M., Higashida H., Shimada A., Iwata N., Ozaki N., Nishikawa K., Shirasawa S., Uchida M., et al. Recombinant human serotonin 5A receptors stably expressed in C6 glioma cells couple to multiple signal transduction pathways. J. Neurochem. 2003;84:222–232. doi: 10.1046/j.1471-4159.2003.01518.x. [DOI] [PubMed] [Google Scholar]
- 22.Morikawa H., Khodakhah K., Williams J. T. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J. Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higashida H., Zhang J. S., Mochida S., Chen X. L., Shin Y., Noda M., Hossain K. Z., Hoshi N., Hashii M., Shigemoto R., et al. Subtype-specific coupling with ADP-ribosyl cyclase of metabotropic glutamate receptors in retina, cervical superior ganglion and NG108-NG115 cells. J. Neurochem. 2003;85:1148–1158. doi: 10.1046/j.1471-4159.2003.01751.x. [DOI] [PubMed] [Google Scholar]
- 24.Schuber F., Lund F. E. Structure and enzymology of ADP-ribosyl cyclases: conserved enzymes that produce multiple calcium mobilizing metabolites. Curr. Mol. Med. 2004;4:249–261. doi: 10.2174/1566524043360708. [DOI] [PubMed] [Google Scholar]
- 25.Glick D. L., Hellmich M. R., Beushausen S., Tempst P., Bayley H., Strumwasser F. Primary structure of a molluscan egg-specific NADase, a second-messenger enzyme. Cell Regul. 1991;2:211–218. doi: 10.1091/mbc.2.3.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.States D. J., Walseth T. F., Lee H. C. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem. Sci. 1992;17:495. doi: 10.1016/0968-0004(92)90337-9. [DOI] [PubMed] [Google Scholar]
- 27.Itoh M., Ishihara K., Tomizawa H., Tanaka H., Kobune Y., Ishikawa J., Kaisho T., Hirano T. Molecular cloning of murine BST-1 having homology with CD38 and Aplysia ADP-ribosyl cyclase. Biochem. Biophys. Res. Commun. 1994;203:1309–1317. doi: 10.1006/bbrc.1994.2325. [DOI] [PubMed] [Google Scholar]
- 28.Koguma T., Takasawa S., Tohgo A., Karasawa T., Furuya Y., Yonekura H., Okamoto H. Cloning and characterization of cDNA encoding rat ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase (homologue to human CD38) from islets of Langerhans. Biochim. Biophys. Acta. 1994;1223:160–162. doi: 10.1016/0167-4889(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 29.Yamada M., Mizuguchi M., Otsuka N., Ikeda K., Takahashi H. Ultrastructural localization of CD38 immunoreactivity in rat brain. Brain Res. 1997;756:52–60. doi: 10.1016/s0006-8993(97)00117-0. [DOI] [PubMed] [Google Scholar]
- 30.Ceni C., Pochon N., Brun V., Muller-Steffner H., Andrieux A., Grunwald D., Schuber F., De Waard M., Lund F. E., Villaz M., Moutin M.-J. CD38-dependent ADP-ribosyl cyclase activity in developing and adult mouse brain. Biochem. J. 2003;370:175–183. doi: 10.1042/BJ20020604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ceni C., Muller-Steffner H., Lund F., Pochon N., Schweitzer A., De Waard M., Schuber F., Villaz M., Moutin M.-J. Evidence for an intracellular ADP-ribosyl cyclase/NAD+-glycohydrolase in brain from CD38-deficient mice. J. Biol. Chem. 2003;278:40670–40678. doi: 10.1074/jbc.M301196200. [DOI] [PubMed] [Google Scholar]
- 32.Smart T. G., Hosie A. M., Miller P. S. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10:432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- 33.Choi D. W., Koh J. Y. Zinc and brain injury. Annu. Rev. Neurosci. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- 34.Bhatnagar S., Taneja S. Zinc and cognitive development. Br. J. Nutr. 2001;85(Suppl. 2):S139–S145. doi: 10.1079/bjn2000306. [DOI] [PubMed] [Google Scholar]
- 35.Phillips G. R., Huang J. K., Wang Y., Tanaka H., Shapiro L., Zhang W., Shan W. S., Arndt K., Frank M., Gordon R. E., et al. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- 36.Graeff R., Lee H. C. A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem. J. 2002;361:379–384. doi: 10.1042/bj3610379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walseth T. F., Wong L., Graeff R. M., Lee H. C. Bioassay for determining endogenous levels of cyclic ADP-ribose. Methods Enzymol. 1997;280:287–294. doi: 10.1016/s0076-6879(97)80120-6. [DOI] [PubMed] [Google Scholar]
- 38.Augustin A., Muller-Steffner H., Schuber F. Molecular cloning and functional expression of bovine spleen ecto-NAD+ glycohydrolase: structural identity with human CD38. Biochem. J. 2000;345:43–52. doi: 10.1042/bj3450043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price S. R., Pekala P. H. Pyridine nucleotide-linked glycohydrolases. In: Dolphin D., Poulson R., Avramovic O., editors. Pyridine Nucleotide Coenzymes: Chemical, Biochemical and Medical Aspects. New York: John Wiley & Sons; 1987. pp. 513–548. [Google Scholar]
- 40.Cakir-Kiefer C., Muller-Steffner H., Oppenheimer N., Schuber F. Kinetic competence of the cADP-ribose–CD38 complex as an intermediate in the CD38/NAD+ glycohydrolase-catalysed reactions: implication for CD38 signalling. Biochem. J. 2001;358:399–406. doi: 10.1042/0264-6021:3580399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cakir-Kiefer C., Muller-Steffner H., Schuber F. Unifying mechanism for Aplysia ADP-ribosyl cyclase and CD38/NAD+ glycohydrolases. Biochem. J. 2000;349:203–210. doi: 10.1042/0264-6021:3490203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodrich S. P., Muller-Steffner H., Osman A., Moutin M.-J., Kusser K., Roberts A., Woodland D. L., Randall T. D., Kellenberger E., LoVerde P. T., et al. Production of calcium-mobilizing metabolites by a novel member of the ADP-ribosyl cyclase family expressed in Schistosoma mansoni. Biochemistry. 2005;44:11082–11097. doi: 10.1021/bi050704r. [DOI] [PubMed] [Google Scholar]
- 43.Dineley K. E., Votyakova T. V., Reynolds I. J. Zinc inhibition of cellular energy production: implications for mitochondria and neurodegeneration. J. Neurochem. 2003;85:563–570. doi: 10.1046/j.1471-4159.2003.01678.x. [DOI] [PubMed] [Google Scholar]
- 44.Mocchegiani E., Bertoni-Freddari C., Marcellini F., Malavolta M. Brain, aging and neurodegeneration: role of zinc ion availability. Prog. Neurobiol. 2005;75:367–390. doi: 10.1016/j.pneurobio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 45.Brailoiu E., Miyamoto M. D. Inositol trisphosphate and cyclic adenosine diphosphate-ribose increase quantal transmitter release at frog motor nerve terminals: possible involvement of smooth endoplasmic reticulum. Neuroscience. 2000;95:927–931. doi: 10.1016/s0306-4522(99)00509-6. [DOI] [PubMed] [Google Scholar]
- 46.Bouwman J., Maia A. S., Camoletto P. G., Posthuma G., Roubos E. W., Oorschot V. M., Klumperman J., Verhage M. Quantification of synapse formation and maintenance in vivo in the absence of synaptic release. Neuroscience. 2004;126:115–126. doi: 10.1016/j.neuroscience.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 47.Kukimoto I., Hoshino S., Kontani K., Inageda K., Nishina H., Takahashi K., Katada T. Stimulation of ADP-ribosyl cyclase activity of the cell surface antigen CD38 by zinc ions resulting from inhibition of its NAD+ glycohydrolase activity. Eur. J. Biochem. 1996;239:177–182. doi: 10.1111/j.1432-1033.1996.0177u.x. [DOI] [PubMed] [Google Scholar]
- 48.Hirata Y., Kimura N., Sato K., Ohsugi Y., Takasawa S., Okamoto H., Ishikawa J., Kaisho T., Ishihara K., Hirano T. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett. 1994;356:244–248. doi: 10.1016/0014-5793(94)01279-2. [DOI] [PubMed] [Google Scholar]
- 49.Takeda A. Movement of zinc and its functional significance in the brain. Brain Res. Rev. 2000;34:137–148. doi: 10.1016/s0165-0173(00)00044-8. [DOI] [PubMed] [Google Scholar]
- 50.Outten C. E., O'Halloran T. V. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]