

Abstract
Hitherto, all enveloped viruses were thought to shed their lipid membrane during entry into cells by membrane fusion. The extracellular form of Vaccinia virus has two lipid envelopes surrounding the virus core, and consequently a single fusion event will not deliver a naked core into the cell. Here we report a previously underscribed mechanism in which the outer viral membrane is disrupted by a ligand-induced nonfusogenic reaction, followed by the fusion of the inner viral membrane with the plasma membrane and penetration of the virus core into the cytoplasm. The dissolution of the outer envelope depends on interactions with cellular polyanionic molecules and requires the virus glycoproteins A34 and B5. This discovery represents a remarkable example of how viruses manipulate biological membranes, solves the topological problem of how a double-enveloped virus enters cells, reveals a new effect of polyanions on viruses, and provides a therapeutic approach for treatment of poxvirus infections, such as smallpox.
Keywords: antiviral therapy, extracellular enveloped virus, membrane dissolution, Vaccinia virus, virus entry
Hitherto, membrane fusion was the only known mechanism by which enveloped viruses overcome the lipid barrier to enter and replicate in cells (1, 2). Here we show that the extracellular enveloped virus (EEV) of Vaccinia virus (VACV) sheds its outer lipid membrane by a ligand-dependent nonfusogenic mechanism.
VACV replication produces several distinct virions: the intracellular mature virus (IMV), intracellular enveloped virus, cell-associated enveloped virus (CEV), and EEV (3, 4). IMV is surrounded by one lipid membrane (5–9) and is physically robust to aid virus transmission between hosts. CEV and EEV are IMV particles wrapped with an additional membrane derived from the trans-Golgi network (10) or endosomes (11) and are responsible for virus dissemination within the host (4). This extra lipid envelope (EEV membrane) and the associated virus and host membrane proteins serve to protect the IMV particle within from immune surveillance and may contribute to a broader cell tropism of the virus (4). Recently, we provided unequivocal electron micrographs showing that IMV enters by fusion with the plasma membrane (8), consistent with previous reports (6, 12, 13), and the recent genetic evidence for entry by fusion (14–17). Once the naked core has entered the cytoplasm, it moves deeper into the cell on microtubules (18). However, for VACV EEV, the additional EEV membrane presents an unexplained topological problem for entry, because fusion of the EEV outer envelope with the plasma membrane or the membrane of an intracellular vesicle will release only an IMV, instead of a naked core, into the cytosol. For a recent review of VACV entry, see ref. 19.
Here we studied the entry of EEV by immuno-EM and demonstrated that the EEV outer membrane is disrupted at the point of cell contact after binding. This enables the IMV within to enter the cell by fusion with the plasma membrane. The ligands required for membrane rupture were identified on EEV (B5 and A34) and the cell surface (glycosaminoglycans, GAGs). Last, we applied this discovery and showed that polyionic compounds such as heparin (HP) synergize with anti-IMV antibody in protecting against orthopoxvirus infection in vivo. These results explain how the topological problem of having two membranes is solved during EEV entry and reiterate the importance of inducing an immune response to the extracellular form of orthopoxviruses in vaccination.
Results
The binding and entry of EEV to cells were studied by immuno-EM. EEV is produced in low concentration, and its purification by gradient centrifugation damages the fragile outer membrane. So, to achieve a high concentration of EEV sufficient for study by EM, we adopted two approaches. EEV was either spinoculated onto cells (8) or concentrated by centrifugation and then disaggregated by gentle sonication for passive binding to cells. The second approach disrupted the outer EEV membrane in only ≈5% of virions. Using either method, the number of bound EEV particles was increased 200-fold compared to passive binding of unconcentrated virus, as shown by immunofluorescent microscopy (20). EEV particles bound to cells on ice were easily distinguished from IMV (<20% of all virions) by the extra membrane that was labeled by the EEV-specific anti-B5 mAb (Fig. 1A). After warming to 37°C and in some virions even at 4°C (Fig. 6, which is published as supporting information on the PNAS web site), the outer EEV envelope was disrupted at the site of cell contact exposing the IMV particle to the cell surface (Fig. 1B). In numerous images (n > 200), the EEV outer envelope did not fuse with the plasma membrane but remained on the outside of the cell over the IMV particle as a shroud (Fig. 1 B–D). The entry of the virus core then continued as for IMV (8). The IMV membrane fused with the plasma membrane (only after warming to 37°C), and direct continuity of these membranes is evident (Fig. 1 C and D). The IMV membrane then flattened into the plane of the plasma membrane (Fig. 1D), the core moved away from the site of entry (Fig. 1E), and the EEV membrane was left outside the cell (Fig. 1F). The entry of EEV studied by spinoculation (Fig. 1) and passive binding of the concentrated VACV strain IHD-J (Fig. 7, which is published as supporting information on the PNAS web site) were indistinguishable. Dissolution of the EEV membrane occurred only at the point of contact with the cell and was not seen with EEV bound to glass substrates at either 4°C or 37°C (Fig. 6), indicating a requirement for interaction with a specific cell surface molecule(s). This also highlights the fact that the EEV membrane protects the IMV within at all times, from release from the previous infected cell up to entry into the new cell. We have called this process ligand-dependent nonfusogenic dissolution of a virus membrane. This mechanism enables entry of a double-enveloped virus.
Fig. 1.

EM study of VACV EEV entry. Fresh EEV of VACV strain WR was spinoculated onto PtK2 cells at 4°C (A) and then incubated at 37°C for 10 min (B–F). The EEV surface was labeled by rat anti-B5 mAb 19C2 (10), rabbit anti-rat IgG followed by 6-nm protein A-gold conjugate, and the samples were processed for EM. (Scale bars, 100 nm.) Arrowheads (C and D) indicate continuity between the cell membrane and IMV membrane.
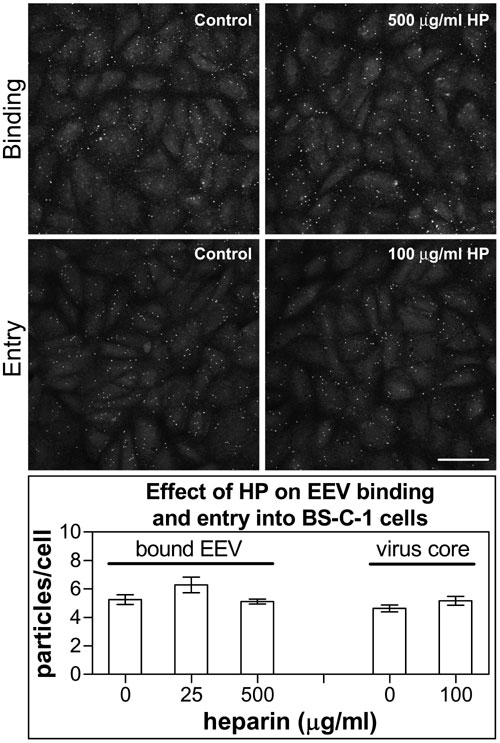
The cellular receptors for poxviruses are unknown, and claims that VACV or Myxoma virus used the epidermal growth factor receptor (21) or chemokine receptors (22) have been refuted (23, 24). In an attempt to identify the binding receptor for EEV, we discovered that, in the presence of the polyanions (PAs), HP, or dextran sulfate (DS), EEV became sensitive to IMV-neutralizing antibody (IMV-NAb) (Fig. 2A). By immunofluorescent staining, it was shown that EEV binding to or penetration into cells was not inhibited by HP (Fig. 8, which is published as supporting information on the PNAS web site). This observation suggested that PAs damage the outer EEV membrane, rendering the virus susceptible to antibody targeting the IMV surface.
Fig. 2.

Inhibition of EEV infectivity by IMV-NAb and PAs. (A) HP (Mr 4,000–6,000) or DS (derived from dextran Mr 5,000) was incubated at 25 μg/ml with virus produced in the supernatant of infected cells in the presence or absence of IMV-neutralizing mAb 2D5 (49) (diluted 1:1,000) at 37°C for 1 h, and infectivity was measured by plaque assay. EEV preparations contained >70% of total infectivity that was resistant to mAb 2D5, representing EEV with intact membranes. Data shown are the mean of two experiments ± SD. (B) Effect of size and charge of PAs. The infectivity of EEV treated with mAb 2D5 (diluted 1:1,000) in the presence of HP, DS, DS-high molecular weight (HMW) (high-Mr 500,000), HP-HMW (Mr 15,000), HP-OverS (over-sulfated HP-HMW) or HP-DeS (desulfated HP-HMW) was determined by plaque assay. Data shown are the mean of two experiments. (C) Anticomet assay. Monolayers of BS-C-1 cells were infected with 25 pfu of VACV for 2 h. The cells were washed and overlaid with medium with or without mAb 2D5 (diluted 1:500) and HP (100 μg/ml), as indicated. The monolayers were stained with crystal violet solution after 3 days.
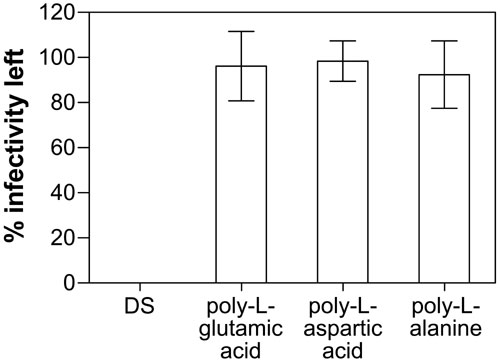
The potency of PAs depends on their charge and size, with larger more negatively charged molecules being more effective (Fig. 2B and Fig. 9, which is published as supporting information on the PNAS web site). The 90% inhibition concentration (IC90) of EEV by HP or DS in the presence of IMV-NAb was ≈7 and 5 μg/ml, respectively, but significantly lower concentrations of high-Mr HP and DS (<1 μg/ml) were able to achieve this level of inhibition. Desulfated HP was nonreactive. Notably, PAs were effective against EEV at concentrations orders of magnitude lower than tested against IMV, where they were largely ineffective (8, 25, 26). Although anionic charges are important, other polyanionic molecules, including poly-l-glutamic and poly-l-aspartic acids, had no effect (Fig. 10, which is published as supporting information on the PNAS web site). This suggests that the carbohydrate structures may also be important for this phenomenon.
The spread of EEV in cell culture, measured by the formation of comet-shaped plaques (27), was also blocked by HP in the presence of IMV-NAb, whereas IMV-NAb or HP alone was ineffective (Fig. 2C).
HP also inhibited the formation of virus-tipped actin tails (28, 29) on the surface of VACV-infected cells dramatically (Fig. 3A), and these structures are important for efficient cell-to-cell spread and VACV virulence (3, 4). To investigate this inhibition, the structure of CEV, especially the integrity of the outer membrane, was studied by EM. When HP was incubated with infected cells, fewer actin tails were found (Fig. 3A), and virions attached to the cell surface (CEV) lacked the outer CEV membrane (Fig. 3B Upper). Similarly, HP disrupted the integrity of the EEV membrane in EEV preparations (Fig. 3B Lower). In the absence of HP, 98.5% (SEM = 0.72) of the circumference of EEV particles (n = 62) was covered with the EEV membrane, whereas after HP treatment, only 46.4% (SEM = 3.44) was covered (n = 46) (a statistically significant difference, P = 0.0001). These data confirm that PAs rupture the outer EEV membrane and suggest that actin tail formation requires intact CEV.
Fig. 3.

Effects of PAs and GAGs on VACV-induced actin tail formation and EEV entry. (A) PtK2 cells were infected with VACV strain WR at 5 pfu per cell for 14 h. HP or DS (50 μg/ml) was added to the medium during or after the incubation. Actin and B5 were labeled with TRITC-phalloidin and rat mAb 19C2, respectively, and the samples were processed for immunofluorescent microscopy as described (20). In the presence of HP and DS during incubation, fewer cells (reduced from 55% to 15%, n = 100) were found to make actin tails. Bar chart shows the mean number of actin tails found in cells that made actin tails. Error bar = standard error. (B) Disruption of CEV/EEV membrane by HP. (Upper) Shown are the virions on the surface of VACV-infected RK13 cells −/+ HP (200 μg/ml). Note intact CEV with fully wrapped double envelopes −HP (Upper Left and Inset) and loss of outer membrane +HP (Upper Right and Inset). (Lower) Shown is isolated EEV treated with or without HP then concentrated by centrifugation. (Scale bar, 100 nm.) (C) EM study of VACV EEV entry into L cells and sog9 cells that lack heparan sulfate and CS. Fresh EEV of VACV strain WR was spinoculated onto cells at 4°C and then incubated at 37°C for 10 min. The EEV surface was labeled by rat anti-B5 mAb as in Fig. 1. In two independent experiments, 100 virions were identified, and bound intact EEV, ruptured EEV, and IMV were scored. No rupture of the EEV membrane was seen on sog9 cells.
Having demonstrated that the EEV membrane is disrupted at point of contact between the virus and cell so as to allow entry of IMV within, we investigated the role of cell surface GAGs in this process by studying EEV entry in GAG-deficient cells by EM. Sog9 cells are derived from L cells and lack cell surface polyanionic heparan sulfate (HS) and chondroitin sulfate (CS) (30). In two independent experiments, virions (n = 100) bound to sog9 cells showed no evidence of EEV membrane rupture after 10-min incubation at 37°C, whereas EEV bound to parental L cells showed rupture of the EEV outer membrane (Fig. 3C), and in some cases, there was fusion of the IMV with the plasma membrane (data not shown). Even after 30-min incubation, no rupture of EEV was seen on sog9 cells (data not shown). Therefore, cell surface GAGs are responsible for the dissolution of the EEV membrane.
To address which EEV surface protein(s) is required for this unusual phenomenon, we studied EEV made by a panel of VACV mutants with individual EEV genes deleted (31–35) (Fig. 4). Loss of the A56 or A33 protein did not affect this phenomenon. The F13 protein has sequence similarity to mammalian phospholipase (PL) D and has PL activity to metabolize phospholipids to phosphatidic acids (36–38) and so was an attractive candidate for involvement in membrane dissolution. However, F13 is important during morphogenesis for intracellular enveloped virus formation, and so its role in entry is difficult to study because without it very little EEV is produced (35). EEV made by a mutant lacking F13L had a partially resistant phenotype, but inhibition of F13 phospholipase activity using butanol-1 (38) did not make wild-type EEV resistant to IMV-NAb and PA treatment (Fig. 11, which is published as supporting information on the PNAS web site). Moreover, that some EEV bound to cells on ice also had a ruptured outer membrane at the site of contact (Fig. 6) argues against an enzymatic reaction. In contrast, EEV from virus mutants lacking protein A34 or B5 were resistant to neutralization by IMV-NAb in the presence of PAs (Fig. 4), showing these proteins, which form a complex (39), are required. EM showed that the outer envelope of CEV/EEV made by these mutants remained intact after addition of HP (data not shown). Interestingly, EEV made by mutants lacking B5 or A34 remains infectious, although with reduced specific infectivity in the latter case (33), and so the mechanism(s) by which these virions enter cells remains to be determined.
Fig. 4.

Genetic analysis of EEV proteins required for sensitivity to PAs. EEV made by WT virus (VACV strain WR) and the deletion mutant viruses vΔA56R (31), vΔA33R (32), vΔA34R (33), vΔB5R (34), and vΔF13L (35) were incubated with mAb 2D5 (diluted 1:1,000) ± 2 μg/ml of HP or DS-HMW for 1 h at 37°C, and virus infectivity was determined by plaque assay. Data shown are the mean of two experiments ± SD. The asterisk indicates virus that produced low levels and poor quality EEV (<25% total infectivity in culture supernatant was resistant to mAb 2D5 versus >70% for other mutants).
Last, we tested whether PAs had therapeutic value against poxvirus infection in vivo. The ability of PAs to render EEV susceptible to neutralization by IMV-NAb is important, because EEV and CEV are responsible for virus dissemination within the host, but they are difficult to neutralize by antibody. Only recently, mAbs that can partially neutralize EEV were identified, but these were 2–3 orders of magnitude weaker than anti-IMV mAbs (40). We and others have shown that EEV-NAb are important for protection (27, 41–43) and are present in humans after smallpox vaccination (43, 44) at lower levels than anti-IMV antibodies (43). To investigate the potential therapeutic value of PAs, mice were injected with rabbit antibody raised against inactivated IMV particles (Rb anti-IMV Ab) and challenged 1 day later with VACV. Two days after challenge, PAs were administrated intranasally. Intranasal infection of mice with VACV strain Western Reserve (WR) causes pneumonia and weight loss. Measurement of body weight and signs of illness (43) showed that Rb anti-IMV Ab provided benefit but was inferior to convalescent antibody (Rb anti-VACV Ab) that contained similar anti-IMV but 25-fold higher anti-EEV activity (Fig. 5A). Interestingly, PAs and IMV-NAb were synergistic, and mice that received Rb anti-IMV Ab and HP were protected as well as mice that received rabbit convalescent antibody (Fig. 5A Left). DS had a similar effect, although it was less potent than HP. The combined treatment with antibody and PAs protected the mice significantly better than antibody alone (P < 0.05; weight change, HP, days 4–10; DS, days 5–10; signs of illness, HP and DS, days 5–10). In control groups receiving irrelevant IgG, animals receiving HP showed a better recovery in signs of illness (P < 0.05, days 7–10), although this treatment did not provide a statistically significant inhibition of weight loss.
Fig. 5.

Synergistic effect of PAs and IMV-NAb in vivo. (A) Mice were injected i.p. with PBS (Mock), rabbit (Rb) anti-VACV Ab, Rb anti-IMV Ab, control Rb IgG, or human (Hu) anti-VACV Ab, 1 day before challenging with 1 × 104 pfu of VACV WR by the intranasal route (43). After 2 days of infection, 20 μl of PBS, HP, or DS (2 mg/mouse) was administered to the mice intranasally. The weight and signs of illness of the mice were recorded daily. Data shown are the mean of six mice ± standard error. Data for Hu anti-VACV Ab are presented separately (Right). (B) Virus titers in infected mice. Groups of mice (n = 9) were treated as in some groups of A, and three mice were killed on days 2, 4, and 6 postinfection for virus titration. Virus titers in lungs, spleens, and brains were determined by plaque assay on RK13 cells. Dashed lines indicate the detection limit of the assay. The statistical significance was analyzed by Student’s t test (one-tailed distribution and two-sample unequal variance) by using Microsoft excel software.
The experiment was extended to study the synergism of PAs with a human VACV-immune antibody (Fig. 5A Right). This antibody (Hu anti-VACV Ab) originated from an individual who had been vaccinated five times against smallpox and contained 52% of anti-IMV and 3% of anti-EEV activity compared with Rb anti-VACV Ab (43). The results demonstrated the benefit of HP in combination with human antibody (versus Hu anti-VACV Ab alone; P < 0.05 for weight loss and signs of illness, days 4–10). This suggested that PAs may be used in conjunction with vaccinia-immune globulin in treating smallpox and vaccine-related complications.
The replication of virus in primary (lungs) and secondary (spleen and brain) tissues was studied (Fig. 5B). The administration of HP 2 days after infection of mice that had been injected with Rb anti-IMV Ab suppressed virus replication in lungs slightly on day 4 and significantly on day 6 (P = 0.005), thus explaining the better protection in this group (Fig. 5A). HP alone caused slight, but statistically insignificant, reduction of virus titers in lungs on days 4 and 6. HP also had a transient effect in preventing virus spreading to spleens on day 4 (P = 0.003), but it is uncertain whether HP acted synergistically with IMV-NAb, because virus spreading to spleens was already inefficient in the presence of the antibody. Reduction of virus replication in brains by HP and antibody was observed on day 6, but it is not statistically significant (P = 0.11). The results are consistent with our previous study showing that protection against weight loss and development of illness in this mouse viral pneumonia model correlates with inhibition of virus replication in lung but not brain (43).
Discussion
In this report, we show that VACV has developed a unique strategy to enable the double-enveloped EEV to enter the cell. The EEV outer envelope is removed by a ligand-dependent nonfusogenic membrane dissolution process, followed by fusion of the IMV membrane with the plasma membrane. Although the molecular mechanism by which the EEV membrane is disrupted remains to be determined, the nonfusogenic removal of a virus membrane is unprecedented in virology and cell biology, for all enveloped viruses had been thought to remove their membrane and enter cells by membrane fusion (1, 2). These results also demonstrated that the cellular factors required for EEV membrane dissolution are highly anionic, and GAGs are likely candidates.
The EEV entry mechanism presented here is consistent with genetic data showing that the VACV IMV surface proteins A28 (14), A21 (17), H2 (15), and L5 (16) are needed for fusion and entry of both IMV and CEV. The observation that both IMV and EEV enter cells by membrane fusion supports previous work by Doms et al. (13), who proposed that both forms of VACV fuse with the plasma membrane, although for EEV, it turns out to be the internal, not external, membrane that fuses. The model for EEV entry presented here has some similarity with that of Ichihashi (45) and Vanderplasschen et al. (46), who proposed that the EEV outer membrane is destroyed by a nonfusogenic mechanism (low pH) within an acidified vesicle followed by fusion of the IMV envelope with the vesicle membrane. Although low pH does disrupt the EEV outer membrane (46), and B5 protein is required for this disruption (O. Krauss and G.L.S., unpublished data), we show here that membrane dissolution actually occurs on the cell surface and at neutral pH and requires neither endocytosis nor acidification.
A useful application of these data is the use of PAs and anti-IMV antibody for treating poxvirus infections. The EEV membrane has been shown to protect the IMV surface from complement and antibody to which it is sensitive (4, 27). Here we showed that this membrane protects IMV from IMV-NAb in vivo but can be overcome by PA-induced rupture of this membrane. PAs ameliorate poxvirus infection by blocking actin tail formation and sensitizing EEV to anti-IMV antibody, therefore providing a significant benefit to the host even after infection has already been established. PAs have an in vitro inhibitory effect on many enveloped viruses, including HIV, herpes, influenza, respiratory syncytial, measles, and parainfluenza viruses (25, 26). However, the undesirable properties such as toxicity, short half-life, and inactivity in plasma had led to failure in clinical trials (25). Our data demonstrated that PAs can be effective if applied directly to the site of virus replication, in this case the respiratory system for viral pneumonia. Recent advances in the design and modification of PAs have reduced toxicity, and several anionic polymers are now in clinical trials as topical and therapeutic antiviral agents against HIV infection (47, 48). These compounds are potentially useful for treating acute respiratory illness caused by enveloped viruses.
Materials and Methods
Immuno-EM.
EEV from the supernatant of 20 T175 flasks of BHK-21 cells was sedimented at 19,000 × g, resuspended in 1 ml, gently sonicated, and bound to chilled PtK2 cells for 1 h on ice. For spinoculation, one flask of EEV supernatant was centrifuged onto chilled PtK2 cells at 650 × g for 1 h at 4°C. Cells were washed with ice-cold PBS, and samples were either fixed for virus-binding analysis or incubated at 37°C for 10 min to permit virus entry. Samples were fixed for 10 min on ice with 4% paraformaldehyde (PFA) in 250 mM Hepes buffer (pH 7.4), followed by 8% PFA at room temperature for 50 min and quenched for 20 min with 20 mM glycine in PBS.
Preembedding immunogold labeling was performed on all samples, as described (7, 8). The EEV membrane was labeled with mouse mAb 19C2 (anti-B5, diluted 1:10) for 1 h, followed by rabbit anti-rat IgG (diluted 1:50). All samples were incubated with protein A conjugated to 6-nm gold particles (diluted 1:100) for 1 h before processing for conventional EM. Samples were viewed by using an FEI Tecnai G2 electron microscope (FEI, Eindhoven, The Netherlands) with a Soft Imaging System Megaview III charge-coupled device camera. Images were collected at 1,376 × 1,032 × 16 pixels by using analysis version docu software (Olympus Soft Imaging Solutions, Münster, Germany).
Plaque-Reduction Assay.
Fresh EEV preparation [diluted to ≈600 plaque-forming units (pfu)/ml] was incubated with an equal volume of diluted PAs with or without the IMV-neutralizing mAb 2D5 (diluted 1:1,000) (49) for 1 h at 37°C. The mixture (0.5 ml, containing ≈150 pfu of input infectivity) was adsorbed onto BS-C-1 cells on six-well plates for 1 h at 37°C. Unbound virus and the antibody mixture were washed away and overlaid with DMEM containing 2.5% FBS and 1.5% carboxymethylcellulose. The cells were incubated for 2 days (for WR and vΔA56R), 5 days (vΔB5R and vΔA34R), and 6 days (vΔA33R and vΔF13L), respectively, and stained with 0.05% crystal violet in 15% ethanol, and the plaques were counted.
Anticomet Assay.
Cells grown on six-well plates were infected with 25 pfu of VACV per well for 2 h at 37°C. Unbound virus was washed away. DMEM (2.5% FBS) containing HP and/or mAb 2D5 was added to the cells and incubated for 44 h before staining with crystal violet solution.
Quantification of EEV, Virus Cores, and Actin Tails by Indirect Immunofluorescent Staining and Confocal Microscopy.
The principles for the detection and quantification of actin tails, EEV, and virus cores were described previously (20, 50). For quantification of EEV binding and entry, EEV ± HP was bound to BS-C-1 cells for 2 h on ice. Unbound virus was washed away by ice-cold PBS, and cells were fixed or incubated at 37°C for 1 h to study virus entry. EEV and virus cores were labeled with mAb 15B6 (specific to protein F13) and rabbit anti-VACV core antibody, and the samples were processed for confocal microscopy (20). For actin tail quantification, PtK2 cells were infected with VACV strain WR at 5 pfu per cell for 14 h. HP or DS (50 μg/ml) was added to the medium during or after incubation. Actin and B5 were labeled with tetramethylrhodamine B isothiocyanate–phalloidin and rat mAb 19C2, respectively, and the samples were quantified as for bound virus.
In Vivo Virus Challenge Studies.
VACV WR was prepared from infected RK13 cells by sedimentation through a 36% (wt/vol) sucrose cushion, and virus infectivity was titrated on BS-C-1 cells (20). The titer of virus administered to animals was redetermined in each experiment. Six- to eight-week-old female BALB/c mice (16–22 g) were injected i.p. with specified antibodies (in 200 μl) 1 day before virus challenge. For intranasal administration of PAs or virus challenge, mice were anesthetized with isoflurane and inoculated intranasally with 20 μl (10 μl each nostril) of fluid. Body-weight change and signs of illness of mice were recorded daily, and mice that reached humane end point (loss of 30% body weight) were killed. Signs of illness were scored as described (43, 51). To measure virus in organs, mice were killed, and the lungs, spleens, and brains were collected. The tissues were stored frozen in culture medium. Thawed tissues were homogenized by using a syringe plunger and filtered through a 70-μm cell strainer. The homogenates were freeze-thawed three times to release virus, which was titrated on RK13 cells. Animal housing and handling followed United Kingdom regulations.
Supplementary Material
Acknowledgments
We thank Bernard Moss (National Institutes of Health, Bethesda) for viruses vΔA33R and vΔF13L; Yasuo Ichihashi (Niigata University, Niigata, Japan) for mAb 2D5; Gareth Griffiths (European Molecular Biology Laboratory, Germany) for mAbs 19C2 (anti-B5), 15B6 (anti-F13), and rabbit anti-core Ab; and Frank Tufaro (Medigene, San Diego) for sog9 cells. This work was supported by the U.K. Medical Research Council and the Wellcome Trust. G.L.S. is a Wellcome Trust Principal Research Fellow, and K.L.R. holds a Biotechnology and Biological Sciences Research Council research studentship.
Abbreviations
- EEV
extracellular enveloped virus
- VACV
Vaccinia virus
- IMV
intracellular mature virus
- CEV
cell-associated enveloped virus
- GAGS
glycosaminoglycans
- HP
heparin
- PA
polyanion
- DS
dextran sulfate
- CS
chondroitin sulfate
- pfu
plaque-forming unit
- IMV-Nab
IMV-neutralizing antibody
- WR
Western Reserve
- HMW
high molecular weight.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Young J. A. T. In: Virology. Fields B. N., Knipe D. M., Howley P. M., Chanock R. M., Melnick J., Monath T. P., Roizman B., Straus S. E., editors. Vol. 1. Philadelphia: Lippincott–Raven; 2001. pp. 87–103. [Google Scholar]
- 2.Earp L. J., Delos S. E., Park H. E., White J. M. Curr. Top. Microbiol. Immunol. 2005;285:25–66. doi: 10.1007/3-540-26764-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moss B. In: Virology. Fields B. N., Knipe D. M., Howley P. M., Chanock R. M., Melnick J., Monath T. P., Roizman B., Straus S. E., editors. Vol. 2. Philadelphia: Lippincott–Raven; 2001. pp. 2849–2883. [Google Scholar]
- 4.Smith G. L., Vanderplasschen A., Law M. J. Gen. Virol. 2002;83:2915–2931. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- 5.Dales S., Siminovitch L. J. Biophys. Biochem. Cytol. 1961;10:475–503. doi: 10.1083/jcb.10.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang A., Metz D. H. J. Gen. Virol. 1976;32:275–282. doi: 10.1099/0022-1317-32-2-275. [DOI] [PubMed] [Google Scholar]
- 7.Hollinshead M., Vanderplasschen A., Smith G. L., Vaux D. J. J. Virol. 1999;73:1503–1517. doi: 10.1128/jvi.73.2.1503-1517.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter G. C., Law M., Hollinshead M., Smith G. L. J. Gen. Virol. 2005;86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- 9.Heuser J. J. Cell Biol. 2005;169:269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmelz M., Sodeik B., Ericsson M., Wolffe E. J., Shida H., Hiller G., Griffiths G. J. Virol. 1994;68:130–147. doi: 10.1128/jvi.68.1.130-147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tooze J., Hollinshead M., Reis B., Radsak K., Kern H. Eur. J. Cell Biol. 1993;60:163–178. [PubMed] [Google Scholar]
- 12.Armstrong J. A., Metz D. H., Young M. R. J. Gen. Virol. 1973;21:533–537. doi: 10.1099/0022-1317-21-3-533. [DOI] [PubMed] [Google Scholar]
- 13.Doms R. W., Blumenthal R., Moss B. J. Virol. 1990;64:4884–4892. doi: 10.1128/jvi.64.10.4884-4892.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senkevich T. G., Ward B. M., Moss B. J. Virol. 2004;78:2357–2366. doi: 10.1128/JVI.78.5.2357-2366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senkevich T. G., Moss B. J. Virol. 2005;79:4744–4754. doi: 10.1128/JVI.79.8.4744-4754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Townsley A. C., Senkevich T. G., Moss B. J. Virol. 2005;79:10988–10998. doi: 10.1128/JVI.79.17.10988-10998.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Townsley A. C., Senkevich T. G., Moss B. J. Virol. 2005;79:9458–9469. doi: 10.1128/JVI.79.15.9458-9469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter G. C., Rodger G., Murphy B. J., Law M., Krauss O., Hollinshead M., Smith G. L. J. Gen. Virol. 2003;84:2443–2458. doi: 10.1099/vir.0.19271-0. [DOI] [PubMed] [Google Scholar]
- 19.Moss B. Virology. 2006;344:48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 20.Law M., Smith G. L. Methods Mol. Biol. 2004;269:187–204. doi: 10.1385/1-59259-789-0:187. [DOI] [PubMed] [Google Scholar]
- 21.Eppstein D. A., Marsh Y. V., Schreiber A. B., Newman S. R., Todaro G. J., Nestor J. J., Jr Nature. 1985;318:663–665. doi: 10.1038/318663a0. [DOI] [PubMed] [Google Scholar]
- 22.Lalani A. S., Masters J., Zeng W., Barrett J., Pannu R., Everett H., Arendt C. W., McFadden G. Science. 1999;286:1968–1971. doi: 10.1126/science.286.5446.1968. [DOI] [PubMed] [Google Scholar]
- 23.Hügin A. W., Hauser C. J. Virol. 1994;68:8409–8412. doi: 10.1128/jvi.68.12.8409-8412.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masters J., Hinek A. A., Uddin S., Platanias L. C., Zeng W., McFadden G., Fish E. N. J. Biol. Chem. 2001;276:48371–48375. doi: 10.1074/jbc.M108019200. [DOI] [PubMed] [Google Scholar]
- 25.Luscher-Mattli M. Antivir. Chem. Chemother. 2000;11:249–259. doi: 10.1177/095632020001100401. [DOI] [PubMed] [Google Scholar]
- 26.De Clercq E. Clin. Microbiol. Rev. 2001;14:382–397. doi: 10.1128/CMR.14.2.382-397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boulter E. A., Appleyard G. Prog. Med. Virol. 1973;16:86–108. [PubMed] [Google Scholar]
- 28.Blasco R., Cole N. B., Moss B. J. Virol. 1991;65:4598–4608. doi: 10.1128/jvi.65.9.4598-4608.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cudmore S., Cossart P., Griffiths G., Way M. Nature. 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- 30.Banfield B. W., Leduc Y., Esford L., Schubert K., Tufaro F. J. Virol. 1995;69:3290–3298. doi: 10.1128/jvi.69.6.3290-3298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanderson C. M., Frischknecht F., Way M., Hollinshead M., Smith G. L. J. Gen. Virol. 1998;79:1415–1425. doi: 10.1099/0022-1317-79-6-1415. [DOI] [PubMed] [Google Scholar]
- 32.Roper R. L., Wolffe E. J., Weisberg A., Moss B. J. Virol. 1998;72:4192–4204. doi: 10.1128/jvi.72.5.4192-4204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIntosh A. A., Smith G. L. J. Virol. 1996;70:272–281. doi: 10.1128/jvi.70.1.272-281.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engelstad M., Smith G. L. Virology. 1993;194:627–637. doi: 10.1006/viro.1993.1302. [DOI] [PubMed] [Google Scholar]
- 35.Blasco R., Moss B. J. Virol. 1991;65:5910–5920. doi: 10.1128/jvi.65.11.5910-5920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung T. C., Roper R. L., Zhang Y., Rudge S. A., Temel R., Hammond S. M., Morris A. J., Moss B., Engebrecht J., Frohman M. A. EMBO J. 1997;16:4519–4530. doi: 10.1093/emboj/16.15.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baek S. H., Kwak J. Y., Lee S. H., Lee T., Ryu S. H., Uhlinger D. J., Lambeth J. D. J. Biol. Chem. 1997;272:32042–32049. doi: 10.1074/jbc.272.51.32042. [DOI] [PubMed] [Google Scholar]
- 38.Husain M., Moss B. J. Virol. 2002;76:7777–7789. doi: 10.1128/JVI.76.15.7777-7789.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Röttger S., Frischknecht F., Reckmann I., Smith G. L., Way M. J. Virol. 1999;73:2863–2875. doi: 10.1128/jvi.73.4.2863-2875.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aldaz-Carroll L., Whitbeck J. C., Ponce de Leon M., Lou H., Hirao L., Isaacs S. N., Moss B., Eisenberg R. J., Cohen G. H. J. Virol. 2005;79:6260–6271. doi: 10.1128/JVI.79.10.6260-6271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galmiche M. C., Goenaga J., Wittek R., Rindisbacher L. Virology. 1999;254:71–80. doi: 10.1006/viro.1998.9516. [DOI] [PubMed] [Google Scholar]
- 42.Fogg C., Lustig S., Whitbeck J. C., Eisenberg R. J., Cohen G. H., Moss B. J. Virol. 2004;78:10230–10237. doi: 10.1128/JVI.78.19.10230-10237.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Law M., Pütz M. M., Smith G. L. J. Gen. Virol. 2005;86:991–1000. doi: 10.1099/vir.0.80660-0. [DOI] [PubMed] [Google Scholar]
- 44.Pütz M. M., Alberini I., Midgley C. M., Manini I., Montomoli E., Smith G. L. J. Gen. Virol. 2005;86:2955–2960. doi: 10.1099/vir.0.81265-0. [DOI] [PubMed] [Google Scholar]
- 45.Ichihashi Y. Virology. 1996;217:478–485. doi: 10.1006/viro.1996.0142. [DOI] [PubMed] [Google Scholar]
- 46.Vanderplasschen A., Hollinshead M., Smith G. L. J. Gen. Virol. 1998;79:877–887. doi: 10.1099/0022-1317-79-4-877. [DOI] [PubMed] [Google Scholar]
- 47.Shaunak S., Davies D. S. Br. J. Clin. Pharmacol. 2002;54:75–80. doi: 10.1046/j.1365-2125.2002.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D’Cruz O. J., Uckun F. M. Curr. Pharm. Des. 2004;10:315–336. doi: 10.2174/1381612043386374. [DOI] [PubMed] [Google Scholar]
- 49.Ichihashi Y., Oie M. Virology. 1996;220:491–494. doi: 10.1006/viro.1996.0337. [DOI] [PubMed] [Google Scholar]
- 50.Law M., Hollinshead M., Lee H. J., Smith G. L. J. Gen. Virol. 2004;85:1279–1290. doi: 10.1099/vir.0.79863-0. [DOI] [PubMed] [Google Scholar]
- 51.Alcami A., Smith G. L. Cell. 1992;71:153–167. doi: 10.1016/0092-8674(92)90274-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}