

Abstract
Tetrahydrobiopterin (BH4) is an essential cofactor for several enzymes, including all three forms of nitric oxide synthases, the three aromatic hydroxylases, and glyceryl-ether mono-oxygenase. A proper level of BH4 is, therefore, necessary for the metabolism of phenylalanine and the production of nitric oxide, catecholamines, and serotonin. BH4 deficiency has been shown to be closely associated with diverse neurological psychiatric disorders. Sepiapterin reductase (SPR) is an enzyme that catalyzes the final step of BH4 biosynthesis. Whereas the number of cases of neuropsychological disorders resulting from deficiencies of other catalytic enzymes involved in BH4 biosynthesis and metabolism has been increasing, only a handful of cases of SPR deficiency have been reported, and the role of SPR in BH4 biosynthesis in vivo has been poorly understood. Here, we report that mice deficient in the Spr gene (Spr−/−) display disturbed pterin profiles and greatly diminished levels of dopamine, norepinephrine, and serotonin, indicating that SPR is essential for homeostasis of BH4 and for the normal functions of BH4-dependent enzymes. The Spr−/− mice exhibit phenylketonuria, dwarfism, and impaired body movement. Oral supplementation of BH4 and neurotransmitter precursors completely rescued dwarfism and phenylalanine metabolism. The biochemical and behavioral characteristics of Spr−/− mice share striking similarities with the symptoms observed in SPR-deficient patients. This Spr mutant strain of mice will be an invaluable resource to elucidate many important issues regarding SPR and BH4 deficiencies.
Tetrahydrobiopterin (BH4) is an essential cofactor for multiple enzymes, including phenylalanine-4-hydroxylase (PAH), tyrosine-3-hydroxylase (TH [MIM 191290]), tryptophan-5-hydroxylase (TPH [MIM 191060]), all three forms of nitric oxide synthase (NOS1 [MIM 163731], NOS2 [MIM 163730], and NOS3 [MIM 163729]), and glyceryl-ether mono-oxygenase (Thöny et al. 2000; Blau et al. 2001b). Deficiencies in BH4 metabolism can affect the function of all these enzymes, leading to a variety of metabolic syndromes. For example, PAH converts phenylalanine to tyrosine in the liver, which is the first step in phenylalanine degradation. When PAH is missing or defective, phenylalanine may reach levels of ⩾1 mM in the blood, leading to phenylketonuria (PKU [MIM 261600]) (Scriver and Kaufman 2001). These levels result in severe damage to early brain development unless strict dietary restriction of phenylalanine is instituted. Lower levels of phenylalanine (0.12–0.99 mM; termed “hyperphenylalaninemia”) are usually considered benign, although dietary modification may be considered at higher levels in this range (Huttenlocher 2000; Scriver and Kaufman 2001). PKU (or “variant PKU”) can also be caused by defects in BH4 biosynthesis (Blau et al. 1992; Thöny and Blau 1997; Thöny et al. 1998a, 1998b). Recent studies have shown that PKU-affected patients with certain BH4 deficiencies as well as PAH mutations benefitted from the supplementation of BH4 (Spaapen and Rubio-Gozalbo 2003; Fiege et al. 2005). In addition, TH and TPH are rate-limiting enzymes for the production of the biogenic amine neurotransmitters dopamine, norepinephrine, and serotonin (Fitzpatrick 1999). Deficiencies in TH or TPH have been implicated in recessive Segawa syndrome (MIM 605407), familial infantile parkinsonism (MIM 168600), and unipolar major depression (MIM 608516) (Ludecke et al. 1995, 1996; Swaans et al. 2000). BH4 deficiency can present with similar phenotypes (Blau et al. 2001b). Nitric oxide (NO) is synthesized by the three forms of NOS (NOS1–NOS3) and has a variety of physiological roles (Mungrue et al. 2003). Deficiencies in NOS function in human patients because of BH4 biosynthetic defects have not been carefully examined but may well exist.
BH4 homeostasis is regulated by the de novo biosynthesis pathway, as well as by the salvage pathway and the de novo regeneration pathway (fig. 1A). BH4 is synthesized from guanosine triphosphate (GTP) through a cascade of enzymatic modifications (Thöny et al. 2000; Blau et al. 2001a, 2001b). The first step of de novo biosynthesis is the conversion of GTP to d-erythro-7,8-dihydroneopterin triphosphate, which is mediated by GTP cyclohydrolase I (GTPCH [MIM 600225]). This reaction is thought to be the rate-limiting step in de novo biosynthesis. d-erythro-7,8-dihydroneopterin triphosphate is then converted to 6-pyruvoyl-tetrahydropterin (PTP) by 6-pyruvoyl-tetrahydropterin synthase (PTPS [MIM 261640]) and subsequently to BH4 by sepiapterin reductase (SPR [MIM 182125]) through reduced nicotinamide adenine dinucleotide phosphate (NADPH)–dependent reduction steps 1–3 (fig. 1A). Although SPR is known to be the major enzyme in these final reduction steps, aldo-keto reductases (AKR) and carbonyl reductases (CBR) can also convert PTP to BH4. AKR can convert PTP to 1′-oxo-2′-hydroxy tetrahydropterin (1′-oxo-TP) (Milstien and Kaufman 1989; Levine et al. 1990) and can also reduce 1′-hydroxy-2′-oxo-tetrahydropterin (2′-oxo-TP) to BH4 (Park et al. 1991). CBR can convert PTP to 1′-oxo-TP or to 2′-oxo-TP (Park et al. 1991). Therefore, in the presence of both AKR and CBR, BH4 can be synthesized from PTP without SPR. Bonafe et al. (2001) showed greatly reduced BH4 levels in the cerebrospinal fluid (CSF) of human patients with SPR deficiency, indicating that such pathways indeed exist and partially compensate for the lack of SPR in vivo. However, a detailed assessment of this compensatory activity in various organ systems has been lacking.
Figure 1.

Biosynthesis of BH4 and generation of Spr-knockout mice. A, Biosynthesis of BH4, including the possible alternative pathways (dashed arrows). B, Mouse Spr genomic structure (wild-type), the knockout (KO) construct, and the resulting targeted mutant allele. The Spr gene consists of three exons. Blackened and unblackened boxes represent exons that encode translated and untranslated regions of SPR, respectively. Restriction-enzyme sites for EcoRI (E), HindIII (H), NcoI (N), and SacI (S) are indicated. Restriction-enzyme sites in parentheses indicate the loss of a recognition sequence during construction of the KO vector. The probes (5′ probe and 3′ probe) for Southern-blot analysis and the expected sizes of fragments from wild-type and mutant Spr loci are shown. To construct the targeting vector, a segment from exon 1 and the entire exon 2 region were replaced with the PGK-neomycin resistance cassette (PGK-Neo). The herpes simplex virus–thymidine kinase cassette (HSV-TK) was inserted at the 3′ end of the construct, for negative selection with FIAU. C, Southern-blot analysis of embryonic stem cell clones by use of the 5′ probe. Genomic DNA isolated from embryonic stem cell clones was digested with EcoRI. The 5′ probe detects the Spr pseudogene (>12 kb) as well as the wild-type (9.5 kb) and mutant (4.4 kb) bands in the authentic Spr locus. D, Southern-blot analysis of pups from Spr+/− matings by use of the 3′ probe. Genomic DNA from pups was digested with NcoI. Southern-blot analysis using the authentic Spr gene-specific 3′ probe showed 13.3-kb and 9.9-kb bands for the wild-type and mutant alleles, respectively. E, Northern-blot analysis showing no detectable Spr transcript in the kidney of Spr−/− mice.
In the salvage pathway, sepiapterin is formed nonenzymatically from 1′-oxo-TP and is converted to 7,8-dihydrobiopterin (BH2) by SPR (step 4 in fig. 1). BH2 is then reduced to BH4 by dihydrofolate reductase (DHFR [MIM 126060]). During the conversion of phenylalanine to tyrosine by PAH, BH4 is oxidized to pterin-4α-carbinolamine (Fitzpatrick 1999). Pterin-4α-carbinolamine dehydratase (PCD [MIM 126090]) converts pterin-4α-carbinolamine to q-dihydrobiopterin, which is regenerated to BH4 by dihydropteridine reductase (DHPR [MIM 261630]). The regeneration of BH4 from pterin-4α-carbinolamine is important because it supplies BH4 and removes the harmful metabolites produced by pterin-4α-carbinolamine (Thöny et al. 2000).
Genetic mutations of all the enzymes involved in BH4 homeostasis have been shown to cause BH4 deficiencies (Thöny et al. 2000; Blau et al. 2001a, 2001b). BH4 deficiencies manifest very heterogeneous symptoms, depending on individual mutations and other modifying genes. In general, patients with BH4 deficiency exhibit progressive neuronal deterioration, mental retardation, convulsions, disturbances of tone and posture, abnormal movements, hypersalivation, swallowing difficulties, and temperature instability (Blau et al. 1996). BH4 deficiencies can be broadly divided into two groups, depending on the presence of a PKU phenotype. Individuals harboring two mutant alleles in GTPCH, PTPS, DHFR, and PCD display symptoms accompanied by a varying (mild to severe) degree of PKU (Blau et al. 2001b), whereas dominantly inherited GTPCH deficiency manifests as dopa-responsive dystonia (MIM 128230) without PKU (Blau et al. 2001a).
SPR deficiency in a human patient was not reported until 2001, and thus it had been believed that the absence of SPR-deficient patients might have been because of the presence of compensatory pathways via AKR and CBR (fig. 1A) and because SPR is dispensable for BH4 biosynthesis in vivo. Since 2001, when a new diagnostic method for detecting SPR enzyme activity in cultured fibroblasts from patients was developed, 15 patients with SPR deficiency have been reported (Bonafe et al. 2001; Elzaouk et al. 2002; Neville et al. 2005; Tetrahydrobiopterin Web site). These patients with SPR deficiency were initially misdiagnosed as having DHPR deficiency. The pterin metabolic profiles of two patients with SPR mutations described by Bonafe et al. (2001) showed differences between brain and peripheral tissues. Bonafe et al. (2001) suggested that these differences might be the result of different levels of enzymes (i.e., AKR, CBR, and DHFR) playing a compensatory role for the lack of SPR. These results indicate that SPR is indeed essential for BH4 homeostasis and that a better understanding of the in vivo role of SPR in the regulation of BH4 homeostasis would be helpful for the correct diagnosis of SPR deficiency.
To investigate the role of SPR in the regulation of BH4 homeostasis and in BH4 deficiency, we generated a mouse strain deficient in the Spr gene by the genetargeting technique. Spr-deficient mice (Spr−/−) were born normal, but they developed dwarfism during the early postnatal period. Spr−/− mice appeared to have greatly reduced levels of BH4 in the brain and liver. Dopamine and serotonin levels were also significantly reduced in the relevant brain areas, and the mice displayed impaired locomotion activities. Interestingly, unlike results for human patients, the serum phenylalanine level in the Spr−/− mice was greatly increased. Dwarfism and high serum phenylalanine levels could be completely reversed by oral supplementation of BH4 and neurotransmitter precursors. This Spr mutant strain will be an invaluable resource to uncover many important issues regarding SPR and BH4 deficiencies.
Material and Methods
The Spr-knockout mouse strain was generated at the University of Florida. The mice were maintained under standard specific-pathogen–free conditions, and all animal procedures performed were reviewed and approved by the University of Florida Institutional Animal Care and Use Committee.
Generation of Spr-Knockout Mice
The mouse Spr gene encodes 261 aa and consists of three exons (fig. 1B) (Ota et al. 1995; Lee et al. 1999). We had previously isolated a phage clone containing the entire Spr gene from a 129/SvJ (129) mouse genomic DNA library (Lee et al. 1999). To construct a targeting vector, a 3.3-kb HindIII-SacI fragment including a part of exon 1 and a 3.8-kb SacI-HindIII fragment containing the exon 3 region were sequentially inserted into the XhoI and XbaI sites, respectively, of the pPNT vector (Tybulewicz et al. 1991). The phosphoglycerate kinase (PGK)–neomycin cassette replaced a portion of exon 1, intron 1, and the entire exon 2 encoding the short-chain dehydrogenase/reductase domain. After electroporation of the linearized construct, G418-resistant and 1-(2-deoxy-2-fluoro-1-β-d-arabinofuranosyl)-5-iodouracil (FIAU)–resistant colonies were selected. Approximately 300 double-resistant colonies were screened by Southern-blot analysis using a 5′ external probe (5′ probe in fig. 1B). One of the three positive clones was microinjected into C57BL/6J (B6) blastocysts to generate chimeras, which were crossed with B6 mice to establish and maintain the Spr+/− mouse line on a mixed 129/B6 hybrid background. The genotype of offspring from the breeding of heterozygous mice was determined with PCR primer sets SprF1 (5′-AAGTGGTGCTGGCAGCCGCCGAT-3′) and NeoP3 (5′-CGGTGCTGTCCATCTGCACGAGAC-3′), for detection of the mutant allele, and srex2F (5′-CCTCCATGCTCTGTTTGACT-3′) and srex2R (5′-GTTCCCCTCCTTGCCTAGC-3′), for detection of the wild-type allele. The genomic region amplified by the srex2F and srex2R primer set was deleted in the mutant allele, and thus no PCR amplification occurs for the Spr−/− mice.
Southern-Blot Analysis
DNA extracted from embryonic stem cells and mouse tails was digested with EcoRI or NcoI, run on a 0.8% agarose gel, blotted onto Gene Screen plus membrane, and hybridized with the radiolabeled 5′ probe (for EcoRI-digested DNA) or 3′ probe (for NcoI-digested DNA) (fig. 1B).
Northern-Blot Analysis
Total RNA was isolated from the kidney and the brain with a Trizol reagent (Invitrogen). A sample of 7 μg of kidney RNA or 20 μg of brain RNA was used for Northern-blot analysis (Pelle and Murphy 1993) with the Spr or Th cDNA probe, respectively.
Replacement Therapy
Replacement therapy was performed in accord with the method described by Elzaouk et al. (2003). To prepare the low-dose solution, 390 mg of BH4-2HCl (Schircks Laboratories), 300 mg of ascorbic acid (Sigma), 44.4 mg of 5-hydroxytryptophan (Sigma), 15 mg of carbidopa (Sigma), and 150 mg of N-acetyl-l-cysteine (Sigma) were dissolved in 5 ml of H2O. Dopa solution was prepared by dissolving 63.6 mg of l-dopa (Sigma) in 1 ml of 0.5 N HCl. The two solutions were mixed and adjusted to pH 5.0 with sodium citrate. To prepare the high-dose solution, 780 mg of BH4-2HCl, 600 mg of ascorbic acid, 57 mg of 5-hydroxytryptophan, 15 mg of carbidopa, and 300 mg of N-acetyl-l-cysteine were dissolved in 5 ml of H2O. Dopa solution was prepared by dissolving 81 mg of l-dopa in 1 ml of 0.5 N HCl. The two solutions were mixed and adjusted to pH 5.0 with sodium citrate and then were frozen until use. The mixed solution (500 μl per kg body weight) was administered orally twice a day by use of micropipettes.
Preparation of Mouse Tissues for Pteridine Measurement
The brain and liver were removed immediately after each mouse was sacrificed and were blended in a cold homogenizing buffer containing 50 mM Tris-HCl (pH 7.5), 0.1 M KCl, 1 mM EDTA, 1 mM dithiothreitol, and proteinase inhibitors (EDTA-free proteinase inhibitor cocktail [Roche]) by use of an electric homogenizer (Biospec Products). For the brain tissue, 4 μl/mg of the homogenizing buffer was added, and, for the liver, 5 μl/mg was added. Homogenates were centrifuged at 15,000 g for 20 min at 4°C, and supernatants were kept frozen at −80°C.
Neopterin, Biopterin, and BH4 Measurements
The BH4 level was measured using high-performance liquid chromatography (HPLC) analysis after iodine oxidation in acidic or alkaline conditions, as described elsewhere (Fiege et al. 2004). To measure the amount of neopterin and biopterin, a volume of either 100 μl of liver or brain homogenates was used for acidic (or basic) oxidation, followed by the addition of 100 μl of 0.2 N HCl (or NaOH). The homogenate was then incubated in the dark for 1 h, after 10 μl of acidic iodine solution was added (0.9% iodine and 1.8% potassium iodine in 0.1 N HCl [or NaOH]). The reaction was terminated by addition of 10 μl of 5% ascorbic acid. Oxidized samples were filtered using an Ultra-free MC filter (10,000-MW cutoff [Millipore]). The filtrates were subjected to HPLC analysis (using Waters 600E Pump). Concentrations are expressed as pmol per mg of protein. BH4 levels were derived by subtracting biopterin levels under a basic oxidative state (biopterin + BH2) from the levels under an acidic oxidative state (i.e., total biopterin = biopterin + BH2 + BH4). The separation was performed on a Spherisorb ODS1 C18 (particle size 5 μm; length × internal diameter = 250×4.6 mm; Waters) by use of 1 mM potassium phosphate (pH 4.6) with 5% (v/v) methanol at a flow rate of 0.6 ml/min. Pterins were detected by their native fluorescence (excitation wavelength 350 nm; emission wavelength 450 nm) by use of a 2475 fluorescence detector (Waters).
Sepiapterin Measurement
Tissue homogenates were filtered with an Ultrafree-MC filter (10,000-MW cutoff [Millipore]) and were subjected to HPLC. The separation was performed on a precolumn (Symmetry C8 [5 μm; 20×3.9 mm; Waters]) and an analytical column (Symmetry C8 [5 μm; 150×4.6 mm; Waters]) by use of the method described by Zorzi et al. (2002), with some modifications. The compositions of solvents A and B for gradient elution were isopropanol:methanol:acetic acid:H2O (0.5:0.5:0.5:98.95 [v/v]) for solvent A and isopropanol:methanol:acetic acid (49:49:2 [v/v]) for solvent B. The flow rate was maintained at 0.6 ml/min by use of the following gradient program: solvent A at 100% for 3.4 min, solvent B at 0%–2% for 8.5 min, solvent B at 2% for 3.4 min, solvent B at 2%–20% for 17 min, solvent B at 20% for 1.7 min, solvent B at 40% for 8.5 min, and solvent A at 100% for 17 min.
Measurement of Catecholamines, Serotonin, and Other Related Metabolites
Dopamine, norepinephrine, 3,4-dihydroxyphenylacetic acid (DOPAC), serotonin, and 5-hydroxyindole acetic acid (HIAA) levels in several brain regions were measured by HPLC with electrochemical detection (Blau et al. 1999). The frontal cortex, caudate putamen, cerebellum, and general area of the cortex were separated from the brain and were immediately frozen in liquid N2. Tissue samples were maintained at −80°C until the neurotransmitters and their metabolites were measured. Tissue was weighed with a minimum of thawing and was then homogenized in 2 ml of 0.1 N perchloric acid by use of a Brinkmann Homogenizer. The homogenate was centrifuged at 7,360 g for 20 min. The supernatant was filtered through a 0.22-μm filter and was injected onto the HPLC column (YMC-Pac Pro C18 [5 μm; 250×4.6 mm; YMC]). The column was connected to an HP 3390A integrator maintained at 0.5–1.0 mA and to an LDC/Milton Roy Minipump. The mobile phase consisted of 12.5 mM citric acid, 7.5 mM sodium phosphate (monobasic), 0.2 mM Na2EDTA, 0.15 mM sodium octyl sulfate (35 mg/liter), and 3.5% acetonitrile. The mobile phase was then passed through a 0.22-μm filter (Micron Separations) before being degassed under helium for at least 15 min. All separations were performed at a flow rate of 2.22 ml/min. The amounts of neurotransmitters and their metabolites in each sample were normalized for the weight of the brain tissue from which they were extracted.
Microplate Serum Phenylalanine Assay
Serum phenylalanine was measured using a fluorimetric assay (McCaman and Robins 1962) modified for small volumes and microtiter plate format. In the assay, 7.5 μl of serum was precipitated with 11.2 μl of 0.3 N trichloroacetic acid and were chilled on ice for 10 min. Using a 96-well PCR plate, 64 μl of assay cocktail were dispensed into each well. The cocktail contained 4.40 ml of 0.3 M succinate (pH 5.8), 1.76 ml of 30 mM ninhydrin, and 0.880 ml of 5 mM l-leucyl-l-alanine. Standards were prepared from 10 mM phenylalanine in 0.3 N trichloroacetic acid. Each standard and sample was run in triplicate. Four microliters of each standard was added to three wells spaced across the microtiter plate. The precipitated serum samples were centrifuged at 16,000 g for 10 min in a microcentrifuge. From each sample, 4 μl of the supernatant was added to three consecutive wells, and the plate was capped and incubated in a thermocycler at 60°C for 2 h. Copper reagent (200 μl; 15 mM Na2CO3, 2.3 mM potassium sodium tartrate, and 2.4 mM CuSO4·5H2O) was added to each well of a 96-well 0.5-ml black fluorimeter plate (Nunc). Additional copper reagent (100 μl) was added to the incubated samples in the PCR plate, and then the entire volume was transferred into the fluorimeter plate. The PCR plate was washed with another 100 μl of copper reagent, and the wash was added to the fluorimeter plate. The plate was read twice on an FLx800 Multidetection Microplate Reader (BioTek) with excitation filter of 380 nm and emission filter of 508 nm. Both readings were averaged to calculate the phenylalanine concentration in each serum sample relative to the standard curve.
Immunohistochemistry
Dopaminergic and noradrenergic neural cell bodies are located in the substantia nigra, ventral tegmental area, and locus ceruleus. The animals were anaesthetized with ketamine, and perfusion fixation was performed by perfusing transcardially with ice-cold 10% neutral buffered formalin for 10 min at a flow rate of 30–40 ml/min. The brains of the animals were immediately removed and then were fixed in the same solution overnight. The fixed tissues were embedded in paraffin. Coronal sections (3-μm thick) and parasagittal sections (3-μm thick) were used for the evaluation of immunoreactivities. Immunohistochemical staining was performed using EnVision anti-mouse or anti-rabbit kits (DAKO). Heat-induced antigen retrieval was required. The sections were placed in a pressure cooker for 4 min at maximum pressure in 10 mM citrate buffer (pH 7.0). The sections were pretreated with 0.3% H2O2 in 0.1 M Tris-buffered saline (TBS) (pH 7.4) for 10 min to quench endogenous peroxidases. Anti-mouse TH (1:50 dilution [Novocastra]), anti-rabbit TPH (1:50 dilution [Novocastra]), anti-mouse calbindin (1:40 dilution [Abcam]), and anti-rabbit AADC (1:50 dilution [BIOMOL]) antibodies were used. The primary antibodies were applied and incubated for 30 min at room temperature (TH, TPH, and calbindin) or overnight at 4°C (AADC) in a humid chamber. After being washed with 0.1 M TBST (0.1 M TBS and 0.01% Tween 20), sections were incubated with EnVision anti-mouse or anti-rabbit (DAKO) polymer for 30 min. Visualization of the antibody complex was achieved by treatment with 3,3′-diaminobenzidine chromogen substrate solution (DAKO) and counterstaining with Meyer’s hematoxylin.
Measurement of Insulin-Like Growth Factor 1 (IGF1)
Total IGF1 (MIM 147440) levels in the serum were measured by radioimmunoassay in accord with the method described by Breier et al. (1991). Serum samples were extracted with acid-ethanol. Then, 25 μl of serum was treated briefly with 100 μl of acid-ethanol (12.5% 2 N HCl and 87.5% ethanol) and was incubated for 30 min at room temperature. After centrifugation at ∼6,000 g for 3 min at room temperature, 50 μl of the clear supernatant was transferred to a new tube. The sample was then neutralized with 250 μl of 0.855 M Tris-base and was centrifuged at 10,000 g for 10 min at 4°C. The supernatant was then assayed for the IGF1 level. To determine the recovery of IGF1 from acid-ethanol extraction, recombinant IGF1 was spiked into the mouse serum at 10, 5, and 2 ng/ml. Finally, the IGF1 concentration was calculated by radioimmunoassay. Two sets of standards were routinely included in each assay to determine IGF1 concentration calculated by a Riasmart program (Packard). The lower limit of detection for this assay was 1 ng/ml. The intra-assay and interassay coefficients of validation were 6.7% and 10.2%, respectively.
Behavioral Test
To analyze locomotor activity, we performed two tests for voluntary movement: four-limb akinesia and turning on a screen (Szczypka et al. 2001). In the four-limb akinesia test, the mice were picked up by the tail and were slowly placed on a surface. The amount of time required for the mice to move all four limbs was measured. In the turning test, the mice were placed head-down on a screen positioned at the edge of a countertop and tilted 60° from the surface. The amount of time required for the mice to reorient themselves in the opposite direction was measured.
Results
Generation of Spr-Knockout Mouse Line
To investigate the role of SPR in mouse development and in the biosynthesis of BH4, we generated a strain of mice deficient in the Spr gene, using conventional knockout technology. The mouse Spr gene encodes 261 aa and consists of three exons (Ota et al. 1995; Lee et al. 1999). As shown in figure 1B, a knockout construct was generated by replacing a Spr genomic region, including a portion of exon 1 and the entire exon 2, with the PGK-neomycin cassette. Since exon 2 encodes the short-chain dehydrogenase/reductase domain essential for SPR function and because the designed deletion leads to a frameshift in the subsequent exon 3, the Spr-knockout allele was expected to be null. Homologous recombination in embryonic stem cells was confirmed by genomic Southern-blot analysis. Using the 5′ probe, we identified three positive embryonic stem cell clones from 300 G418- and FIAU-resistant clones (fig. 1C). One of three correctly targeted embryonic stem cell clones was microinjected into B6 blastocysts to generate chimeras. The chimeras were crossed with B6 mice to establish and maintain the Spr heterozygous (Spr+/−) mouse line on a mixed 129/B6 hybrid background. Homozygous mice (Spr−/−) were obtained from heterozygote breeding. The 5′ probe detects the Spr pseudogene (Spr-ps1), which lacks the promoter region and exon 3 that are present in the authentic Spr gene (Lee et al. 1999). Using the 3′ probe, which detects the Spr locus but not Spr-ps1, we confirmed that the gene targeting was made on the authentic Spr locus (fig. 1D). We showed the absence of Spr transcripts in the kidneys of the Spr−/− mice by Northern-blot analysis (fig. 1E).
Growth Retardation and Reduced Serum IGF1 Levels in Spr−/− Mice
Spr+/− mice are viable and fertile and do not display any detectable abnormalities. Spr−/− mice were indistinguishable from their littermates at birth, but all Spr−/− mice (n>60) showed discernible growth retardation within a few days after birth, and almost no gain of body weight was recorded beyond 2 wk after birth (fig. 2A and 2B). Most Spr−/− mice died within 1–2 mo, whereas a few survived for 2–6 mo. To test whether dwarfism was affected by littermate competition, Spr−/− mice were quarantined from their normal siblings. However, no significant weight gain was observed in these mice, in comparison with those kept together with their normal siblings. The growth curve of Spr−/− mice was remarkably similar to that of Pts−/− mice that were rescued by replacement therapy with high doses of BH4 and neurotransmitter precursors (Elzaouk et al. 2003). Since the serum IGF1 levels were significantly reduced in the rescued Pts−/− mice with dwarfism, we examined the serum IGF1 levels in Spr−/− mice, using a radioimmunoassay. As shown in figure 2C, the serum IGF1 levels were severely reduced in Spr−/− mice, compared with the levels in the wild-type and Spr+/− littermates. This finding suggests that reduced IGF1 is a probable cause of the growth retardation in the Spr−/− mice.
Figure 2.

Growth of Spr−/− mice and serum IGF1 levels. A, A 45-d-old Spr−/− mouse with dwarfish compared with an Spr+/+ littermate. B, Growth curves of Spr+/+ (red), Spr+/− (blue), and Spr−/− (green) mice. Each line represents an individual mouse. C, Serum IGF1 levels. Three 4-wk-old mice of each genotype were tested. Values are means (and SE is indicated above each bar). *P<.01, compared with Spr+/+ mice, as determined by Student’s t test.
Severely Impaired BH4 Biosynthesis in Spr−/− Mice
It has been postulated that, unlike GTPCH and PTPS function, SPR function can be compensated by AKR and CBR (Milstien and Kaufman 1989; Levine et al. 1990; Park et al. 1991). However, the extent to which SPR deficiency affects BH4 homeostasis in vivo has been unknown. To address this issue, we measured the neopterin, total biopterin, BH4, and sepiapterin levels in the liver and brain of 4-wk-old Spr+/+ (wild-type), Spr+/−, and Spr−/− mice (fig. 3). Eight brain samples from each genotype of mice, six liver samples from Spr+/+ and Spr+/− mice, and eight liver samples from Spr−/− mice were analyzed. We found a significant decrease in BH4 levels in both liver and brain, but the magnitude of the decrease was different in these two organs. In the liver, the BH4 level was dramatically reduced (to 1.1% of wild type) in Spr−/− mice compared with the controls, whereas a relatively mild decrease (40.5% of wild type) was detected in the brain of Spr−/− mice. We also found dramatic sepiapterin accumulation in the brain and liver of Spr−/− mice, compared with that found in the brains and livers of the Spr+/+ and Spr+/− mice. The amount of neopterin, a 7,8-dihydroneopterin triphosphate metabolite, increased moderately in the liver (to ∼3 times that found in Spr+/+ mice) and in the brain (to 5 times). These data show that AKR and CBR partially compensate for the loss of SPR in both the brain and liver and that the brain has a greater degree of compensation than the liver does.
Figure 3.
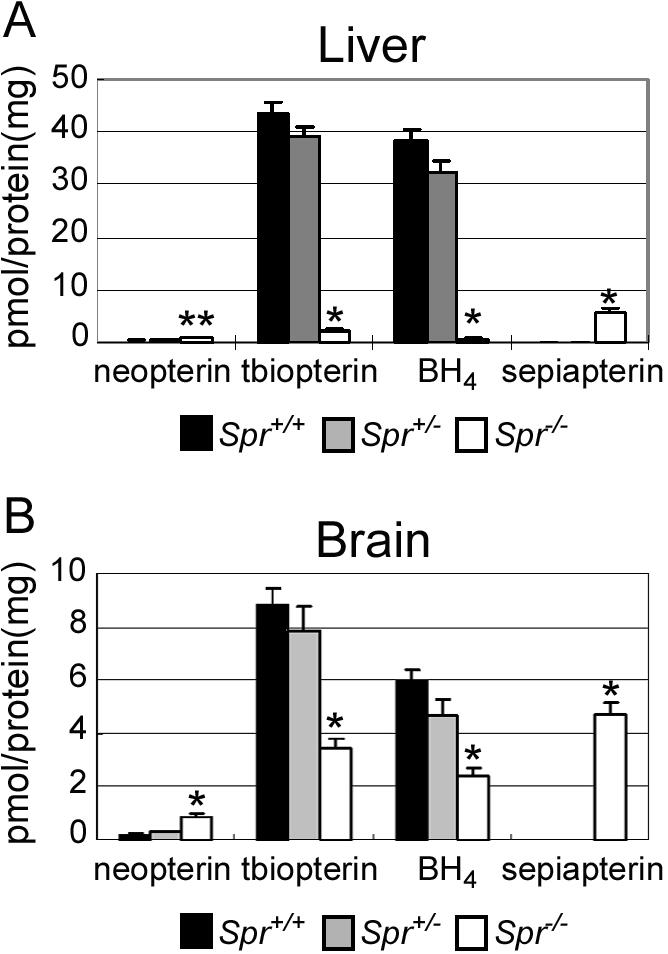
Pteridine levels. Neopterin, total biopterin (tbiopterin), BH4, and sepiapterin levels in the liver (A) and brain (B) are shown. Six Spr+/+, six Spr+/−, and eight Spr−/− mice were used for the liver tests. Eight mice of each genotype were used for the brain tests. Values are means (and SE is indicated above each bar). *P<.001 and **P<.01, compared with Spr+/+ mice, as determined by Student’s t test.
Severely Impaired Phenylalanine Metabolism and Biosynthesis of Catecholamines and Serotonin in Spr−/− Mice
Because BH4 is an essential cofactor for PAH, TH, and TPH, we then investigated the extent to which the lowered BH4 level impacted the function of these enzymes. With regard to TH and TPH enzyme activities, we measured the levels of dopamine, DOPAC (a metabolite of dopamine), and norepinephrine, for TH, and the levels of serotonin and 5-HIAA (a metabolite of serotonin), for TPH, in the caudate putamen, cortex, and cerebellum of the wild-type (Spr+/+), Spr+/−, and Spr−/− mice. We found that the amount of neurotransmitters (i.e., dopamine, norepinephrine, and serotonin) was markedly reduced in the relevant areas of the Spr−/− brain. The dopamine levels in the caudate putamen and cortex of Spr−/− mice was 10-fold lower than those in the caudate putamens and cortexes of Spr+/+ and Spr+/− mice (fig. 4A). DOPAC in the caudate putamen and cortex was below the detection limit in Spr−/− mice (fig. 4B). Norepinephrine was also dramatically reduced in the cortex and cerebellum of Spr−/− mice (fig. 4C). The serotonin level was greatly reduced in the cortex of Spr−/− mice, whereas the 5-HIAA level was comparable to that in Spr+/+ and Spr+/− mice (fig. 4D and 4E). Spr−/− mice showed remarkably higher serum phenylalanine levels (2,732 μM; n=5) than did the Spr+/+ and Spr+/− controls (∼63 μM; n=5) (fig. 4F), and the phenylalanine levels were even higher than those of the Pahenu2 mice (1,701 μM; n=10), a PKU animal model widely used for Pah mutation studies (Shedlovsky et al. 1993). These data clearly demonstrate that BH4 deficiency in Spr-knockout mice is sufficient to cause impaired activity of BH4-dependent enzymes, including TH, TPH, and PAH.
Figure 4.

Activities of TH, TPH, and PAH. To determine the TH enzyme activity, dopamine (A), DOPAC (B), and norepinephrine (C) levels were measured in the caudate putamen (CPu), cortex (CTX), and cerebellum (CBL). For TPH activity, serotonin (D) and 5-HIAA (E) levels were measured in the CTX and CBL. Three Spr+/+, five Spr+/−, and three Spr−/− mice were analyzed. Values are means (and SE is indicated above each bar). *P<.01 and **P<.05, compared with Spr+/+ mice, as determined by Student’s t test. F, Phenylalanine levels of 28–44-d-old mice were measured to determine the functional PAH enzyme activity. Pahenu2 mice harbor a mutation in the Pah gene that is generated by N-ethyl-N-nitrosourea (ENU) mutagenesis. Five Spr+/+ or Spr+/− mice (controls), 5 Spr−/− mice, and 10 Pahenu2 mice were used. Values are means (and SE is indicated above each bar). *P<.001, compared with Spr+/+ mice, as determined by Student’s t test.
Reduced TH Protein Level in Nerve Terminals of Spr−/− Mice
TH-immunoreactivity in Pts−/− mice declined considerably in the nerve terminals but not in the cell bodies (Sumi-Ichinose et al. 2001). As described above, the BH4 and dopamine levels in Spr−/− mice were comparable to those found in Pts−/− mice, and thus we examined the localization of TH proteins in cell bodies as well as in the nerve terminals of Spr−/− brains. Whereas TH staining in the dopaminergic cells of wild-type mouse brains stretches from the substantia nigra into the caudate putamen along the dopaminergic fibers, TH staining of the Spr−/− brain is strongly detected in the substantia nigra but gradually becomes faint along the dopaminergic fibers and finally disappears near the caudate putamen region (fig. 5A and 5B). The coronal sections of the brains in the striatum area further demonstrate the striking difference between the two groups. Whereas the nerve fibers were heavily stained with the TH antibody in the striatum of the wild-type brain (fig. 5C), almost no staining was detected in the corresponding region of the Spr−/− brain (fig. 5D). A few unusual TH-immunopositive cells (red arrowheads in fig. 5F) and weakly positive fibers (black arrowheads in fig. 5F) were found in the striatum of the Spr−/− brain. To test whether this difference was the result of the degeneration of dopaminergic neurons in the striatum of the Spr−/− brains, we stained the brains of both wild-type and Spr−/− mice, using aromatic amino acid decarboxylase (AADC [MIM 107930]) antibodies, which detect dopaminergic neurons. The AADC antibody-staining pattern in the striatum areas of the Spr−/− brain was comparable to that of the wild-type brain, indicating normal development of dopaminergic neurons in Spr−/− mice (fig. 5G and 5H). The immunostaining pattern using calbindin antibodies yielded the same result (data not shown). Northern-blot analysis showed normal expression of the Th transcript in the Spr−/− brain (data not shown). These data are consistent with the results in studies of Pts−/− mice (Sumi-Ichinose et al. 2001) and suggest that a normal level of BH4 is required for the stability and/or localization of TH proteins at nerve fibers. We also stained the Spr−/− brain sections with TPH antibodies and found no differences in staining pattern or intensity between the mutant brains compared with controls, showing that (unlike for TPH function), the expression, stability, and localization of TPH are unaffected by the lower level of BH4 (data not shown).
Figure 5.

Expression of TH in the brain. Brain sections from Spr+/+ (A, C, E, and G) and Spr−/− (B, D, F, and H) mice were immunostained with anti-TH antibody (A, B, C, D, E, and F) or anti-AADC antibody (G and H). A few unusual TH-immunopositive cells (red arrowheads) and weakly positive fibers (black arrowheads) are indicated in the caudate putamen (CPu) of the Spr−/− brain (F). Images E and F are magnified (×400) views of the CPu regions in images C and D (original magnification ×5), respectively. All mice were 4 wk old. SN = substantia nigra; VTA = ventral tegmental area.
Abnormal Locomotion Activities Exhibited by Spr−/− Mice
Dopaminergic neurons are essential for the regulation of body movement. Dramatically reduced dopamine levels and the abnormal expression of TH in the striatum predict impairment of locomotive activities in Spr−/− mice. To test this, we used four-limb akinesia and turning tests to monitor voluntary movement (fig. 6). The Spr−/− mice scored significantly worse on both tests than did the wild-type group, a finding that presents further evidence of dysfunction in the nigrostrial pathway and of reduced dopamine and TH levels in the striatum of Spr−/− mice.
Figure 6.

Behavioral defects in Spr−/− mice. A, Four-limb akinesia test. The mice were picked up by the tail and slowly placed on a surface. The time that it took for the mice to move all four limbs was recorded. B, Turning test. The mice were placed head-down on a screen positioned at the edge of a countertop and tilted 60° from the surface. The time required for the mice to reorient themselves in the opposite direction was recorded. The maximum time for each set of these tests was 2 min. Four mice of each genotype were used. Values are means (and SE is indicated above each bar). *P⩽.001, compared with the Spr+/+ mice, as determined by Student’s t test.
Restoration of Spr-Null Phenotypes by Oral Supplementation of BH4 and Neurotransmitter Precursors
To investigate whether the supplementation of BH4 and neurotransmitter precursors can rescue the growth retardation and other related phenotypes of Spr−/− mice, we used the same oral treatment methods and doses used by Elzaouk et al. (2003) to rescue the Pts−/− mice. We used only two dose ranges (designated as “high” and “low” doses; see the “Material and Methods” section for details) instead of three. We administered supplements orally twice a day, beginning at the newborn stage. Body weights were recorded every day for 75 d, and the brains, livers, and serum samples were collected to measure the levels of BH4 and its derivatives, IGF1, and phenylalanine after 4 wk of treatment. Figure 7 shows body weights of controls and experimental Spr−/− mice after 4 wk and 75 d of treatment (fig. 7A and 7B, respectively). Although Spr−/− mice treated with either high- or low-dose supplementation gained more body weight and developed glossier coats than did nontreated controls, those administered a low dose gained considerably less than did those given a high dose (fig. 7A–7D). The serum IGF1 level was almost completely restored in the treated mutant mice (fig. 7E). We also found that the phenylalanine levels in rescued Spr−/− mice were restored to the control level (data not shown).
Figure 7.

Rescue of Spr−/− mice with BH4 and neurotransmitter precursors. A and B, Body weights of rescued 4-wk-old mice (A) and 75-d-old mice (B). Values are means (and SE is indicated above each bar). *P<.01, as determined by Student’s t test. C and D, Representative 4-wk-old rescued mice with low-dose (C) or high-dose (D) supplementation. E, Serum IGF1 levels in rescued mice. The nontreated group (Non) comprised three mice of each genotype; the low-dose–treated group (Low) comprised two Spr+/+, three Spr+/−, and three Spr−/− mice; and the high-dose–treated group (High) comprised two Spr+/+, two Spr+/−, and six Spr−/− mice. Serum was collected 6–8 h after the final treatment. Values are means (and SE is indicated above each bar).
In contrast to this rescue of body weight and of serum levels of IGF1 and phenylalanine by oral supplementation, there was no rescue of BH4 level in the brain and little increase in BH4 level in the liver, in comparison with untreated controls (table 1). In addition, we stained the high-dose–treated Spr−/− brain sections with TH antibodies to test whether the treatment recovered TH localization in the striatum. The brains, isolated 3 h after the administration of two high doses at once, showed no changes in the TH-staining patterns in the rescued Spr−/− brain compared with those of untreated controls (data not shown).
Table 1.
Effects of Repeated Oral Administration of BH4 and Neurotransmitters[Note]
|
Level in Brain(pmol/mg of protein) |
Level in Liver(pmol/mg of protein) |
|||||||
|
Treatment and Mice |
Neopterin | Total Biopterin |
BH4 | Sepiapterin | Neopterin | Total Biopterin |
BH4 | Sepiapterin |
| None: | ||||||||
| Spr+/+ | .17 ± .06 | 8.85 ± .58 | 5.97 ± .42 | ND | .21 ± .06 | 43.41 ± 2.49 | 38.57 ± 1.80 | ND |
| Spr+/− | .29 ± .04 | 15.81 ± .94 | 4.67 ± .60 | ND | .35 ± .08 | 39.28 ± 1.87 | 32.36 ± 1.92 | ND |
| Spr−/− | .89 ± .08 | 3.42 ± .40 | 2.42 ± .31 | 4.75 ± .43 | .71 ± .10 | 1.94 ± .44 | .42 ± .32 | 5.47 ± 1.04 |
| Low dose: | ||||||||
| Spr+/+ | .77 | 8.69 | 5.76 | ND | .37 ± .04 | 39.44 ± 5.12 | 34.89 ± 5.74 | ND |
| Spr+/− | .41 | 13.44 | 8.64 | ND | .5 ± .07 | 49.23 ± 1.3 | 45.65 ± 1.19 | ND |
| Spr−/− | .91 ± .16 | 6.30 ± 2.34 | .58 ± .05 | 6.22 ± .88 | .5 | 1.89 | .33 | 9.05 |
| High dose: | ||||||||
| Spr+/+ | .2 ± .03 | 8.99 ± .33 | 7.24 ± .42 | ND | .33 ± .01 | 41.41 ± 2.71 | 36.92 ± 4.16 | ND |
| Spr+/− | .36 ± .22 | 8.58 ± 1.01 | 6.50 ± 2.01 | ND | .31 ± .06 | 48.90 ± 1.62 | 45.64 ± 1.34 | ND |
| Spr−/− | .53 ± .06 | 4.00 ± 1.17 | 1.77 ± .37 | 5.68 ± .58 | .34 ± .03 | 3.82 ± 1.1 | 1.86 ± .60 | 8.72 ± .46 |
Note.— ND = not detectable.
Discussion
Here, we report the generation and characterization of mice deficient in the Spr gene, which is involved in BH4 biosynthesis and metabolism. We found that homeostasis of BH4, dopamine, norepinephrine, serotonin, and phenylalanine are greatly disturbed in Spr−/− mice, which exhibit dwarfism and impaired body movement. Oral supplementation of BH4 and neurotransmitter precursors did not restore BH4 levels but did completely rescue dwarfism and phenylalanine metabolism.
Pterin profile data in the liver and brain of Spr−/− mice showed elevated neopterin and sepiapterin as well as reduced biopterin and BH4, in comparison with the control littermates (table 1). The implications of these data are as follows. First, the BH4 level was markedly reduced, but not abolished, in Spr−/− mice, which demonstrates that SPR is essential for BH4 homeostasis in mouse tissues. This result is consistent with recent findings in SPR-deficient humans (Bonafe et al. 2001; Elzaouk et al. 2002; Tetrahydrobiopterin Web site). Second, the alternative pathway of BH4 biosynthesis from PTP, most likely mediated by AKR and CBR, can only partially compensate for the role of SPR. The degree of BH4 reduction in the liver and brain was significantly different—that is, there was more reduction in the liver than in the brain. This result is consistent with differences in pterin profiling among the brain (CSF), peripheral tissues, and fibroblasts of SPR-deficient patients (Bonafe et al. 2001). Such a variance is likely the result of different expression and activities of the enzymes involved in the alternative pathway of BH4 biosynthesis (i.e., AKR and CBR) and the salvage pathway (i.e., CBR and DHFR) among different organs and tissues. It may also be the result of greater use of BH4 in the liver for conversion of phenylalanine to tyrosine from dietary phenylalanine. Third, the sepiapterin level was significantly elevated in the liver and brain of Spr−/− mice, which suggests that SPR is the major reductase from sepiapterin to BH2 in the salvage pathway (fig. 1A). To date, eight SPR-deficient patients’ pterin profiles are available (Bonafe et al. 2001; Elzaouk et al. 2002; Tetrahydrobiopterin Web site). The sepiapterin level was reported for two of these patients, both of whom showed a high level. This elevated level of sepiapterin could be a signature of the pterin profile in SPR deficiency.
We found dramatically reduced levels of dopamine, norepinephrine, and serotonin in the relevant brain areas of Spr−/− mice, suggesting that the enzymatic functions of TH and TPH were severely compromised in these mutant mice. We also showed, using anti-TH immunostaining, that TH localization was normal in the cell body (substantia nigra) but undetectable in the projected nerve terminals (caudate putamen) of Spr−/− mice (fig. 5). Northern-blot analysis showed no differences in the TH transcript level among different genotypes. This result is in accordance with that obtained from Pts−/− mice (Sumi-Ichinose et al. 2001) and suggests that a normal BH4 level is required for the subcellular localization and/or stability of TH proteins in dopaminergic neurons. BH4 was reported to prevent the nitration of tyrosine residues on TH mediated by reactive oxygen species (ROS) such as peroxynitrite or nitrogen dioxide (Kuhn and Geddes 2003). Such protein modifications by ROS are known to be irreversible (Agar and Durham 2003), and nitrated TH has reduced function and stability (Kuhn and Geddes 2003). It was also reported that low levels of BH4 can produce ROS rather than NO. Altogether, we postulate that reduced BH4 in the striatum of Spr−/− mice elevates ROS and reduces the prevention of nitration on TH, which in turn decreases the stability and function of TH. In contrast, whereas serotonin levels were severely reduced, TPH enzyme appeared at normal levels in the Spr−/− brain sections. This may suggest that—although the reduction in amine neurotransmitters such as serotonin, norepinephrine, and dopamine might be in part the result of high serum phenylalanine levels interfering with either amino acid transport in the brain or the activities of the two biosynthetic enzymes—different modes of regulation may be employed for the enzyme activities of TH and TPH, as proposed by Sumi-Ichinose et al. (2001).
Spr−/− mice exhibited severe dwarfism from the early postnatal period. The growth curve of Spr−/− mice is similar, if not identical, to that of Pts−/− mice supplemented with the “high” dose (Elzaouk et al. 2003). IGF1 has been shown to play a pivotal role in body growth (Liu et al. 1993, 1998; Powell-Braxton et al. 1993). Rescued Pts−/− mice as well as Spr−/− mice showed significantly reduced serum IGF1, which suggests a causative relationship between IGF1 deficiency and growth retardation in these mutant mice. It has been shown, in D2 dopamine receptor–deficient mice, that dopamine is an essential factor for growth-hormone release (Brambilla et al. 1997; Diaz-Torga et al. 2002). In the absence of the dopamine signal, growth hormone and hypothalamic growth-hormone–releasing hormone (GHRH)–induced IGF1 production is impaired, resulting in dwarfism. Elzaouk et al. (2003) have shown normal growth hormone and thyroxine levels in the rescued Pts−/− mice and have suggested chronic malnutrition (due to swallowing difficulties or reduced appetite) as a potential cause of the IGF1 deficiency and dwarfism. Our Spr−/− mice exhibited low dopamine and IGF1 levels that were comparable to those of the rescued Pts−/− mice. Interestingly, oral supplementation completely rescued the dwarfism phenotype of Spr−/− mice and restored the IGF1 level, without any apparent change in BH4 levels. At least one SPR-deficient patient displayed growth retardation (Bonafe et al. 2001). Since the fostering environments for mice and humans are obviously different, it remains to be investigated whether growth retardation is a common feature of SPR deficiency between these two species. More detailed studies are also required for deciphering the signaling pathway from lowered dopamine to lowered IGF1 for the dwarfism phenotype of the Spr mutants.
The Spr−/− serum contained a high level of phenylalanine that exceeded the levels found in Pahenu2 mice, a PKU mouse model (Shedlovsky et al. 1993). SPRdeficient patients displayed abnormal responses in the phenylalanine loading test, indicating that the PAH function is somewhat impaired, yet the phenylalanine levels in these patients appeared to be normal (Bonafe et al. 2001). This suggests that the lowered BH4 is sufficient for PAH function in the liver of SPR-deficient patients to maintain the normal phenylalanine homeostasis. The reason for this discrepancy between SPR-deficient patients and Spr−/− mice is unknown. One simple explanation is that mice and humans have different levels of the alternative enzyme activities that compensate for the loss of SPR, so a higher level of BH4 (a level sufficient for the function of PAH) is produced in human livers than in mouse livers. Alternatively, a higher metabolic rate in mice than in humans (Terpstra 2001) may lead to faster exhaustion of BH4 in mouse livers. Nonetheless, our data raise some caution about classifying SPR deficiency as a “BH4-deficiency without PKU” until consistent data are established from a larger patient pool.
We have shown, using the four-limb akinesia and turning tests, that Spr−/− mice displayed impaired body movement. Additionally, the prolonged crouching posture and the neglect of grooming habits demonstrated by the mice indicated depression and social detachment. The Spr−/− mice also displayed a tremor phenotype, a symptom associated with dystonia and Parkinson disease. Although these symptoms could be the result of small body mass and muscle weakness, the behavioral characteristics observed in Spr−/− mice share striking similarities with the symptoms observed in SPR-deficient patients, who present psychomotor retardation, spasticity, dystonia, tremors, and growth retardation (Bonafe et al. 2001; Elzaouk et al. 2002; Neville et al. 2005; Tetrahydrobiopterin Web site). Patients with dystonia present dopamine deficiency and locomotive impairment while their dopaminergic neurons are intact. The same phenomenon was observed in Spr−/− mice. In a previous study, movement abnormalities observed in a 15-year-old SPR-heterozygous patient developed into dystonia and tremor 8 years later (Steinberger et al. 2004), which suggests that Spr+/− mice may also exhibit dystonic symptoms in the future; 1-year-old Spr+/− mice are normal, but it is possible that they will develop additional symptoms. In addition, SPR was recently reported to be associated with the PARK3 locus, which is associated with early-onset Parkinson disease (Karamohamed et al. 2003). Consequently, the fact that a 15-year-old SPR-heterozygous patient developed a tremor and that SPR is included in the PARK3 locus suggests that SPR may be involved in modulating the age at onset of Parkinson disease.
We succeeded in rescuing phenotypes observed in Spr−/− mice with the high dose of BH4 and neurotransmitter precursors. After the treatment, the body weight and serum IGF1 and phenylalanine levels of the mice were restored to normal. TH expression in the brain, however, remained the same after 4 wk of either high- or low-dose treatment. In addition, both high- and low-dose treatment resulted in a prolonged average lifespan. We have observed normal behavior, vitality, and fertility in 8-mo-old Spr−/− mice treated with the high dose. It would be of great interest to investigate the change in the biochemical and behavioral characteristics of the rescued Spr−/− mice after cessation of the supplementation. It would also be fascinating to examine whether dopamine alone, without BH4 and serotonin, can produce complete or partial rescue of the Spr mutant phenotypes.
Recently, hph-1 mice, in which the GTPCH activity is partially impaired, displayed pulmonary arterial hypertension (MIM 178600) with decreased NOS3 activity in the lungs (Khoo et al. 2005; Nandi et al. 2005). Because of technical limitations, we have not studied NO and NOS activities in the Spr mutant. Our Spr knockout mice would be an additional animal model for studying the impact of lowered BH4 biosynthesis on NOS functions and pulmonary hemodynamics.
We believe that the Spr-knockout mice, as well as tissue-specific conditional Spr-knockout mice and transgenic mice expressing Spr in specific tissues for rescue experiments, will be an invaluable resource to address many important questions regarding SPR and BH4 deficiencies. Such studies would provide crucial information not only for better diagnostic criteria and treatment options for human SPR deficiency but also for a broad field of neuropathogenesis, including neurotransmitter disorders, dopamine-responsive dystonia, mental retardation, PKU, and Parkinson disease.
Acknowledgments
We thank Dr. E. Li for J1 embryonic stem cell lines, Dr. Ed Grace for technical assistance, and Marya Park for editorial assistance. This study was supported in part by National Institutes of Health grant HL64024 (to S.P.O.) and by Basic Research Program of Korea Science and Engineering Foundation grant R01-2004-000-10128-0 (to J.H.C.).
Web Resources
The URLs for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for TH, TPH, NOS1, NOS2, NOS3, PKU, Segawa syndrome, Parkinson disease, major depression, GTPCH, PTPS, SPR, DHFR, PCD, DHPR, dopa-responsive dystonia, IGF1, AADC, and pulmonary arterial hypertension)
- Tetrahydrobiopterin Web site, http://www.BH4.org/
References
- Agar J, Durham H (2003) Relevance of oxidative injury in the pathogenesis of motor neuron diseases. Amyotroph Lateral Scler Other Motor Neuron Disord 4:232–242 10.1080/14660820310011278 [DOI] [PubMed] [Google Scholar]
- Blau N, Barnes I, Dhondt JL (1996) International database of tetrahydrobiopterin deficiencies. J Inherit Metab Dis 19:8–14 10.1007/BF01799342 [DOI] [PubMed] [Google Scholar]
- Blau N, Bonafe L, Thöny B (2001a) Tetrahydrobiopterin deficiencies without hyperphenylalaninemia: diagnosis and genetics of doparesponsive dystonia and sepiapterin reductase deficiency. Mol Genet Metab 74:172–185 10.1006/mgme.2001.3213 [DOI] [PubMed] [Google Scholar]
- Blau N, Heizmann CW, Sperl W, Korenke GC, Hoffmann GF, Smooker PM, Cotton RG (1992) Atypical (mild) forms of dihydropteridine reductase deficiency: neurochemical evaluation and mutation detection. Pediatr Res 32:726–730 [DOI] [PubMed] [Google Scholar]
- Blau N, Thöny B, Cotton RGH, Hyland K (2001b) Disorders of tetrahydrobiopterin and related biogenic amines. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. Vol. II. McGraw-Hill, New York, pp 1725–1776 [Google Scholar]
- Blau N, Thöny B, Renneberg A, Penzien JM, Hyland K, Hoffmann GF (1999) Variant of dihydropteridine reductase deficiency without hyperphenylalaninaemia: effect of oral phenylalanine loading. J Inherit Metab Dis 22:216–220 10.1023/A:1005584627797 [DOI] [PubMed] [Google Scholar]
- Bonafe L, Thöny B, Penzien JM, Czarnecki B, Blau N (2001) Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet 69:269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla F, Bellodi L, Perna G, Arancio C, Bertani A (1997) Dopamine function in obsessive-compulsive disorder: growth hormone response to apomorphine stimulation. Biol Psychiatry 42:889–897 10.1016/S0006-3223(96)00549-5 [DOI] [PubMed] [Google Scholar]
- Breier BH, Gallaher BW, Gluckman PD (1991) Radioimmunoassay for insulin-like growth factor-I: solutions to some potential problems and pitfalls. J Endocrinol 128:347–357 [DOI] [PubMed] [Google Scholar]
- Diaz-Torga G, Feierstein C, Libertun C, Gelman D, Kelly MA, Low MJ, Rubinstein M, Becu-Villalobos D (2002) Disruption of the D2 dopamine receptor alters GH and IGF-I secretion and causes dwarfism in male mice. Endocrinology 143:1270–1279 10.1210/en.143.4.1270 [DOI] [PubMed] [Google Scholar]
- Elzaouk L, Leimbacher W, Turri M, Ledermann B, Burki K, Blau N, Thöny B (2003) Dwarfism and low insulin-like growth factor-1 due to dopamine depletion in Pts−/− mice rescued by feeding neurotransmitter precursors and H4-biopterin. J Biol Chem 278:28303–28311 10.1074/jbc.M303986200 [DOI] [PubMed] [Google Scholar]
- Elzaouk L, Osmani H, Romstad A, Friedman J, Maccollin M, Thöny B, Blau N (2002) Sepiapterin reductase deficiency: molecular analysis in a new case presenting with neurotransmitter deficiency without hyperphenylalaninemia. In: Milstien S, Kapatos G, Shane B, Levine RA (eds) Chemistry and biology of pteridines and folates. Kluwer Academic Publishers, Norwell, MA, pp 277–285 [Google Scholar]
- Fiege B, Ballhausen D, Kierat L, Leimbacher W, Goriounov D, Schircks B, Thöny B, Blau N (2004) Plasma tetrahydrobiopterin and its pharmacokinetic following oral administration. Mol Genet Metab 81:45–51 10.1016/j.ymgme.2003.09.014 [DOI] [PubMed] [Google Scholar]
- Fiege B, Bonafe L, Ballhausen D, Baumgartner M, Thöny B, Meili D, Fiori L, Giovannini M, Blau N (2005) Extended tetrahydrobiopterin loading test in the diagnosis of cofactor-responsive phenylketonuria: a pilot study. Mol Genet Metab 86:91–95 10.1016/j.ymgme.2005.09.014 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick PF (1999) Tetrahydropterin-dependent amino acid hydroxylases. Annu Rev Biochem 68:355–381 10.1146/annurev.biochem.68.1.355 [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR (2000) The neuropathology of phenylketonuria: human and animal studies. Eur J Pediatr Suppl 2 159:S102–S106 [DOI] [PubMed] [Google Scholar]
- Karamohamed S, DeStefano AL, Wilk JB, Shoemaker CM, Golbe LI, Mark MH, Lazzarini AM, et al (2003) A haplotype at the PARK3 locus influences onset age for Parkinson’s disease: the GenePD study. Neurology 61:1557–1561 [DOI] [PubMed] [Google Scholar]
- Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, Wilkins MR, Channon KM (2005) Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation 111:2126–2133 10.1161/01.CIR.0000162470.26840.89 [DOI] [PubMed] [Google Scholar]
- Kuhn DM, Geddes TJ (2003) Tetrahydrobiopterin prevents nitration of tyrosine hydroxylase by peroxynitrite and nitrogen dioxide. Mol Pharmacol 64:946–953 10.1124/mol.64.4.946 [DOI] [PubMed] [Google Scholar]
- Lee SW, Park IY, Hahn Y, Lee JE, Seong CS, Chung JH, Park YS (1999) Cloning of mouse sepiapterin reductase gene and characterization of its promoter region. Biochim Biophys Acta 1445:165–171 [DOI] [PubMed] [Google Scholar]
- Levine RA, Kapatos G, Kaufman S, Milstien S (1990) Immunological evidence for the requirement of sepiapterin reductase for tetrahydrobiopterin biosynthesis in brain. J Neurochem 54:1218–1224 [DOI] [PubMed] [Google Scholar]
- Liu JL, Grinberg A, Westphal H, Sauer B, Accili D, Karas M, LeRoith D (1998) Insulin-like growth factor-I affects perinatal lethality and postnatal development in a gene dosage-dependent manner: manipulation using the Cre/loxP system in transgenic mice. Mol Endocrinol 12:1452–1462 10.1210/me.12.9.1452 [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A (1993) Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75:59–72 10.1016/0092-8674(93)90679-K [DOI] [PubMed] [Google Scholar]
- Ludecke B, Dworniczak B, Bartholome K (1995) A point mutation in the tyrosine hydroxylase gene associated with Segawa's syndrome. Hum Genet 95:123–125 [DOI] [PubMed] [Google Scholar]
- Ludecke B, Knappskog PM, Clayton PT, Surtees RAH, Clelland JD, Heales SJR, Brand MP, Bartholome K, Flatmark T (1996) Recessively inherited L-DOPA-responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum Mol Genet 5:1023–1028 10.1093/hmg/5.7.1023 [DOI] [PubMed] [Google Scholar]
- McCaman MW, Robins E (1962) Fluorimetric method for the determination of phenylalanine in serum. J Lab Clin Med 59:885–890 [Google Scholar]
- Milstien S, Kaufman S (1989) Immunological studies on the participation of 6-pyruvoyl tetrahydropterin (2′-oxo) reductase, an aldose reductase, in tetrahydrobiopterin biosynthesis. Biochem Biophys Res Commun 165:845–850 10.1016/S0006-291X(89)80043-9 [DOI] [PubMed] [Google Scholar]
- Mungrue IN, Bredt DS, Stewart DJ, Husain M (2003) From molecules to mammals: what’s NOS got to do with it? Acta Physiologica Scandinavica 179:123–135 10.1046/j.1365-201X.2003.01182.x [DOI] [PubMed] [Google Scholar]
- Nandi M, Miller A, Stidwill R, Jacques TS, Lam AA, Haworth S, Heales S, Vallance P (2005) Pulmonary hypertension in a GTP-cyclohydrolase 1-deficient mouse. Circulation 111:2086–2090 10.1161/01.CIR.0000163268.32638.F4 [DOI] [PubMed] [Google Scholar]
- Neville BG, Parascandalo R, Farrugia R, Felice A (2005) Sepiapterin reductase deficiency: a congenital dopa-responsive motor and cognitive disorder. Brain 128:2291–2296 10.1093/brain/awh603 [DOI] [PubMed] [Google Scholar]
- Ota A, Ichinose H, Nagatsu T (1995) Mouse sepiapterin reductase: an enzyme involved in the final step of tetrahydrobiopterin biosynthesis. Primary structure deduced from the cDNA sequence. Biochim Biophys Acta 1260:320–322 [DOI] [PubMed] [Google Scholar]
- Park YS, Heizmann CW, Wermuth B, Levine RA, Steinerstauch P, Guzman J, Blau N (1991) Human carbonyl and aldose reductases: new catalytic functions in tetrahydrobiopterin biosynthesis. Biochem Biophys Res Commun 175:738–744 10.1016/0006-291X(91)91628-P [DOI] [PubMed] [Google Scholar]
- Pelle R, Murphy NB (1993) Northern hybridization: rapid and simple electrophoretic conditions. Nucleic Acids Res 21:2783–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA (1993) IGF-I is required for normal embryonic growth in mice. Genes Dev 7:2609–2617 [DOI] [PubMed] [Google Scholar]
- Scriver CR, Kaufman S (2001) Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. Vol. II. McGraw-Hill, New York, pp 1667–1724 [Google Scholar]
- Shedlovsky A, McDonald JD, Symula D, Dove WF (1993) Mouse models of human phenylketonuria. Genetics 134:1205–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaapen LJ, Rubio-Gozalbo ME (2003) Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency, state of the art. Mol Genet Metab 78:93–99 10.1016/S1096-7192(02)00229-9 [DOI] [PubMed] [Google Scholar]
- Steinberger D, Blau N, Goriuonov D, Bitsch J, Zuker M, Hummel S, Müller U (2004) Heterozygous mutation in 5′-untranslated region of sepiapterin reductase gene (SPR) in a patient with dopa-responsive dystonia. Neurogenetics 5:187–190 10.1007/s10048-004-0182-3 [DOI] [PubMed] [Google Scholar]
- Sumi-Ichinose C, Urano F, Kuroda R, Ohye T, Kojima M, Tazawa M, Shiraishi H, Hagino Y, Nagatsu T, Nomura T, Ichinose H (2001) Catecholamines and serotonin are differently regulated by tetrahydrobiopterin—a study from 6-pyruvoyltetrahydropterin synthase knockout mice. J Biol Chem 276:41150–41160 10.1074/jbc.M102237200 [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, Palmiter RD (2001) Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron 30:819–828 10.1016/S0896-6273(01)00319-1 [DOI] [PubMed] [Google Scholar]
- Terpstra AH (2001) Differences between humans and mice in efficacy of the body fat lowering effect of conjugated linoleic acid: role of metabolic rate. J Nutr 131:2067–2068 [DOI] [PubMed] [Google Scholar]
- Thöny B, Auerbach G, Blau N (2000) Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J 347:1–16 10.1042/0264-6021:3470001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thöny B, Blau N (1997) Mutations in the GTP cyclohydrolase I and 6-pyruvoyl-tetrahydropterin synthase genes. Hum Mutat 10:11–20 [DOI] [PubMed] [Google Scholar]
- Thöny B, Neuheiser F, Kierat L, Blaskovics M, Arn PH, Ferreira P, Rebrin I, Ayling J, Blau N (1998a) Hyperphenylalaninemia with high levels of 7-biopterin is associated with mutations in the PCBD gene encoding the bifunctional protein pterin-4a-carbinolamine dehydratase and transcriptional coactivator (DCoH). Am J Hum Genet 62:1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thöny B, Neuheiser F, Kierat L, Rolland MO, Guibaud P, Schlüter T, Germann R, Heidenreich RA, Duran M, de Klerk JB, Ayling JE, Blau N (1998b) Mutations in the pterin-4α-carbinolamine dehydratase (PCBD) gene cause a benign form of hyperphenylalaninemia. Hum Genet 103:162–167 10.1007/s004390050800 [DOI] [PubMed] [Google Scholar]
- Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65:1153–1163 10.1016/0092-8674(91)90011-M [DOI] [PubMed] [Google Scholar]
- Zorzi G, Redweik U, Trippe H, Penzien JM, Thöny B, Blau N (2002) Detection of sepiapterin in CSF of patients with sepiapterin reductase deficiency. Mol Genet Metab 75:174–177 10.1006/mgme.2001.3273 [DOI] [PubMed] [Google Scholar]