

Abstract
A population pharmacokinetic model of liposomal amphotericin B (L-AmB) in pediatric patients with malignant diseases was developed and evaluated. Blood samples were collected from 39 pediatric oncology patients who received multiple doses of L-AmB with a dose range from 0.8 to 5.9 mg/kg of body weight/day. The patient cohort had an average age of 7 years (range, 0.2 to 17 years) and weighed an average of 28.8 ± 19.8 kg. Population pharmacokinetic analyses were performed with NONMEM software. Pharmacokinetic parameters, interindividual variability (IIV), between-occasion variability (BOV), and intraindividual variability were estimated. The influence of patient characteristics on the pharmacokinetics of L-AmB was explored. The final population pharmacokinetic model was evaluated by using a bootstrap sampling technique. The L-AmB plasma concentration-time data was described by a two-compartment pharmacokinetic model with zero-order input and first-order elimination. The population mean estimates of clearance (CL) and volume of distribution in the central compartment (V1) were 0.44 liters/h and 3.12 liters, respectively, and exhibited IIV (CL, 10%) and significant BOV (CL, 46% and V1, 56%). The covariate models were CL (liters/h) = 0.44 · e0.0152 ·(WT − 21) and V1 (liters) = 3.12 · e0.0241 · (WT − 21), where WT is the patient's body weight (kg) centered on the study population cohort median of 21 kg. Model evaluation by the bootstrap procedure indicated that the full pharmacokinetic model was robust and parameter estimates were accurate. In conclusion, the pharmacokinetics of L-AmB in pediatric oncology patients were adequately described by the developed population model. The model has been evaluated and can be used in the design of rational dosing strategies for L-AmB antifungal therapy in this special population.
The application of liposomes as drug carriers, which was originally proposed by Gregoriadis in 1981 (10), offers a potential means of manipulating drug pharmacokinetics (PKs) to improve antimicrobial efficacy and reduce toxicity. Amphotericin B (AMB) deoxycholate (Fungizone, D-AmB) has been the “gold standard” therapy for invasive fungal infection for decades due to its broad spectrum of antifungal activity. However, clinical treatment with D-AmB is limited by drug-related nephrotoxicity (7). The encapsulation of AmB in liposomes (AmBisome, L-AmB), which are composed of hydrogenated soy phosphatidylcholine, cholesterol, and distearoylphosphatidylglycerol in a molar ratio of 0.4:2:1:0.8 (7), has been found in preclinical studies to be as effective or, in some cases, more effective but less nephrotoxic than D-AmB in the treatment of a broad spectrum of medically important fungal pathogens, including Candida, Aspergillus, and Cryptococcus species (8, 17, 23). The results of two multicenter, randomized clinical trials in patients with persistent fever and neutropenia (21, 25) and in patients with invasive fungal infections (16) demonstrated the equivalent efficacy and superior safety of L-AmB relative to D-AmB. The reduced toxicity of the liposomal formulation allows for the administration of much higher doses of AmB than those that can be safely administered when given as conventional AmB, leading to the expanding therapeutic potential of L-AmB in comparison with D-AmB.
Children with malignant diseases who experience prolonged periods of myelosuppression due to cytotoxic chemotherapy or hematopoietic stem cell transplant are highly susceptible to invasive fungal infections (11). The uses of indwelling vascular catheters and broad-spectrum antibiotics further increase the risk of immunosuppressed children developing invasive fungal infections. The broad-spectrum antifungal activity and improved therapeutic properties of L-AmB may provide an important therapeutic advance in the treatment and prophylaxis of invasive fungal infections in pediatric oncology patients. However, there remains debate about the appropriate dosing regimens in children. No single study has prospectively investigated the pharmacokinetics of L-AmB that might guide rational design of dosing regimens in this special population.
The clinical pharmacokinetics of L-AmB have been investigated in healthy adult volunteers after a single intravenous dose (5) and in adult patients after multiple doses (26). All of the studies utilized a traditional pharmacokinetic data analysis approach. However, in the clinical setting where pediatric patients or those with severe illness are studied, only limited numbers of samples can be obtained due to ethical, logistical, and medical considerations. Therefore, the population approach was employed in the present study as it provides not only a solution for combining concentration information from both intensive and sparse sampling but also the valuable knowledge of the population distribution of pharmacokinetic parameters and factors influencing the variability in these parameters.
The aim of this study was to explore the pharmacokinetics of L-AmB in pediatric oncology patients by using a nonlinear mixed effects modeling approach in order to estimate the typical population pharmacokinetic parameters, the variability between patients, and the variability between occasions and within patients and to identify the covariates that are significant predictors of variability in L-AmB pharmacokinetics. As the developed population pharmacokinetic model will be used for the dosing strategy optimization, the stability and predictive performance of the model were evaluated using a bootstrap sampling technique.
MATERIALS AND METHODS
Study design.
This study was designed as a prospective, single-center, open-label, observational clinical trial for the investigation of the pharmacokinetics of L-AmB. The study protocol was approved by the Ethics Committee at the Children's Hospital at Westmead (Sydney, NSW, Australia) and the Human Ethics Committee at the University of Sydney (Sydney, NSW, Australia). A total of 40 pediatric patients who were receiving L-AmB for antifungal therapy or prophylaxis were entered in the study. All patients had malignant disease, were neutropenic, and were experiencing fever. However, only 39 patients were included in the population pharmacokinetic analysis as the dosing history for 1 patient was not complete. Written informed consents were obtained from the parents or legal guardians of all the children.
Pharmacokinetic sampling.
L-AmB (AmBisome; Gilead Sciences, VIC, Australia) was administered once daily as a 1-h infusion through one of the lumens of a central line catheter for each patient. The first day of L-AmB administration was defined as day 1. Heparinized whole-blood samples (1 to 2 ml) were collected from the opposite lumen to which L-AmB was delivered and after 5 ml blood was withdrawn and discarded to avoid contamination. The times of dosing and sample collection were accurately recorded on a sample collection sheet. Multiple blood samples, to characterize the full pharmacokinetic profile, were taken in 22 out of 48 courses at specific times in one dosing interval, including immediately before the infusion and 1, 2, 4, 7, 12, and 24 h after the infusion. Trough and peak samples were obtained from 25 patients on day 7 of therapy or, if those samples were not available, on day 6 or day 8. The remaining pharmacokinetic samples were collected with those for routine hospital care throughout the period of L-AmB therapy in order to minimize the risk of infection due to blood sampling from the central line catheter.
Plasma fractions were separated by centrifugation at 1,500 × g for 10 min at 4°C (Beckman CS-15R; Beckman Instruments, California) and were stored at −80°C until analysis.
Drug assay.
High-performance liquid chromatography was employed for the quantification of AmB in plasma (5). Briefly, the internal standard p-nitroaniline at a concentration of 27 μg/ml (30 μl) and methanol (200 μl) were added to an aliquot of plasma sample (100 μl) in a microcentrifuge tube. The tube was vortex mixed for 1 min and stood in the dark for 10 min to allow protein precipitation. After centrifugation at 12,100 × g for 10 min (Eppendorf MiniSpin; Biolab, Australia), an aliquot of 20 μl of methanol extract was injected directly into the high-performance liquid chromatography system for analysis. During the extraction procedure, the liposomal bilayer was disrupted so that the total AmB concentration could be measured.
The separation was performed isocratically on a Phenomenex Spherex C18 (150 by 4.6 mm, 5 μm) analytical column, which was coupled by an Alltech Alltima C18 (7.5 by 4.6 mm, 5 μm) guard column. The mobile phase, consisting of 10 mM sodium acetate buffer, including 10 mM EDTA (pH 3.6) and acetonitrile (650:350, vol/vol), was delivered at 1.0 ml/min. Peaks were detected at a wavelength of 405 nm. The assay was linear up to 80 μg/ml, with a limit of quantitation of 0.1 μg/ml. The overall deviation from the true concentration was less than 11.0% for within run and 6.0% for between runs, and the precision was less than 4.5% for within run and 5.0% for between runs. The average recovery was 88.4% at the concentrations of quality control samples with a standard deviation of 6.2%.
Population pharmacokinetic analysis.
A population pharmacokinetic model was developed and fitted to AmB concentration-time data by using NONMEM (version 5.1.1; NONMEM project group, University of California, San Francisco, CA). A Compaq visual Fortran compiler (version 6.6) was used. The first-order conditional estimation method was used throughout the model-building procedure. The modeling approach was implemented in a series of steps which are outlined below.
(i) Development of base population pharmacokinetic model.
The base pharmacokinetic model was defined as the model that best described the L-AmB concentration-time data without consideration of covariate effects. One-compartment and two-compartment open pharmacokinetic models with zero-order input were evaluated using subroutines from the NONMEM library (6). The exponential error model was employed to model the interindividual variability (IIV) for the pharmacokinetic parameters defined as follows:
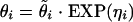 |
where θi represents the pharmacokinetic parameter for the ith individual, θ̃ is the typical value of pharmacokinetic parameter θ in the population (e.g., population mean), ηi quantifies the deviation of θi from θ̃ with a distribution of (0, ω2). The difference between the jth observed concentration (Y) in the ith individual and its respective prediction (Ŷ) was modeled with a combined additive and proportional error model:
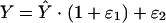 |
where ɛ1 (proportional component) and ɛ2 (additive component) are random effects quantifying the errors between Y and Ŷ with a distribution of (0, σ2). Between-occasion variability (BOV) for the parameter θ was evaluated as an additional level of random effect and expressed as follows:
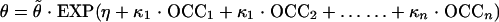 |
where OCCn has the value of 1 for the nth occasion and 0 otherwise; κ1…n are random variables assumed to be normally distributed with a mean of 0, and variance is denoted by π2 (15). In the present modeling analysis, each “occasion” was defined as a period of time in which patients were administered L-AmB on a daily basis and was therefore also referred to as a “course” of therapy. The time interval between two occasions was at least two weeks. BOV was tested on clearance (CL) and volume of distribution in the central compartment (V1) and was estimated using NONMEM's BLOCK (2) SAME option, as it was assumed that BOV was the same for all occasions.
(ii) Development of full population pharmacokinetic model.
Factors, including body weight (WT) and age, were investigated by using a covariate model. Taking the covariate WT as an example, the typical model would be
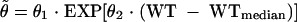 |
where θ̃ is the model predicted value for θ given the covariate value for WT; θ1 represents the population central tendency for the pharmacokinetic parameter θ and corresponds to θ̃ evaluated at the value of WTmedian for WT; θ2 represents the effect of WT on θ; WTmedian is the median weight of the study population. The possible influence of various covariates on the pharmacokinetic parameter of L-AmB was modeled in a multiplicative fashion (14). Parameters were centered on the typical value (median) of each covariate in the study population. The relationships between pharmacokinetic parameters and covariates were evaluated using univariate analysis in NONMEM. Discrimination between hierarchical models was based on the objective function value (OFV) by using the log-likelihood ratio test (22). A P value of 0.001, corresponding to the decrease in OFV by more than 10.83 (df = 1), was chosen to decide the full model.
(iii) Full model evaluation.
The accuracy and robustness of the full model were evaluated by using a bootstrap procedure, which was performed in an automated fashion by using the bootstrap option in the software package Wings for NONMEM (WFN, version 408; N. Holford, University of Auckland) (20). The results from 1,000 successful runs were obtained. The mean and 95% predictive interval (PI) were reported, and the difference between the bootstrap mean and the estimate obtained from original data set for all the population parameters was calculated.
Pharmacodynamic analysis of L-AmB in antifungal therapy.
Two PK-pharmacodynamic (PD) parameters, peak concentration at steady state in relation to the MIC (Cmax,ss/MIC) and area under the concentration-time curve at steady state in relation to the MIC (AUCss/MIC), were employed to evaluate their relationship with clinical antifungal efficacy of L-AmB (1). Cmax,ss was predicted using the final population pharmacokinetic model performed in NONMEM by assuming that the steady state of multiple daily dosing of L-AmB is achieved by day 7 (24). AUCss was estimated as dose/CL, where CL is the individual posterior Bayesian estimate of L-AmB clearance and dose is the dose of L-AmB on the 7th day of the course. The determination of MICs of fungal pathogen was performed in the Pacific Laboratory Medicine Services (Royal North Shore Hospital, NSW, Australia) by testing fungal species, which were isolated from individual patients who had proven invasive fungal infections, against D-AmB. The assessment of proven invasive fungal infection and antifungal efficacy were based upon the criteria proposed by the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group (EORTC/IFICG) (4). SPSS (version 11.5; SPSS, Inc., Illinois) was used to carry out the statistical analyses. A P value of less than 0.05 was considered to be statistically significant.
RESULTS
Population characteristics.
A total of 39 patients treated with L-AmB for a total of 48 courses were included in the population pharmacokinetic analysis. The patient characteristics on entry into the study are summarized in Table 1. The patient population consisted of 26 males and 13 females. Patient ages ranged from 2 months to 17 years, and the median body weight was 21.1 kg, ranging from 6.1 to 84.1 kg. Sixteen patients had received a bone marrow transplant.
TABLE 1.
Patient characteristics in population pharmacokinetic study of L-AmBa
| Variable | Mean (SD) | Median | Range |
|---|---|---|---|
| No. of patients | 39 | ||
| No. of courses | 48 | ||
| No. of observations | 637 | ||
| No. of observations/course | 13.3 (7.0) | 12 | 3-30 |
| Sex (male/female) | 26/13 | ||
| Age, yr | 7.1 (5.1) | 6.5 | 0.17-17 |
| Weight, kg | 28.8 (19.8) | 21.1 | 6.1-84.1 |
| Height, cm | 119.2 (33.7) | 118.5 | 61.5-190.0 |
| BSA, m2b | 0.96 (0.46) | 0.82 | 0.32-2.11 |
| Creatinine, μmol · l−1 | 74 (108) | 47 | 19-659 |
| GGT, U · l−1c | 105 (211) | 39 | 11-1398 |
In the cohort, 15 patients had been diagnosed with acute lymphoblastic leukemia, 4 had been diagnosed with acute myloid leukemia, and 20 had received other diagnoses. Sixteen patients had received bone marrow transplants (12 allogeneic and 4 autologous).
BSA, body surface area (18); BSA(m2) = 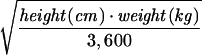 .
.
GGT, gamma-glutamyl transferase.
In total, 637 plasma concentration-time data were available for the pharmacokinetic analyses, representing an average of 13 samples per course. The doses of L-AmB that were given to patients ranged from 0.8 to 5.9 mg/kg of body weight/day. While the typical dosing regimen of L-AmB (1 to 3 mg/kg/day) is given to achieve the prophylactic benefits in persistent febrile neutropenia patients, the L-AmB dose is escalated to 5 to 6 mg/kg/day when signs of invasive fungal infections emerge.
Population pharmacokinetic modeling.
The population pharmacokinetic model-building process is presented in Table 2. A two-compartment pharmacokinetic model with zero-order input and first-order elimination was found to describe L-AmB-observed concentration-time data better than a one-compartment model as it markedly decreased the OFV (ΔOFV, −673.8), leading to its use for the following model-building procedures.
TABLE 2.
Pharmacokinetic model-building summary for L-AmB
| Model no. | Modela | Compared against | OFV | ΔOFV | SIG (P)b |
|---|---|---|---|---|---|
| One compartment | |||||
| 1 | CL = θ1 · EXP(η) | 1,158.2 | |||
| V = θ2 · EXP(η) | |||||
| 2 | CL = θ1 · EXP(η + κ) | 1 | 1,102.6 | −55.6 | <0.001 |
| V = θ2 · EXP(η + κ) | |||||
| Two compartment | |||||
| 3 | CL = θ1 · EXP(η) | 484.40 | |||
| V1 = θ2 · EXP(η) | |||||
| 4 | CL = θ1 · EXP(η + κ) | 3 | 398.94 | −85.5 | <0.001 |
| V1 = θ2 · EXP(η + κ) | |||||
| 5 | CL = θ1 · EXP(θ5 · (WT − 21)) · EXP(η + κ) | 4 | 394.63 | −4.31 | <0.05 |
| V1 = θ2 · EXP(η + κ) | |||||
| 6 | CL = θ1 · EXP(θ5 · (age − 6.5)) · EXP(η + κ) | 4 | 396.20 | −2.74 | |
| V1 = θ2 · EXP(η + κ) | |||||
| 7 | CL = θ1 · EXP(η + κ) | 4 | 394.74 | −4.20 | <0.05 |
| V1 = θ2 · EXP(θ5 · (WT − 21)) · EXP(η + κ) | |||||
| 8 | CL = θ1 · EXP(η + κ) | 4 | 395.04 | −3.90 | <0.05 |
| V1 = θ2 · EXP(θ5 · (age − 6.5)) · EXP(η + κ) | |||||
| 9 | CL = θ1 · EXP(θ5 · (WT − 21)) · EXP(η + κ) | 5 | 380.93 | −13.7 | <0.001 |
| V1 = θ2 · EXP(θ6 · (WT − 21)) · EXP(η + κ) | |||||
| 10 | CL = θ1 · EXP(θ5 · (WT − 21)) · EXP(η + κ) | 9 | 382.10 | 1.16 | |
| V1 = θ2 · EXP(θ6 · (WT − 21)) · EXP(θ7 · (age − 6.5)) · EXP(η + κ) |
η, random variable of interindividual variability; κ, random variable of between occasion variability. WT is centered to the cohort median value of 21 kg; age is centered to the cohort median value of 6.5 years.
SIG, Significance of adding covariates into a model, assessed by P value.
The development of the base model by inclusion of BOV into two fixed-effect parameters, CL and V1, provided a significant decrease in the OFV of 85.5 points. Furthermore, the IIV of CL and V1 decreased from 53 to 24% and 56 to 28%, respectively, after the incorporation of BOV, indicating the importance of BOV in the pharmacokinetic variability of L-AmB. With respect to the residual variability, a combined additive and proportional error model was chosen. The estimate of the additive error component of residual variability (0.025 mg/liter) was smaller relative to the limit of quantitation of the assay (0.1 mg/liter) but added stability to the model. This was evidenced by the minimization problems caused when other error models were explored.
The individual posterior Bayesian estimates of pharmacokinetic parameters that were generated from the base model were plotted against weight, height, age, and sex. The Mann-Whitney U test indicated that sex did not significantly affect the pharmacokinetics of L-AmB. Since weight and height were highly correlated (r2 = 0.93) and weight alone yielded a better correlation with CL and V1 than did height, weight and age were evaluated as potential covariates.
In the first step of the model-building procedure, each single combination of a covariate with one of the primary pharmacokinetic parameters (CL and V1) was included sequentially into the base model. The covariate effects on Q (intercompartmental clearance) and V2 were not evaluated because of a lack of a physiological rationale and clinical meaning. At the significance level of P < 0.05, weight had significant effect on both CL and V1 and age was found to significantly influence V1. The base model with the addition of weight as a covariate for CL was used as the reference model for the next step of the forward addition procedure.
In the second step of the model selection procedure, further inclusion of weight as a covariate for V1 produced a significant decrease in the OFV by 13.7 points compared to that produced by the reference model (P < 0.001). When this model was used as the reference model, the inclusion of the covariate age on V1 increased the OFV. Therefore, the inclusion of patient weight rather than age in the analysis provided the largest effect on the goodness-of-fit of the model. The structure of the full model is presented in Table 2 as model 9.
Table 3 summarizes the population parameter estimates obtained from the full population pharmacokinetic model. Figure 1 displays the diagnostic plots for the full model. All of the pharmacokinetic parameters were reliably estimated as the percentage of relative standard errors (RSE) was less than 50%. No trend was observed in the weighted residual plots, and the population-predicted concentrations were symmetrically distributed around the line of identity, indicating that the model adequately describes the pharmacokinetic profile of L-AmB in all the doses studied. The representative concentration-time profile in one patient treated with L-AmB at a dose of 1.8 is shown in Fig. 2, in which the model fit appears to be adequate.
TABLE 3.
L-AmB population parameter estimates from full model
| Parameter | Estimate (RSEa) | IIVb (RSE) | BOVc (RSE) |
|---|---|---|---|
| Structural | |||
| CL (l · h−1) | 0.44 (27) | 10 (40) | 46 (54) |
| V1 (l) | 3.12 (40) | 56 (64) | |
| Q (l · h−1) | 0.73 (18) | 77 (27) | |
| V2 (l) | 18.0 (40) | 74 (37) | |
| θ5d | 0.0152 (12) | ||
| θ6e | 0.0241 (29) | ||
| Residual variabilityf | |||
| Proportional (%) | 27 (7) | ||
| Additive (mg · l−1) | 0.034 (24) |
Relative standard error was calculated as a standard error/population estimate and is expressed as a percentage.
Interindividual variability is expressed as percent coefficient of variation (CV).
Between-occasion variability is expressed as percent CV.
θ5, effect of weight on CL.
θ6, effect of weight on V1.
The proportional error component of residual variability is expressed as percent CV; the additive error component of residual variability is expressed as standard deviation.
FIG. 1.

Diagnostic plots of full population pharmacokinetic model. Upper panel, observed AmB concentration (conc) versus population-predicted concentration (mg/liter) (r2 = 0.7159; the dash lined is the line of identity). Lower panel, weight residuals (WRES) versus time after first dose (h). Time is presented with reference to the first L-AmB dose in each study course.
FIG. 2.

Representative concentration (conc)-time profile of a patient treated with a 1-h intravenous infusion of L-AmB at a dose of 1.8 mg/kg over a dosing interval. Closed circles represent the individual observed concentrations, and the solid line represents the population-predicted concentrations obtained from the full pharmacokinetic model.
The final covariate models for CL and V1 of L-AmB were presented as follows:
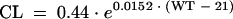 |
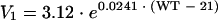 |
For the population investigated, the typical value of CL and V1 was 0.44 liters/h and 3.12 liters for a patient weighing 21 kg. CL and V1 increased with every 10 kg of weight by 16 and 27%, respectively. Compared with the base model, the IIV of CL was decreased from 24 to 10% but the IIV for V1 failed to be well estimated, as its ω2 output was very low. This should not be interpreted as an absence of variability in V1 but instead was a consequence of the fact that NONMEM had difficulty in characterizing the differences in mean parameter values for individuals when BOV was much higher than IIV (15). The estimate of the proportional term for the residual variability, expressed as the percentage coefficient of variation, was 27% and the estimate of the additive component of residual variability was 0.034 mg/liter.
Full model evaluation.
From the original data set, 1,000 replicate data sets were generated and used for the evaluation of the stability of the full model and accuracy of parameter estimates. Minimization successful and minimization terminated due to rounding errors with numbers of significance above two were included in the bootstrap calculations. Table 4 lists the results of the bootstrap procedure, presented as mean and 95% PI. The mean values of the bootstrap procedure were comparable to the parameter estimates from the original data set, indicating that the accuracy and robustness of the full model was acceptable. The secondary pharmacokinetic parameters of L-AmB, including t1/2β (59.4 ± 36.5 h) and Vss (1.14 ± 0.68 liters/kg), were generated using the individual posterior Bayesian estimates of parameters obtained from the full model.
TABLE 4.
Evaluation of the full model using the bootstrap procedure
| Parameter | Full modela | Bootstrapb | 95% PI | Difference (%)c |
|---|---|---|---|---|
| Structural model | ||||
| θCL (l · h−1) | 0.44 | 0.44 | 0.36-0.51 | −0.9 |
| θV1 (l) | 3.12 | 3.34 | 2.29-4.39 | 7.1 |
| θQ (l · h−1) | 0.73 | 0.73 | 0.53-0.93 | 0.1 |
| θV2 (l) | 18.0 | 18.7 | 13.4-23.9 | 3.6 |
| θCL ≈ WT (kg−1)d | 0.0152 | 0.0151 | 0.0067-0.0236 | −0.7 |
| θV1 ≈ WT (kg−1)e | 0.0241 | 0.0223 | 0.0118-0.0329 | −7.5 |
| Statistical modelf | ||||
| ω2CL | 0.0108 | 0.0103 | 0.0005-0.0201 | −4.6 |
| ω2V1 | 0.0450 | 0.0001-0.0900 | ||
| ω2Q | 0.593 | 0.591 | 0.320-0.861 | −0.4 |
| ω2V2 | 0.546 | 0.537 | 0.184-0.889 | −1.7 |
| π2CL | 0.209 | 0.189 | 0.067-0.312 | −9.5 |
| π2V1 | 0.319 | 0.312 | 0.062-0.561 | −2.3 |
| σ2add | 0.0011 | 0.0014 | 0.0002-0.0025 | 22.1 |
| σ2prop | 0.0709 | 0.0707 | 0.0556-0.0857 | −0.3 |
Mean estimates from the original data set.
Mean estimates from 1,000 bootstrap analyses.
Results are shown as (bootstrap value − full model value)/full model value × 100.
Effect of WT on CL.
Effect of WT on V1.
ω2, random effect parameter that represents interindividual variance; π2, random effect parameter that represents between occasion variance; σ2, random effect parameter that represents residual variance.
Clinical efficacy of L-AmB in patients with proven fungal infection.
A total of nine patients had proven fungal infection. The mycological, pharmacokinetic, and pharmacodynamic information are displayed in Table 5. For patients with possible fungal infection or for those undergoing empirical antifungal therapy, the clinical outcomes of L-AmB were not incorporated due to the lack of MICs. All of the nine patients achieved a clinical response (either complete or partial), with the exception of one patient having Scedosporium prolificans isolated from cerebrospinal fluid, which is a filamentous fungus intrinsically resistant to AmB with the MIC as high as 4 μg/ml. The Cmax,ss/MIC for patients with complete clinical response was statistically higher than that of the patients with partial response (P = 0.021) (Fig. 3). However, in these patients, there was no significant correlation between AUCss/MIC and response (P = 0.285).
TABLE 5.
Microbiological, pharmacokinetic, and pharmacodynamic information for patients with proven fungal infection
| IDa | Dose (mg/kg/day) | Pathogen (n = 11 cases)
|
Cmax,ss/ MIC | Efficacyc | |
|---|---|---|---|---|---|
| Fungi | MICb | ||||
| 3 | 5.4 | C. albicans | 0.50 | 88.4 | C |
| A. fumigatus | 1.00 | 44.2 | P | ||
| A. niger | 1.00 | 44.2 | P | ||
| 8 | 2.9 | A. fumigatus | 1.00 | 22.5 | P |
| 10b | 3.1 | A. fumigatus | 1.00 | 36.5 | P |
| 20b | 3.5 | C. krusei | 1.00 | 32.1 | P |
| 22 | 3.0 | Rhodotorula rubra | 0.25 | 68.8 | C |
| 32 | 4.8 | Scedosporium prolificans | 4.00 | 4.3 | F |
| 36a | 4.9 | C. albicans | 0.25 | 45.6 | C |
| 38 | 5.3 | C. albicans | 0.50 | 61.6 | P |
| 40 | 4.1 | C. albicans | 0.25 | 68.8 | C |
ID, patient identification number; a, the first course of L-AmB therapy; b, the second course of L-AmB therapy.
MIC of D-AmB (μg·ml−1).
C, complete response; P, partial response; F, failure to respond.
FIG. 3.

The ratio of peak concentration at steady state to the MIC of the patients with partial response (left box, 40.2 ± 13.3, n = 6 cases) versus that of the patients with complete clinical response (right box, 67.9 ± 17.5, n = 4 cases).
DISCUSSION
This study is the first investigation in which a population pharmacokinetic modeling approach was applied to assess the pharmacokinetics of L-AmB in pediatric patients. A two-compartment pharmacokinetic model with zero-order input and first-order elimination was appropriate to describe the concentration-time data of L-AmB. Weight was found to be a covariate that significantly influenced the clearance and volume of distribution of L-AmB. The incorporation of a term to describe BOV into the pharmacokinetic model reflected the clinical situation of L-AmB therapy and favorably influenced the model-building process. The full model predicted the population mean estimates of L-AmB clearance and volume of distribution in the central compartment to be 0.44 liters/h and 3.12 liters, respectively. The mean pharmacokinetic parameter estimates that were obtained from the 1,000 bootstrap replicates were within 15% of those from the full model of the original data set, indicating the validity of the developed full pharmacokinetic model.
The inclusion of the potential covariates in the model (e.g., patient demographic characteristics) was performed according to a stepwise forward addition procedure. Patient clinical characteristics, including the status of fungal infections and the clinical responses to L-AmB treatment, were not included into the covariate model-building process as these characteristics putatively have no relevance to the pharmacokinetics of L-AmB. Since renal elimination plays a minor role in the excretion of L-AmB (5), markers of renal function were not considered as potential covariates. It was found that body weight was an influential covariate, as it explained part of the variability in L-AmB pharmacokinetics with statistical significance. The incorporation of weight into the pharmacokinetic model indicated that weight-related changes in pharmacokinetic parameters would result in the alteration of drug exposure in patients receiving a given dose. A series of simulations were performed using the population pharmacokinetic parameters and variability to explore the Cmax,ss of L-AmB in a range of doses from 1 to 12.5 mg/kg for patients with weight ranges from 10 to 70 kg. It was found that achieving a comparable Cmax,ss in younger patients whose weight was less than 20 kg would necessitate the administration of a higher dose, suggesting that the current dosing regimens of L-AmB are likely to be suboptimal in this patient cohort.
The clearance and volume of distribution of L-AmB in the central compartment exhibited significant BOV with estimated values of 46 and 56%, respectively. The reason for this observation is unclear but is likely to reflect physiological changes that are occurring in this population over the period of the study. Two mechanisms may contribute to the observed BOV. Firstly, lipid-based formulations influence the association of drugs with lipoproteins in blood, which in turn may be significantly influenced by disease (27). Therefore, it is possible that changes in the lipoprotein profile of patients between occasions could lead to alterations in the pharmacokinetics of AmB. Secondly, dose-dependent hepatic uptake of L-AmB, which we have observed in the isolated perfused rat liver (13), suggests the total amount of drug a patient receives on occasion may impact on the pharmacokinetics of L-AmB. Karlsson and Sheiner (15) indicated that the relative sizes of IIV and BOV have implications for therapeutic drug monitoring (TDM) and concluded that the BOV of pharmacokinetic parameters needed to be lower than those of IIV for carrying out meaningful TDM. The observation that a drug in a particular patient cohort has a larger BOV than IIV suggests that the concentrations measured in the first occasion are not likely to inform the dose individualization in the subsequent occasions. A study which (i) has sufficient patients contributing data for more than one occasion and (ii) has the data on each occasion being informative about the parameter of interest would improve the estimate of IIV when BOV is higher than IIV. Further work is needed to clarify the possible impact of this observation on the TDM of L-AmB in children.
The encapsulation of AmB into liposomes significantly alters the pharmacokinetics of AmB. The clearance and volume of distribution in the central compartment decreased compared with those reported by our research group in pediatric patients who were treated with D-AmB (CL, 0.44 versus 0.88 liters/h and V1, 3.12 versus 9.97 liters) (19), suggesting a more rapid distribution of D-AmB into the tissues than L-AmB. Consistent with these observations, L-AmB exhibits higher plasma peak concentrations and prolonged retention in plasma compared with those that were attained when clinically relevant doses of D-AmB are administered. Taken together, the unique pharmacokinetics of L-AmB represent a potential clinical advantage for antifungal therapy with L-AmB as these pharmacokinetic properties can facilitate the localization of the drug to the sites of infection (9).
Three pharmacokinetic-pharmacodynamic parameters, including the percentage of time that drug concentration exceeds the MIC (%T > MIC), the Cmax/MIC, and the AUC/MIC, have been used to describe the association between drug exposure and antifungal effect (1, 3). For antifungal drugs exhibiting concentration-dependent killing and long postantifungal effects, such as polyenes, the PK-PD parameters that characterize this type of dosing strategy include Cmax/MIC and AUC/MIC. However, in this study, because the patients with proven fungal infection received a range of dosing regimens on each occasion, it was not possible to provide one meaningful estimate of AUC and AUC/MIC. In this relatively small patient cohort, Cmax/MIC, rather than AUC/MIC, was chosen as a potential PK-PD parameter which is predictive of clinical outcomes. A previous PK-PD study in neutropenic disseminated-candidiasis mice treated with D-AmB has demonstrated that Cmax/MIC was the PK-PD parameter best predictive of antifungal efficacy (as measured by a log reduction in CFU) relative to AUC/MIC and %T > MIC, and the breakpoint of Cmax/MIC equal to 10 was associated with the achievement of maximum antifungal efficacy (2). Furthermore, Groll et al. investigated the relationship between drug concentrations and the antifungal efficacy of each of four formulations of AmB, including D-AmB, AMB colloidal dispersion, AMB lipid complex, and L-AmB in a rabbit model of central nervous system candidiasis. A strong inverse correlation between Cmax/MIC (62.0 ± 1.3) and residual fungus burden in brain tissue was observed in L-AmB-treated group (r = −0.792; P < 0.0001) (12). Consistent with the animal studies, the present study demonstrated an impact of Cmax,ss/MIC on the clinical antifungal efficacy of L-AmB in patients with proven fungal infections with a higher Cmax,ss/MIC associated with the desired clinical response.
Using the population pharmacokinetic data and the preliminary pharmacodynamic data that were derived from this study, it is possible to evaluate the suitability of selected L-AmB dose regimens in typical patients by using a simulation approach. Based on the predicted Cmax,ss, the PK-PD parameter (Cmax,ss/MIC) was generated for typical patients over the weight range of 10 to 70 kg who were treated with L-AmB at doses ranging from 1 mg/kg to 12.5 mg/kg by assuming that MIC for Candida spp. and Aspergillus spp. was 0.5 and 1.0 mg/liter, respectively. Using the a priori criterion of achieving no less than 85% of patients attaining the proposed pharmacodynamic target (Cmax,ss/MIC > 50) following L-AmB antifungal therapy, it was found that patients weighing greater than 30 kg attained the target Cmax,ss/MIC against Candida and Aspergillus when receiving an L-AmB daily dose of 4 mg/kg and 7.5 mg/kg, respectively, whereas lighter patients should be given even higher doses. However, further clinical studies that are sufficiently powered and appropriately designed are warranted to define the PD target of L-AmB antifungal efficacy, which can be used to inform the dosing strategy optimization in conjunction with the validated population pharmacokinetic model. It should be noted also that these metrics have utilized plasma concentrations of L-AmB which combine unbound AmB, plasma protein-bound AmB, and liposomal encapsulated AmB. The implications of using the unbound concentration of AmB in these calculations have yet to be elucidated.
In conclusion, a population pharmacokinetic model was developed and evaluated for L-AmB in pediatric oncology patients. The clearance and volume of distribution were influenced by body weight and showed significant between-occasion variability. The application of the population pharmacokinetic model in designing the optimal dosing strategy is worthy of further investigation in an attempt to improve the antifungal management in pediatric patients with malignant diseases.
Acknowledgments
Y.H. is supported by a University of Sydney International Postgraduate Research Scholarship. C.E.N. is supported by the Leukemia Research & Support Fund at the Children’s Hospital at Westmead.
REFERENCES
- 1.Andes, D. 2004. Antifungal pharmacokinetics and pharmacodynamics: understanding the implications for antifungal drug resistance. Drug Resist. Updat. 7:185-194. [DOI] [PubMed] [Google Scholar]
- 2.Andes, D., T. Stamsted, and R. Conklin. 2001. Pharmacodynamics of amphotericin B in a neutropenic-mouse disseminated-candidiasis model. Antimicrob. Agents Chemother. 45:922-926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andes, D., and W. A. Craig. 2002. Animal model pharmacokinetics and pharmacodynamics: a critical review. Int. J. Antimicrob. Agents 19:261-268. [DOI] [PubMed] [Google Scholar]
- 4.Ascioglu, S., J. H. Rex, B. de Pauw, J. E. Bennett, J. Bille, F. Crokaert, D. W. Denning, J. P. Donnelly, J. E. Edwards, Z. Erjavec, D. Fiere, O. Lortholary, J. Maertens, J. F. Meis, T. F. Patterson, J. Ritter, D. Selleslag, P. M. Shah, D. A. Stevens, and T. J. Walsh. 2002. Defining opportunistic invasive fungal infections in immunocompromised patients with cancer and hematopoietic stem cell transplants: an international consensus. Clin. Infect. Dis. 34:7-14. [DOI] [PubMed] [Google Scholar]
- 5.Bekersky, I., R. M. Fielding, D. E. Dressler, J. W. Lee, D. N. Buell, and T. J. Walsh. 2002. Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob. Agents Chemother. 46:828-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boeckmann, A. J., L. B. Sheiner, and S. L. Beal. 1992. NONMEM users guide, part V. Introductory guide. NONMEM Project Group, University of California, San Francisco, Calif.
- 7.Boswell, G. W., D. Buell, and I. Bekersky. 1998. AmBisome (liposomal amphotericin B): a comparative review. J. Clin. Pharmacol. 38:583-592. [DOI] [PubMed] [Google Scholar]
- 8.Clemons, K. V., and D. A. Stevens. 1998. Comparison of fungizone, Amphotec, AmBisome, and Abelcet for treatment of systemic murine cryptococcosis. Antimicrob. Agents Chemother. 42:899-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fielding, R. M., R. O. Lewis, and L. Moon-McDermott. 1998. Altered tissue distribution and elimination of amikacin encapsulated in unilamellar, low-clearance liposomes (MiKasome). Pharm. Res. 15:1775-1781. [DOI] [PubMed] [Google Scholar]
- 10.Gregoriadis, G. 1981. Targeting of drugs: implications in medicine. Lancet ii:241-246. [DOI] [PubMed] [Google Scholar]
- 11.Groll, A. H., M. Kurz, W. Schneider, V. Witt, H. Schmidt, M. Schneider, and D. Schwabe. 1999. Five-year-survey of invasive aspergillosis in a paediatric cancer centre. Epidemiology, management and long-term survival. Mycoses 42:431-442. [DOI] [PubMed] [Google Scholar]
- 12.Groll, A. H., N. Giri, V. Petraitis, R. Petraitiene, M. Candelario, J. S. Bacher, S. C. Piscitelli, and T. J. Walsh. 2000. Comparative efficacy and distribution of lipid formulations of amphotericin B in experimental Candida albicans infection of the central nervous system. J. Infect. Dis. 182:274-282. [DOI] [PubMed] [Google Scholar]
- 13.Hong, Y., I. Ramzan, and A. J. McLachlan. 2005. Hepatobiliary disposition of liposomal amphotericin B in the isolated perfused rat liver. J. Pharm. Sci. 94:169-176. [DOI] [PubMed] [Google Scholar]
- 14.Jonsson, E. N., and M. O. Karlsson. 1998. Automated covariate model building within NONMEM. Pharm. Res. 15:1463-1468. [DOI] [PubMed] [Google Scholar]
- 15.Karlsson, M. O., and L. B. Sheiner. 1993. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J. Pharmacokinet. Biopharm. 21:735-750. [DOI] [PubMed] [Google Scholar]
- 16.Leenders, A. C., S. Daenen, R. L. Jansen, W. C. Hop, B. Lowenberg, P. W. Wijermans, J. Cornelissen, R. Herbrecht, H. van der Lelie, H. C. Hoogsteden, H. A. Verbrugh, and S. de Marie. 1998. Liposomal amphotericin B compared with amphotericin B deoxycholate in the treatment of documented and suspected neutropenia-associated invasive fungal infections. Br. J. Haematol. 103:205-212. [DOI] [PubMed] [Google Scholar]
- 17.Leenders, A. C., S. de Marie, M. T. ten Kate, I. A. Bakker-Woudenberg, and H. A. Verbrugh. 1996. Liposomal amphotericin B (AmBisome) reduces dissemination of infection as compared with amphotericin B deoxycholate (Fungizone) in a rate model of pulmonary aspergillosis. J. Antimicrob. Chemother. 38:215-225. [DOI] [PubMed] [Google Scholar]
- 18.Mosteller, R. D. 1987. Simplified calculation of body-surface area. N. Engl. J. Med. 317:1098. [DOI] [PubMed] [Google Scholar]
- 19.Nath, C. E., A. J. McLachlan, P. J. Shaw, R. Gunning, and J. W. Earl. 2001. Population pharmacokinetics of amphotericin B in children with malignant diseases. Br. J. Clin. Pharmacol. 52:671-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parke, J., N. H. Holford, and B. G. Charles. 1999. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput. Methods Programs Biomed. 59:19-29. [DOI] [PubMed] [Google Scholar]
- 21.Prentice, H. G., I. M. Hann, R. Herbrecht, M. Aoun, S. Kvaloy, D. Catovsky, C. R. Pinkerton, S. A. Schey, F. Jacobs, A. Oakhill, R. F. Stevens, P. J. Darbyshire, and B. E. Gibson. 1997. A randomized comparison of liposomal versus conventional amphotericin B for the treatment of pyrexia of unknown origin in neutropenic patients. Br. J. Haematol. 98:711-718. [DOI] [PubMed] [Google Scholar]
- 22.Sheiner, L. B., and T. M. Ludden. 1992. Population pharmacokinetics/dynamics. Annu. Rev. Pharmacol. Toxicol. 32:185-209. [DOI] [PubMed] [Google Scholar]
- 23.van Etten, E. W., C. van den Heuvel-de Groot, and I. A. Bakker-Woudenberg. 1993. Efficacies of amphotericin B-desoxycholate (Fungizone), liposomal amphotericin B (AmBisome) and fluconazole in the treatment of systemic candidosis in immunocompetent and leucopenic mice. J. Antimicrob. Chemother. 32:723-739. [DOI] [PubMed] [Google Scholar]
- 24.Walsh, T. J., J. L. Goodman, P. Pappas, I. Bekersky, D. N. Buell, M. Roden, J. Barrett, and E. J. Anaissie. 2001. Safety, tolerance, and pharmacokinetics of high-dose liposomal amphotericin B (AmBisome) in patients infected with Aspergillus species and other filamentous fungi: maximum tolerated dose study. Antimicrob. Agents Chemother. 45:3487-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh, T. J., R. W. Finberg, C. Arndt, J. Hiemenz, C. Schwartz, D. Bodensteiner, P. Pappas, N. Seibel, R. N. Greenberg, S. Dummer, M. Schuster, and J. S. Holcenberg. 1999. Liposomal amphotericin B for empirical therapy in patients with persistent fever and neutropenia. National Institute of Allergy and Infectious Diseases Mycoses Study Group. N. Engl. J. Med. 340:764-771. [DOI] [PubMed] [Google Scholar]
- 26.Walsh, T. J., V. Yeldandi, M. McEvoy, C. Gonzalez, S. Chanock, A. Freifeld, N. I. Seibel, P. O. Whitcomb, P. Jarosinski, G. Boswell, I. Bekersky, A. Alak, D. Buell, J. Barret, and W. Wilson. 1998. Safety, tolerance, and pharmacokinetics of a small unilamellar liposomal formulation of amphotericin B (AmBisome) in neutropenic patients. Antimicrob. Agents Chemother. 42:2391-2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wasan, K. M., A. L. Kennedy, S. M. Cassidy, M. Ramaswamy, L. Holtorf, J. W. Chou, and P. H. Pritchard. 1998. Pharmacokinetics, distribution in serum lipoproteins and tissues, and renal toxicities of amphotericin B and amphotericin B lipid complex in a hypercholesterolemic rabbit model: single-dose studies. Antimicrob. Agents Chemother. 42:3146-3152. [DOI] [PMC free article] [PubMed] [Google Scholar]