

Abstract
We conducted a 10-centimorgan linkage autosomal genome scan in a set of 19 extended American pedigrees (219 subjects) ascertained through probands with panic disorder. Several anxiety disorders—including social phobia, agoraphobia, and simple phobia—in addition to panic disorder segregate in these families. In previous studies of this sample, linkage analyses were based separately on each of the individual categorical affection diagnoses. Given the substantial comorbidity between anxiety disorders and their probable shared genetic liability, it is clear that this method discards a considerable amount of information. In this article, we propose a new approach that considers panic disorder, simple phobia, social phobia, and agoraphobia as expressions of the same multivariate, putatively genetically influenced trait. We applied the most powerful multipoint Haseman-Elston method, using the grade of membership score generated from a fuzzy clustering of these phenotypes as the dependent variable in Haseman-Elston regression. One region on chromosome 4q31-q34, at marker D4S413 (with multipoint and single-point nominal P values < .00001), showed strong evidence of linkage (genomewide significance at P<.05). The same region is known to be the site of a neuropeptide Y receptor gene, NPY1R (4q31-q32), that was recently connected to anxiolytic-like effects in rats. Several other regions on four chromosomes (4q21.21-22.3, 5q14.2-14.3, 8p23.1, and 14q22.3-23.3) met criteria for suggestive linkage (multipoint nominal P values < .01). Family-by-family analysis did not show any strong evidence of heterogeneity. Our findings support the notion that the major anxiety disorders, including phobias and panic disorder, are complex traits that share at least one susceptibility locus. This method could be applied to other complex traits for which shared genetic-liability factors are thought to be important, such as substance dependencies.
Anxiety disorders are serious illnesses that cause substantial morbidity at the population level. Anxiety disorders, including simple or specific phobia (SimP [MIM %608251]), social phobia (SocP), agoraphobia (AgP), and panic disorder (PD [MIM %167870]), are common disorders, with lifetime prevalences estimated by the U.S. National Comorbidity Survey Replication study to be 4.7%, 1.4%, 12.5%, and 12.1% for PD, AgP without PD, SimP, and SocP, respectively (Kessler et al. 2005). The DSM-IIIR (American Psychiatric Association 1987) provides standard criteria for the diagnoses of AgP, SocP, SimP, and PD. AgP is discomfort and anxiety from being in public places or places from which escape might be difficult, such as highways or bridges. SocP, on the other hand, is fear of or great discomfort from being in situations that might involve the scrutiny of others, such as public speaking, to the extent that specific situations are avoided or endured with great distress. This condition disrupts a person’s ability to function at work or school and causes withdrawal from social activities and/or relationships. A person with SimP experiences excessive or irrational fear of a specific object or situation, such that the fear causes impairment or that exposure to the object or situation causes an immediate anxiety response. The person may have distress about having the phobia and may realize that the fear is excessive or irrational. The object or situation is endured with distress or avoided altogether. Finally, PD is characterized by sudden episodes of acute anxiety or intense fear that may occur without any apparent reason or stimulus. In general, anxiety disorders are chronic disorders, and their clinical courses are variable.
Anxiety disorders are genetically influenced (Kendler et al. 1999) and frequently co-occur (Magee et al. 1996; Curtis et al. 1998). There is also frequent comorbidity with other disorders, particularly psychiatric ones, including, for example, nicotine dependence (MIM #188890); elsewhere, we reported on linkage to nicotine dependence in the present study sample (Gelernter et al. 2004a). A review of the studies that examine the genetic etiology of anxiety and PD reveals the complexity of these disorders, their multifactorial nature, and the fact that they are greatly influenced by genetic factors (van den Heuvel et al. 2000). Nevertheless, the influence of the family on anxiety is covaried by both genetic and environmental mechanisms. Hettema et al. (2001) showed that the role of nonshared environmental experience is significant in the etiology of anxiety. Epidemiological studies worldwide have consistently reported higher rates of anxiety disorders in women (whereas men consistently show higher rates of substance abuse and antisocial disorders), thus indicating sex as a risk factor for anxiety. However, the underlying structure of the genetic and environmental risk factors for anxiety disorders is similar for men and women (Hettema et al. 2005). Age at first onset of anxiety disorders varies from adolescence to early adulthood, with later onsets being mostly of comorbid conditions (Kessler et al. 2005), and diagnostic variations for anxiety disorders attributed to age have been pointed out by Jeste et al. (2005).
Several studies suggest that alterations in neurotransmitter balance, in the function of neurotransmitter receptors (Sand et al. 2000) or transporters (Mazzanti et al. 1998; Ohara et al. 1998; Nakamura et al. 1999), or in enzymes involved in their regulation (Hamilton et al. 2002) may contribute to anxiety disorders. However, peripheral systems are also important for the perception and experience of anxiety, as recently demonstrated in a study by Stein et al. (2004) that showed association of a variant of the β1 adrenergic receptor gene (ADRB1 [MIM *109630]) with the social anxiety–related traits of shyness and extroversion.
Several total-genome scans have been performed for anxiety phenotypes, both including and excluding PD, with variable results. Most of the previous studies focused on PD have identified only “suggestive” linkages (Knowles et al. 1998; Crowe et al. 2001; Gelernter et al. 2001), but genomewide-significant linkage results were reported by Thorgeirsson et al. (2003) for anxiety and PD at chromosome 9q31. Significant linkages were also reported for a “panic disorder syndrome” that includes PD, bladder or kidney problems, headache, and thyroid problems (Weissman et al. 2000). Additionally, suggestive-linkage regions were reported by Gelernter et al. (2001) for AgP, by Smoller et al. (2001) for PD and AgP, and by Gelernter et al. (2004b) for SocP, and genomewide-significant linkage to chromosome 14 markers was reported for SimP by Gelernter et al. (2003).
However, despite the considerable evidence that genetic factors play a major role in the etiology of phobias and other anxiety disorders, to our knowledge no research has been completed that unequivocally (i.e., confirmed via either replication in an independent sample or identification of a variant from a linked region associated with the trait) establishes the locations of genes contributing to these disorders. Clearly, one impediment to gene mapping is the complex nature of the trait, which can translate into misspecification of the mode of inheritance, which in turn reduces the power of any genetic model–based analysis (Clerget-Darpoux et al. 1986; Risch and Giuffra 1992; Dizier et al. 1996). This can be addressed through the use of genetic model–free analyses, but this approach may involve a sacrifice in power. Also, these traits are commonly analyzed univariately, one (binary) phenotype at a time, which (given that there is an underlying genetic relationship between these traits) can weaken the linkage or association signal. This highlights an additional dilemma of genetic model–based analyses that focus on a single phenotype of this multivariate trait; for example, in an analysis focused on PD, individuals with only a diagnosis of SocP would be defined as unaffected, which obviously could be inaccurate in the situation of a shared risk locus. Furthermore, these methods are related to hard clustering and assign each person to one cluster in a set of defined clusters (spectra of diagnoses or combinations of them), often assuming well-defined boundaries between the clusters and not taking into account the potential phenotype heterogeneity that characterizes most complex diseases. Also, the diagnostic criteria for anxiety disorders are constructed in such a way that they can be met with different (although usually overlapping) sets of symptoms, which is an issue that was not usually considered by previous studies.
Thus, more-accurate investigations (to obtain more-powerful tests) should properly model the multivariate nature and variable expressivity of the anxiety phenotype, which is the focus of this study. An approach using fuzzy clustering may be better suited than a categorical classification approach to anxiety data. In fuzzy clustering, grade of membership (GoM) scores between 0 and 1 are assigned to every data element (individual), and this should better model the variable expression of the genetic factors.
We therefore based our analysis on GoM scores, which aim to summarize the whole phenotype, reflecting the multivariate nature and heterogeneity of the set of anxiety disorders. The analysis of these GoM scores is equivalent to an analysis of all the traits together and has been shown to be a powerful form of multivariate analysis (Kaabi and Elston 2003). This method of linkage analysis, based on the use of GoM scores resulting from fuzzy clustering to define a new dependent variable for the various Haseman-Elston approaches, provides a means of data reduction (which implies fewer dependent variables and fewer df) and data mining (i.e., it seeks a hidden structure that may be linked to a specific marker). Thus, it addresses a common problem in genetic linkage analysis of complex traits, both by allowing for the inclusion of more diagnostic information and by accounting for the uncertainty about that information. The inclusion of all individual phenotypes and the flexibility in categorizing them results in increased power for a given sample size.
Material and Methods
Pedigree Identification and Collection of DNA
Families were identified through the Anxiety Clinic at the Connecticut Mental Health Center at Yale University or through advertisements. All families were ascertained through probands with PD. Family inclusion criteria were a family history of known symptoms of panic attacks or AgP, generalized anxiety, or SocP (at least two family members, in addition to the proband, with symptoms of anxiety disorder and one member with PD). Exclusion criteria and the source of DNA are described extensively elsewhere (Gelernter et al. 2001). All subjects gave informed consent as approved by the appropriate institutional review boards. The diagnostic process was identical for all phobia variables (i.e., the anxiety disorders PD, AgP, SimP, and SocP) (Gelernter et al. 2001, 2003, 2004a, 2004b). In total, the sample includes 19 families (219 subjects), comprising 61 sibships among whom 200 subjects were given diagnoses, and 162 of these subjects were genotyped. For more-detailed descriptive statistics of the data set used in this analysis, see tables 1 and 2.
Table 1.
Summary of Descriptive Statistics of the Data Set
| Statistic | Value |
| No. of pedigrees | 19 |
| Mean ± SD size of pedigrees (range) | 11.53±5.83 (3–21) |
| No. of sibships | 61 |
| Mean ± SD size of sibships (range) | 2.28±1.33 (1–8) |
| No. of sibships of size >1 | 42 |
| Mean ± SD size of sibships of size >1 (range) | 2.86±1.23 (2–8) |
| No. of sibships with: | |
| 0 Parents with data | 21 |
| 1 Parent with data | 22 |
| 2 Parents with data | 18 |
| No. of sibships of size >1 with: | |
| 0 Parents with data | 16 |
| 1 Parent with data | 15 |
| 2 Parents with data | 11 |
| No. of male subjects | 99 |
| No. of female subjects | 120 |
| No. of pairs of type: | |
| Parent-offspring | 278 |
| Sib-sib | 143 |
| Sister-sister | 51 |
| Brother-brother | 23 |
| Brother-sister | 69 |
Table 2.
Frequency of Anxiety Phenotypes among Subjects
|
No. (%) of Subjects |
||||
| Phenotype | Definitely Affected |
Probably or Partially Affected |
Definitely Unaffected |
Missing or Unknown |
| AgP | 76 (38) | 4 (2) | 115 (57.5) | 5 (2.5) |
| SimP | 74 (37) | 5 (2.5) | 116 (58) | 5 (2.5) |
| SocP | 65 (32.5) | 5 (2.5) | 125 (62.5) | 5 (2.5) |
| PD | 55 (27.5) | 7 (3.5) | 133 (66.5) | 5 (2.5) |
Phenotypic Evaluation and Diagnosis
The Schedule for Affective Disorders and Schizophrenia–Lifetime (modified to permit DSM-IIIR diagnosis) or the Structured Clinical Interview for DSM-IV Axis I Disorders (First et al. 1997) was used for diagnostic evaluation. Assignment of the diagnoses is described elsewhere (Gelernter et al. 2001). All included individuals received direct interviews. Those not interviewed were coded as unknown or missing for the anxiety variables considered. On the basis of a review of the interview by a doctoral-level expert and the interviewer's assessment, subjects were grouped into three classifications: definitely affected, probably or partially affected, and definitely unaffected (table 2). Each of the variables was scored 2 for definitely affected, 1 for probably affected, or 0 for definitely unaffected.
Laboratory Methods
Genotypes from 400 autosomal markers were analyzed. These markers included 372 from ABI PRISM LD-MD10 Linkage Mapping Set version 2.0 in addition to 6 markers used as substitutes for failed markers from the ABI PRISM set. In addition, polymorphisms at several candidate loci were genotyped. Twelve markers, mostly from the ABI PRISM marker set, were added to increase marker density in regions of interest for phobias or PD on the basis of previous linkage results for these phenotypes (Gelernter et al. 2001, 2003). PCR and genotyping techniques are comprehensively described elsewhere (Gelernter et al. 2001). In construction of the genetic map, sex-averaged distances were used for markers on the Marshfield genetic map, whereas the nucleotide position was converted to centimorgans (1 Mb ≈ 1 cM) for the others (mostly candidate polymorphisms) by use of the NCBI Map Viewer (Cooperative Human Linkage Center).
Statistical Analyses
We checked for genotyping errors, which, if ignored, may either lead to false-positive results (i.e., increase type I error [Seaman and Holman 2005]) or diminish evidence of linkage (i.e., yield less power [Abecasis et al. 2001]). When parents or additional sibs are available, genotyping errors and new mutations can often be detected as Mendelian incompatibilities or apparent double recombinants by use of common programs, such as GENEHUNTER (Kruglyak Lab Web site) and the S.A.G.E. package procedure MARKERINFO (S.A.G.E. v5.0). In cases of identified Mendelian errors, the genotypes of the child, parents, and other siblings were classified as missing for that marker. Moreover, the results of linkage studies may be compromised if a substantial number of putative sib pairs are not actually sib pairs. We used the programs SIBMED (Douglas et al. 2000, 2002) and RELTEST (Olson 1999) for the purpose of identifying any such misclassified individuals.
Fuzzy Clustering
By use of the anxiety variables described above, a fuzzy clustering was performed using the S-PLUS (Insightful Corporation 2003) procedure FANNY (Kaufman and Rousseeuw 1990). This procedure is based on the similarity or dissimilarity of individuals with respect to the variables considered. As input for the procedure FANNY, we used the anxiety variables (SocP, SimP, AgP, and PD) coded as quantitative variables with possible values of 0, 1, or 2. We prespecified the number of clusters as two, and the rest of the FANNY parameters (including membership exponent = 2) were set to default values. The output contained the GoM scores for each cluster, the clustering vector of the nearest crisp clustering or the closest hard (i.e., crisp) clustering, and Dunn’s partition coefficient F(k) of the clustering (Dunn 1977), where k is the number of clusters. F(k) is the sum of all squared membership coefficients, divided by the number of observations. Its value is always between 1/k and 1. The normalized form of the coefficient is defined as
 |
and ranges between 0 and 1. A low value of Dunn’s coefficient indicates a very fuzzy clustering, whereas a value close to 1 indicates a near-crisp clustering. We identified two groups (clusters), and every data element (individual) was assigned a GoM to these groups. By construction, the sum of these GoM scores for each person equals 1. We also performed a post hoc characterization in terms of the initial anxiety variables of the automatically determined clusters.
Linkage Analysis
Genotype data from all family members were used to estimate multipoint and single-point identity-by-descent allele sharing, with the GENIBD program of the S.A.G.E. package. Then, we used SIBPAL, a model-free linkage program from the same package, to perform linkage analysis using all possible sib pairs. The dependent variable studied was the GoM score, which ranged from 0 to 1, precomputed using the FANNY procedure. Evidence of linkage was investigated by the Haseman-Elston regression method (Haseman and Elston 1972; Elston et al. 2000). Other adaptations and modifications of this method have been proposed and implemented in the SIBPAL program through a variety of options (given in the S.A.G.E. v5.0 user manual). We used the most powerful and newest adaptation of the method, which transforms the sib-pair trait values to a weighted combination of the squared trait difference and squared trait mean-corrected sum and which allows for nonindependent sib pairs (Shete et al. 2003). It is implemented as option W4 in the SIBPAL program. In addition to the straightforwardness of the analysis, the advantage of restricting the analysis to full sib pairs is the simplicity with which permutation-test P values can be obtained while correctly allowing for the correlations between pairs of sib pairs. Hence, all multipoint results that were nominally (asymptotically) significant at the 1% level were checked by comparison with the null permutation distribution, by use of a sample of up to 100,000 replicate permutations of the allele-sharing data—permutations both within sibships and across sibships of the same size, as implemented in SIBPAL. Single-point P values were also computed for specific markers that showed strong significance, to verify the genomewide significance level directly. Multipoint nominal and empirical P values are reported in table 3 for specific markers. We should emphasize that these single-point and empirical P values are not adjusted for multiple testing. However, the empirical P values can be interpreted in terms of genomewide significance levels by use of the locus-counting method of Wiltshire et al. (2002). Two regression models were investigated: one that did not include the sib-pair age difference as a covariate (model 1) and one that did include it (model 2). Unlike ordered-subset analysis (Hauser et al. 2004), in which mean family age is computed and used to order families and to compute LOD scores or other statistics sequentially, this adjustment for age considers only the age difference between sibs.
Table 3.
Genetic Locations and Multipoint P Values for Markers Showing Possible Linkage (P⩽.01)[Note]
|
Model 1(Without Adjustment for Age) |
Model 2(With Adjustment for Age) |
||||
|
Chromosome and Marshfield Map Position (cM) |
Marker | Nominal P | Empirical P | Nominal P | Empirical P |
| Chromosome 2: | |||||
| 27.06 | D2S168 | 3.830 × 10−3 | 2.16 × 10−2 | 6.921 × 10−2 | NC |
| Chromosome 3: | |||||
| 22.33 | D3S1304 | 9.682 × 10−3 | 3.12 × 10−2 | 2.87 × 10−1 | NC |
| Chromosome 4: | |||||
| 88.35 | D4S2964 | 2.048 × 10−3 | 8.32 × 10−3 | 1.122 × 10−4 | 8.64 × 10−3 |
| 95.09 | D4S1534 | 6.990 × 10−4 | 4.48 × 10−3 | 6.557 × 10−4 | 6.510 × 10−3 |
| 100.75 | D4S414 | 2.020 × 10−3 | 1.01 × 10−2 | 1.231 × 10−2 | NC |
| 107.95 | D4S1572 | 1.150 × 10−3 | 7.32 × 10−3 | 8.56 × 10−3 | 2.24 × 10−2 |
| 144.56 | D4S424 | 2.170 × 10−3 | 1.12 × 10−2 | 2.38 × 10−3 | 8.5 × 10−3 |
| 157.99 | D4S413 | 5.437 × 10−6 | 5.6 × 10−4 | 6.749 × 10−6 | 4.3 × 10−4 |
| 169.42 | D4S1597 | 1.203 × 10−3 | 8.35 × 10−3 | 7.412 × 10−4 | 6.08 × 10−3 |
| Chromosome 5: | |||||
| 92.38 |
D5S641 |
8.681 × 10−3 |
4.956 × 10−2 |
3.506 × 10−3 |
2.529 × 10−2 |
| 95.40 |
D5S428 |
1.376 × 10−2 |
NC | 7.322 × 10−3 |
3.205 × 10−2 |
| Chromosome 6: | |||||
| 190.14 | D6S281 | 2.950 × 10−3 | 12.19 × 10−2 | 2.447 × 10−2 | NC |
| Chromosome 7: | |||||
| 41.69 | D7S516 | 1.037 × 10−6 | 1.5 × 10−4 | 2.681 × 10−2 | NC |
| 53.50 | D7S484 | 8.693 × 10−4 | 1.01 × 10−2 | 1.286 × 10−1 | NC |
| Chromosome 8: | |||||
| 8.34 |
D8S277 |
1.440 × 10−4 |
1.32 × 10−3 |
3.808 × 10−4 |
2.200 × 10−4 |
| 21.33 |
D8S550 |
4.105 × 10−4 |
2.32 × 10−3 |
7.425 × 10−4 |
4.800 × 10−4 |
| 31.73 | D8S549 | 4.986 × 10−4 | 5.23 × 10−3 | 3.111 × 10−2 | NC |
| Chromosome 9: | |||||
| 159.61 | D9S1826 | 2.086 × 10−3 | 1.625 × 10−2 | 4.283 × 10−1 | NC |
| Chromosome 11: | |||||
| 141.91 | D11S1320 | 9.548 × 10−3 | 3.88 × 10−2 | 3.118 × 10−1 | NC |
| Chromosome 14: | |||||
| 40.11 | D14S70 | 3.954 × 10−3 | 1.51 × 10−2 | 4.379 × 10−1 | NC |
| 63.25 |
D14S274 |
1.911 × 10−1 |
NC | 7.30 × 10−3 |
5.57 × 10−3 |
| 84.69 | D14S1036 | 5.406 × 10−3 | 2.12 × 10−2 | 1.081 × 10−1 | NC |
| 105.0 | D14S280 | 5.491 × 10−3 | 3.83 × 10−2 | 6.85 × 10−2 | NC |
| Chromosome 15: | |||||
| 82.28 | D15S201 | 9.133 × 10−3 | 4.32 × 10−2 | 3.38 × 10−2 | NC |
Note.— Up to 100,000 permutations were used to compute empirical P values. Bold italics indicate strong linkage, underlining indicates suggestive linkage, and all other entries show suggestive or strong linkage for only model 1 or only model 2 (not both). NC = not computed.
The dependent variable in these regression models was based on the GoM score precomputed using the FANNY procedure (Kaabi and Elston 2003). It is difficult to test for heterogeneity and interaction in the context of fuzzy clustering. However, linkage analysis for the whole data set was followed by analysis of each pedigree individually, to the extent possible, with the aim of distinguishing families linked to a specific marker from those eventually linked to another marker.
Results
Clustering Results
The FANNY algorithm identified two clusters based on individuals' assigned GoM scores (GoM>0.5 was denoted as cluster 1, and GoM⩽0.5 was denoted as cluster 2). Dunn’s coefficient and its normalized version were 0.6071 and 0.2142, respectively, which implies a quite fuzzy partition (more fuzzy than crisp). Figure 1 shows a two-dimensional illustration of the clusters along the two principal components of the SocP, SimP, AgP, and PD diagnoses, which account for 66.83% of the total data variance (Pison et al. 1999). The area of overlap represents persons who cannot be unequivocally assigned to either cluster; their GoM scores are close to 0.5. In general, the smaller this area of overlap is, the closer the resulting partition is to ordinary (crisp) clustering. The triangles and circles distinguish between the clusters in the figure.
Figure 1.

Clusterplot graph. Cluster 1 (GoM>0.5) is the ellipse on the right, and cluster 2 (GoM⩽0.5) is the ellipse on the left; the graph depicts the clusters in the two dimensions given by the two first principal components.
The density of the clusters (the number of points per area of ellipse) reflects the number of subjects per cluster. A good clustering result would have a small area of overlap between dense clusters. In figure 1, the two clusters show a small area of complete overlap. We also present, in figure 2, a silhouette graph (Rousseeuw 1987), which is a graphical means of viewing the clustering. The silhouette shows the subjects in the two clusters, one above the other. Observations with a large width (almost 1) are very well clustered, those with a small width (∼0) lie between two clusters, and those with a negative width are probably placed in the wrong cluster. Figure 2 shows the two clusters with a few reversals in cluster 1; the average silhouette width is 0.36. For post hoc cluster characterization in terms of the initial anxiety variables, we provide, in table 4, the number of subjects in each cluster; the P values are for a test of equality of the proportions within each cluster, of a total of 111 subjects in cluster 1 and 84 subjects in cluster 2.
Figure 2.

Silhouette plot for the fuzzy partition of anxiety. Cluster 1 is above cluster 2. See “Clustering Results” section for details.
Table 4.
Cluster Characterization
|
No. of Subjects |
|||
|
Status (Score) and Phenotype |
Cluster 1 (GoM>0.5) |
Cluster 2 (GoM⩽0.5) |
Pa |
| Affected (2): | |||
| PD | 49 | 6 | 3.2×10-8 |
| SocP | 47 | 18 | .0035 |
| SimP | 74 | 0 | <<10−8 |
| AgP | 76 | 0 | <<10−8 |
| Probably affected (1): | |||
| PD | 6 | 1 | .23 |
| SocP | 1 | 4 | .218 |
| SimP | 2 | 3 | .7515 |
| AgP | 2 | 2 | 1 |
| Unaffected (0): | |||
| PD | 56 | 77 | <<10−8 |
| SocP | 63 | 62 | .0210 |
| SimP | 35 | 81 | <<10−8 |
| AgP | 33 | 82 | <<10−8 |
| Total | 111 | 84 | |
P values are for a test of equality of the proportions within each cluster, of a total of 111 subjects in cluster 1 and 84 subjects in cluster 2.
The mean (±SD) age in each cluster is 45.26 (±15.50) years in cluster 1 and 50.72 (±18.32) years in cluster 2. The age difference between the two clusters, when compared using a t test, yields P=.029. The sex distribution in the two clusters is 34 males and 74 females in cluster 1 and 40 males and 44 females in cluster 2. This difference yields P=.030. However, the distribution of sex in cluster 1 (69.3% female) is close to that in the whole data sample (62% female).
Linkage Analysis Results
With GoM scores obtained from the FANNY procedure as the dependent variable (model 1) and also with sib-pair age difference as a covariate (model 2), many markers showed suggestive evidence of linkage, especially on chromosome 4 (table 3 and fig. 3).
Figure 3.

Multipoint results of genomewide linkage scan for phobia using GoM scores as the dependent variable. The dashed lines represent model 1; the bold solid lines represent model 2. For each chromosome, genetic distance (in cM) is plotted on the X-axis against 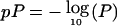 on the Y-axis. The horizontal line in each graph corresponds to P=.01; the zones above that line are suggestive of linkage.
on the Y-axis. The horizontal line in each graph corresponds to P=.01; the zones above that line are suggestive of linkage.
Model 1 identified 14 regions with suggestive and strong linkage signals: 2p25.1-24.3, 3p26.2-25.3, 4q21.21-24, 4q31.21-32.3, 5q14.2, 6q27, 7p15.2-14.2, 8p23.1-22, 9q34.3, 11q25, 14q12-13.1, 14q24.3, 14q32.12, and 15q25.3. Suggestive linkage signals are defined by multipoint nominal P⩽.01 (fig. 3). The strongest linkage signals are on region 4q31.21-32.3 (multipoint nominal P=5.4×10-6; empirical P=5.6×10-4) and region 7p15.2-14.2 (multipoint nominal P=1.0×10-6; empirical P=1.5×10-4). However, whereas the pointwise P value for marker D4S413 is 1.3×10-7 (which is < 2×10-5, the genomewide significance threshold of Lander and Kruglyak [1995]), the pointwise P values for markers on 7p15.2-14.2 (D7S516 and D7S484) are 5.1×10-2 and .14 (>.01), which do not meet the criteria of genomewide significance (figs. 3 and 4). At this stage, two explanations are possible. The first is that the observed signals represent a true linkage signal, but the gene in question covers both markers (D7S516 and D7S484), in the sense that both markers are needed to explain the variation observed. In other words, the observed within-family association is between the haplotype of the two markers and the GoM scores. The second explanation is that the observed signals are not true linkage signals, and we should subsequently eliminate this region as a potential linkage site. By use of model 2, only five regions, on chromosomes 4, 5, 8, and 14, displayed signals of suggestive or strong linkage. These regions are 4q21.21-22.3, 4q31.21-32.3, 5q14.2-14.3, 8p23.1, and 14q22.3-23.3. Signals previously obtained at 2p25.1-24.3, 3p26.2-25.3, 4q21.1-22.3, 6q27, 7p15.2-14.2, 8p22, 9q34.3, 11q25, 14q12-13.1, 14q24.3, 14q32.12, and 15q25.3 by use of model 1 disappear when model 2 (with adjustment for the sib-pair age difference) is used, which suggests that the information given by these markers may be confounded with the information contained in the sib-pair age difference; therefore, their signals cannot yet be regarded as true linkage signals. Similar to the results obtained using model 1, there is a strong signal at 4q31.21-32.3 (multipoint nominal P=6.7×10-6; empirical P=4.3×10-4) with model 2. In summary, the regions of interest identified by both models 1 and 2 (with P values < .01, or LOD⩾1.18) are 4q21.21-22.3, 4q31.21-32.3, 5q14.2-14.3, 8p23.1, and 14q22.3-23.3, with the strongest signal on chromosome 4 (marker D4S413; 4q31.21-32.3). The single-point analysis of these regions gives results consistent with those obtained using model 2, which excludes linkage disequilibrium as a possible false-positive error inflator (Boyles et al. 2005).
Figure 4.

Multipoint results for chromosome 7. The dashed line represents model 1, the bold solid line represents model 2, and the vertical lines represent single-point analysis. Genetic distance (in cM) is plotted on the X-axis against  on the Y-axis. The horizontal lines correspond to P=.01 (bottom line) and P=2×10-5 (top line); the zones above those lines are suggestive of linkage and strongly indicative of linkage, respectively.
on the Y-axis. The horizontal lines correspond to P=.01 (bottom line) and P=2×10-5 (top line); the zones above those lines are suggestive of linkage and strongly indicative of linkage, respectively.
The strong statistical support for linkage at the 4q31.21-32.3 region is novel. Weak linkage signals were observed for this region in previous genome scans for some anxiety disorders (observed by Thorgeirsson et al. [2003] for anxiety and PD by use of a model-based approach). In our previous linkage analyses using these same families—but based on DSM-IIIR diagnosis definitions—we observed results of interest in some locations similar to those identified in the present study. For the same chromosome 4 regions, we obtained a LOD score >1.0 in an analysis using a recessive mode of inheritance for SimP (Gelernter et al. 2003), and, in a linkage scan for SocP (Gelernter et al. 2004b), we obtained a Z score of 2. The regions 5q14.2-14.3 and 14q22.3-23.3 have been reported in previous linkage studies; specifically, 5q14.2-14.3 was reported for PD or AgP, with heterogeneity LOD >1 (Gelernter et al. 2001). The region 14q22.3-23.3 has been reported as a signal of suggestive or significant linkage by several linkage-scan studies (e.g., Gelernter et al. 2001, 2003). The results mentioned above were obtained using the present clinical sample or a subset of it, so some convergence on chromosomal regions possibly linked to phenotype is to be expected.
The family-by-family analysis was performed using both models with the eight pedigrees having enough sibs to compute the regression. No single family is responsible for the P values observed in the suggestive and strong regions of linkage noted above. However, some families have excess sharing of markers in these regions or at markers flanking these regions. The results showed no strong evidence of heterogeneity, because the markers identified by individual pedigrees for each chromosome (at the level P<.001) either belong to or are adjacent to the regions of suggestive and strong linkage identified using the overall data (all pedigree sib pairs). The pedigrees have a mixture of sibships with 0, 1, and 2 parents available; however, the sibships with no genotyped parents tend to be the largest, and so their parental genotypes can be largely inferred.
Discussion
We have presented genomewide linkage results for a set of anxiety disorders considered as phenotypes of a multivariate trait, and we allowed for some flexibility in the characterization of each individual by using a fuzzy-clustering approach. Fuzzy clustering can be viewed as a generalization of ordinary partitioning procedures, allowing for some vagueness in the data. This is made possible through GoM scores that describe the “distance” from each data element to the generated clusters, that provide detailed information on the data structures, and that allow modeling of data heterogeneity. Hence, GoM models present a means for data mining and data reduction—every element in the data set originally characterized by a number of variables (i.e., a multivariate response) can be identified through fuzzy clusters and its GoM scores for those clusters. Analyzing the GoM scores will yield similar information, compared with analyzing multiple correlated trait phenotypes jointly, but will result in a more powerful analysis, as long as the generated partition distinguishes phenotypes related to a specific set of deleterious alleles at the linked trait locus (or loci). It is also possible to consider more than two clusters when analyzing complex traits, but the gain in power that results from reducing the many variables to a single GoM score will diminish as the number of clusters is increased.
We analyzed nuclear and extended families, using a sib-pair allele-sharing method with and without adjusting for sib-pair age differences. We detected a strong linkage signal for anxiety and PD on chromosome 4 (4q31-q34) at marker D4S413, with a pointwise P value of 1.3×10-7, which clearly meets the criterion for significant linkage suggested by Lander and Kruglyak (1995), even with adjustment allowing for the multiple testing arising from our previous linkage studies for the individual disease diagnoses. Interestingly, this same region is the site of the NPY1R gene (MIM *162641), which is functionally related to anxiety-related neurotransmission and has been connected to anxiolytic-like effects in rats (Sørensen et al. 2004). This linkage finding is consistent with, but much stronger than, previously reported results and is very promising because of the functional role played by NPY (MIM *162640) (Heilig et al. 1989) and, by inference, NPY1R in modulating anxiety. On the basis of our results and the physiology literature, we hypothesize that it is very likely that a gene or genes underlying this linkage peak (possibly NPY1R) plays a role in the susceptibility to anxiety in general, rather than to a particular phenotype of phobia or to PD—that is, it relates to one of the hypothesized “common” risk factors. The second peaks observed on chromosome 4, at D4S1534 for model 1 and D4S2964 for model 2, are suggestive linkage signals. Their multipoint nominal P values are 7.0×10-4 and 1.1×10-4, respectively, and are not significant genomewide; the pointwise P values are 3.0×10-3 and 0.20, respectively, which exclude the D4S2964 area as a true “suggestive” region on the basis of our data (see fig. 5).
Figure 5.

Multipoint results for chromosome 4. The dashed line represents model 1, the bold solid line represents model 2, and the vertical lines represent single-point analysis. Genetic distance (in cM) is plotted on the X-axis against 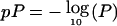 on the Y-axis. The horizontal lines correspond to P=.01 (bottom line) and P=2×10-5 (top line); the zones above those lines are suggestive of linkage and strongly indicative of linkage, respectively.
on the Y-axis. The horizontal lines correspond to P=.01 (bottom line) and P=2×10-5 (top line); the zones above those lines are suggestive of linkage and strongly indicative of linkage, respectively.
The family-by-family analysis did not provide evidence of heterogeneity, nor did it exclude it. The other regions of “suggestive” linkage that overlap with regions identified in previous studies using one or more of the anxiety phenotypes could be responsible for the phenotype diversity (heterogeneity) observed in anxiety disorders. One region of interest is on chromosome 14, which has been reported to show suggestive evidence of linkage in several studies of anxiety disorders.
Broadly speaking, although we used a different approach to the problem of phenotype definitions for anxiety, our finding presents several points of overlap with previous scans for PD or other anxiety disorders. However, none of these previous studies, which used only one or, at most, two of the anxiety phenotypes as the response variable, reported comparably strong evidence of linkage. This may be because of several factors. First, the present analysis allowed the simultaneous use of phenotype information from all individuals in the sample, which by itself provided an increase in power. Second, most of the previous studies were based on the model-based LOD score or conservative nonparametric linkage (NPL) statistic. With regard to LOD-score analyses, misspecification of the mode of inheritance is known to reduce the power of the test, and it is very likely that such misspecification occurred in previous studies, which modeled monogenic inheritance with no allowance for familial association, other than that caused by segregation at the one linked locus. The NPL statistic, although it is a genetic model–free test, only uses information from affected individuals, and this leads to a smaller sample size and hence less power. Third, the weaker findings seen in most previous studies seem to be related to the fact that each analysis considered each anxiety phenotype to be a distinct trait and analyzed it as such, despite that all indications suggest that these phenotypes are correlated and should be more ideally analyzed as a multivariate trait. Finally, the flexibility in cluster identification was of great use, because the affection status of an individual for a psychiatric trait is sometimes difficult to define unequivocally. Using fuzzy clustering, we were able to allow flexibility in the individual affection status and thereby avoid false-positive and false-negative results due to misclassification. The GoM scores are defined for all individuals, and analyzing them as a continuous trait allows us to include in the analysis all individuals, rather than affected persons only.
There are many possible explanations for the loss of linkage signals after adjustment for sib-pair age differences. The first possibility is that it is due to chance (in the context of power loss, since an additional variable is being considered) and that the linkage results are correct. The second possibility is that these genes are differentially expressed with age, and thus age is a confounder; therefore, adjusting for it actually does provide a more accurate answer. Although a P value <.01 is far from significant in a genomewide scan, it is possible that the regions of suggestive linkage represent true anxiety-disorder susceptibility loci if they overlap with other regions indicated in other linkage or association studies. We have three such regions in addition to the strong linkage region 4q31-q34; it is possible that these linkages are to specific forms of phobia and anxiety. Using this multivariate approach, we have provided additional support for the idea that anxiety disorders are complex traits that share some, but not all, of their susceptibility genes. We hypothesize that a susceptibility gene for all the anxiety disorders investigated in the present study is located in the region 4q31-q34.
Finally, we can state that our results, on the basis of the multifactorial nature of anxiety disorders, are consistent with such a hypothesis. We have identified a major locus for anxiety pathology. In the future, we hope to provide further support for this by fine-mapping and association studies. In addition, we hope to demarcate the roles of the genes that have been suggested here, through the construction of gene-pathway models that may reflect their functions, as well as the characteristics and pathology of phobia.
Acknowledgments
We thank the families who participated in this study. G. Kay and A. Lacobelle provided excellent technical assistance. Drs. Susan Kruger and Susan Goodson assisted with diagnoses. This work was supported in part by U.S. Public Health Service grants (resource grant RR03655 from the National Center for Research Resource and research grant GM-28356 from the National Institute of General Medical Sciences), by funds from the U.S. Department of Veterans Affairs (the VA Medical Research Program Merit Review [to J.G.] and funds from the VA Connecticut-Massachusetts Mental Illness Research, Education and Clinical Center), by the VA-CT Research Enhancement Award Program Center, by National Institute on Drug Abuse grants DA12690, DA12849, and DA15105, and by the Ministry of Science, Technology and Competencies Development in Tunisia (Program Contract 2004–2008).
Web Resources
The URLs for data presented herein are as follows:
- Cooperative Human Linkage Center, http://lpgws.nci.nih.gov/ABI/ (for ABI PRISM reference maps)
- Kruglyak Lab, http://www.fhcrc.org/labs/kruglyak/Downloads/ (for GENEHUNTER 2.0)
- NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/mapview/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for SimP, PD, nicotine dependence, ADRB1,NPY1R, and NPY)
- S.A.G.E.—Statistical Analysis for Genetic Epidemiology, http://darwin.cwru.edu/sage/
References
- Abecasis GR, Cherny SS, Cardon LR (2001) The impact of genotyping error on family-based analysis of quantitative traits. Eur J Hum Genet 9:130–134 [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (1987) Diagnostic and statistical manual of mental disorders, third edition, revised. American Psychiatric Press, Washington, DC [Google Scholar]
- Boyles AL, Scott WK, Martin ER, Schmidt S, Li YJ, Ashley-Koch A, Bass MP, Schmidt M, Pericak-Vance MA, Speer MC, Hauser ER (2005) Linkage disequilibrium inflates type I error rates in multipoint linkage analysis when parental genotypes are missing. Hum Hered 59:220–227 10.1159/000087122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerget-Darpoux F, Bonaiti-Pellie C, Hochez J (1986) Effects of misspecifying genetic parameters in lod score analysis. Biometrics 42:393–399 [PubMed] [Google Scholar]
- Crowe RR, Goedken R, Samuelson S, Wilson R, Nelson J, Noyes R Jr (2001) Genomewide survey of panic disorder. Am J Med Genet 105:105–109 [DOI] [PubMed] [Google Scholar]
- Curtis GC, Magee WJ, Eaton WW, Wittchen HU, Kessler RC (1998) Specific fears and phobias: epidemiology and classification. Br J Psychiatry 173:212–217 [PubMed] [Google Scholar]
- Dizier MH, Babron MC, Clerget-Darpoux F (1996) Conclusion of LOD-score analysis for family data generated under two-locus models. Am J Hum Genet 58:1338–1346 [PMC free article] [PubMed] [Google Scholar]
- Douglas JA, Boehnke M, Lange K (2000) A multipoint method for detecting genotyping errors and mutations in sibling-pair linkage data. Am J Hum Genet 66:1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas JA, Skol AD, Boehnke M (2002) Probability of detection of genotyping errors and mutations as inheritance inconsistencies in nuclear-family data. Am J Hum Genet 70:487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JC (1977) Indices of partition fuzziness and the detection of clusters in large data sets. In: Gupta M, Saradis G (eds) Fuzzy automata and decision process. Elsevier, New York, pp 271–284 [Google Scholar]
- Elston RC, Buxbaum S, Jacobs KB, Olson JM (2000) Haseman and Elston revisited. Genet Epidemiol 19:1–17 [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW (1997) Structured clinical interview for DSM-IV axis I disorders, research version, non-patient edition (SCID-NP) version 2.0. Biometrics Research, New York State Psychiatric Institute, New York [Google Scholar]
- Gelernter J, Bonvicini K, Page G, Woods SW, Goddard AW, Kruger S, Pauls DL, Goodson S (2001) Linkage genome scan for loci predisposing to panic disorder or agoraphobia. Am J Med Genet 105:548–557 10.1002/ajmg.1496 [DOI] [PubMed] [Google Scholar]
- Gelernter J, Liu X, Hesselbrock V, Page GP, Goddard A, Zhang H (2004a) Results of a genomewide linkage scan: support for chromosomes 9 and 11 loci increasing risk for cigarette smoking. Am J Med Genet B Neuropsychiatr Genet 128:94–101 10.1002/ajmg.b.30019 [DOI] [PubMed] [Google Scholar]
- Gelernter J, Page GP, Bonvicini K, Woods SW, Pauls DL, Kruger S (2003) A chromosome 14 risk locus for simple phobia: results from a genomewide linkage scan. Mol Psychiatry 8:71–82 10.1038/sj.mp.4001224 [DOI] [PubMed] [Google Scholar]
- Gelernter J, Page GP, Stein MB, Woods SW (2004b) Genome-wide linkage scan for loci predisposing to social phobia: evidence for a chromosome 16 risk locus. Am J Psychiatry 161:59–66 10.1176/appi.ajp.161.1.59 [DOI] [PubMed] [Google Scholar]
- Hamilton SP, Slager SL, Heiman GA, Deng Z, Haghighi F, Klein DF, Hodge SE, Weissman MM, Fyer AJ, Knowles JA (2002) Evidence for a susceptibility locus for panic disorder near the catechol-O-methyltransferase gene on chromosome 22. Biol Psychiatry 51:591–601 10.1016/S0006-3223(01)01322-1 [DOI] [PubMed] [Google Scholar]
- Haseman JK, Elston RC (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19 10.1007/BF01066731 [DOI] [PubMed] [Google Scholar]
- Hauser ER, Watanabe RM, Duren WL, Bass MP, Langefeld CD, Boehnke M (2004) Ordered subset analysis in genetic linkage mapping of complex traits. Genet Epidemiol 27:53–63 10.1002/gepi.20000 [DOI] [PubMed] [Google Scholar]
- Heilig M, Soderpalm B, Engel JA, Widerlov E (1989) Centrally administered neuropeptide Y (NPY) produces anxiolytic-like effects in animal anxiety models. Psychopharmacology (Berl) 98:524–529 [DOI] [PubMed] [Google Scholar]
- Hettema JM, Neale MC, Kendler KS (2001) A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry 158:1568–1578 10.1176/appi.ajp.158.10.1568 [DOI] [PubMed] [Google Scholar]
- Hettema JM, Prescott CA, Myers JM, Neale MC, Kendler KS (2005) The structure of genetic and environmental risk factors for anxiety disorders in men and women. Arch Gen Psychiatry 62:182–189 10.1001/archpsyc.62.2.182 [DOI] [PubMed] [Google Scholar]
- Insightful Corporation (2003) S-PLUS software: S-PLUS® 6.2 for Windows Professional Edition. Insightful Corporation, Seattle [Google Scholar]
- Jeste DV, Blazer DG, First M (2005) Aging-related diagnostic variations: need for diagnostic criteria appropriate for elderly psychiatric patients. Biol Psychiatry 58:265–271 10.1016/j.biopsych.2005.02.004 [DOI] [PubMed] [Google Scholar]
- Kaabi B, Elston RC (2003) New multivariate test for linkage, with application to pleiotropy: fuzzy Haseman-Elston. Genet Epidemiol 24:253–264 10.1002/gepi.10234 [DOI] [PubMed] [Google Scholar]
- Kaufman L, Rousseeuw PJ (1990) Finding groups in data: an introduction to cluster analysis. John Wiley & Sons, New York, chapter 4 [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA (1999) Fears and phobias: reliability and heritability. Psychol Med 29:539–553 10.1017/S0033291799008429 [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE (2005) Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 62:593–602 10.1001/archpsyc.62.6.593 [DOI] [PubMed] [Google Scholar]
- Knowles JA, Fyer AJ, Vieland VJ, Weissman MM, Hodge SE, Heiman GA, Haghighi F, de Jesus GM, Rassnick H, Preud’homme-Rivelli X, Austin T, Cunjak J, Mick S, Fine LD, Woodley KA, Das K, Maier W, Adams PB, Freimer NB, Klein DF, Gilliam TC (1998) Results of a genome-wide genetic screen for panic disorder. Am J Med Genet 81:139–147 [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 10.1038/ng1195-241 [DOI] [PubMed] [Google Scholar]
- Magee W, Eaton WW, Wittchen H-U, McGonagle KA, Kessler RC (1996) Agoraphobia, simple phobia, and social phobia in national comorbidity survey. Arch Gen Psychiatry 53:159–168 [DOI] [PubMed] [Google Scholar]
- Mazzanti CM, Lappalainen J, Long JC, Bengel D, Naukkarinen H, Eggert M, Virkkunen M, Linnoila M, Goldman D (1998) Role of the serotonin transporter promoter polymorphism in anxiety-related traits. Arch Gen Psychiatry 55:936–940 10.1001/archpsyc.55.10.936 [DOI] [PubMed] [Google Scholar]
- Nakamura M, Ueno S, Sano A, Tanabe H (1999) Polymorphisms of the human homologue of the Drosophila white gene are associated with mood and panic disorders. Mol Psychiatry 4:155–162 10.1038/sj.mp.4000515 [DOI] [PubMed] [Google Scholar]
- Ohara K, Nagai M, Suzuki Y, Ochiai M, Ohara K (1998) Association between anxiety disorders and a functional polymorphism in the serotonin transporter gene. Psychiatry Res 81:277–279 10.1016/S0165-1781(98)00100-0 [DOI] [PubMed] [Google Scholar]
- Olson JM (1999) Relationship estimation by Markov-process models in a sib-pair linkage study. Am J Hum Genet 64:1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pison G, Struyf A, Rousseeuw PJ (1999) Displaying a clustering with CLUSPLOT. Comput Stat Data Analysis 30:381–392 10.1016/S0167-9473(98)00102-9 [DOI] [Google Scholar]
- Risch N, Giuffra L (1992) Model misspecification and multipoint linkage analysis. Hum Hered 42:77–92 [DOI] [PubMed] [Google Scholar]
- Rousseeuw PJ (1987) Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math 20:53–65 10.1016/0377-0427(87)90125-7 [DOI] [Google Scholar]
- Sand PG, Godau C, Riederer P, Peters C, Franke P, Nothen MM, Stober G, Fritze J, Maier W, Propping P, Lesch KP, Riess O, Sander T, Beckmann H, Deckert J (2000) Exonic variants of the GABA(B) receptor gene and panic disorder. Psychiatr Genet 10:191–194 [DOI] [PubMed] [Google Scholar]
- Seaman SR, Holmans P (2005) Effect of genotyping error on type-I error rate of affected sib pair studies with genotyped parents. Hum Hered 59:157–164 10.1159/000085939 [DOI] [PubMed] [Google Scholar]
- Shete S, Jacobs KB, Elston RC (2003) Adding further power to the Haseman and Elston method for detecting linkage in larger sibships: weighting sums and differences. Hum Hered 55:79–85 10.1159/000072312 [DOI] [PubMed] [Google Scholar]
- Smoller JW, Acierno JS Jr, Rosenbaum JF, Biederman J, Pollack MH, Meminger S, Pava JA, Chadwick LH, White C, Bulzacchelli M, Slaugenhaupt SA (2001) Targeted genome screen of panic disorder and anxiety disorder proneness using homology to murine QTL regions. Am J Med Genet 105:195–206 10.1002/ajmg.1209 [DOI] [PubMed] [Google Scholar]
- Sørensen G, Lindberg C, Wortwein G, Bolwig TG, Woldbye DP (2004) Differential roles for neuropeptide Y Y1 and Y5 receptors in anxiety and sedation. J Neurosci Res 77:723–729 10.1002/jnr.20200 [DOI] [PubMed] [Google Scholar]
- Stein MB, Schork NJ, Gelernter J (2004) A polymorphism of the β1-adrenergic receptor is associated with low extraversion. Biol Psychiatry 56:217–224 10.1016/j.biopsych.2004.05.020 [DOI] [PubMed] [Google Scholar]
- Thorgeirsson TE, Oskarsson H, Desnica N, Kostic JP, Stefansson JG, Kolbeinsson H, Lindal E, Gagunashvili N, Frigge ML, Kong A, Stefansson K, Gulcher JR (2003) Anxiety with panic disorder linked to chromosome 9q in Iceland. Am J Hum Genet 72:1221–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel OA, van de Wetering BJ, Veltman DJ, Pauls DL (2000) Genetic studies of panic disorder: a review. J Clin Psychiatry 61:756–766 [DOI] [PubMed] [Google Scholar]
- Weissman MM, Fyer AJ, Haghighi F, Heiman G, Deng Z, Hen R, Hodge SE, Knowles JA (2000) Potential panic disorder syndrome: clinical and genetic linkage evidence. Am J Med Genet 96:24–35 [DOI] [PubMed] [Google Scholar]
- Wiltshire S, Cardon LR, McCarthy MI (2002) Evaluating the results of genomewide linkage scans of complex traits by locus counting. Am J Hum Genet 71:1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]