

Abstract
Orf20 of the conjugative transposon Tn916 was purified as a chimeric protein fused to maltose binding protein (MBP-Orf20). The chimeric protein possessed endonucleolytic activity, cleaving both strands of the Tn916 origin of conjugal transfer (oriT) at several distinct sites and favoring GT dinucleotides. Incubation of the oriT DNA with purified Tn916 integrase (Int) and MBP-Orf20 resulted in strand- and sequence-specific cleavage of oriT at a TGGT motif in the transferred strand. This motif lies immediately adjacent to a sequence in oriT previously shown to be protected from DNase I cleavage by Int. The endonucleolytic cleavages produced by Orf20 generated a 3′ OH group that could be radiolabeled by dideoxy ATP and terminal transferase. The production of a 3′ OH group distinguished these Orf20-dependent cleavage events from those catalyzed by Int at the ends of Tn916. Thus, Orf20 functions as the relaxase of Tn916, nicking oriT as the first step in conjugal DNA transfer. Remarkably for a tyrosine recombinase, Tn916 Int acts as a specificity factor in the reaction, conferring both strand and sequence specificities on the endonucleolytic cleavage activity of Orf20.
Tn916 is an 18-kb conjugative transposon originally discovered in Enterococcus faecalis DS16 (9). This element confers tetM tetracycline resistance on its host (8), catalyzes its own excision and integration (17, 25), and carries conjugal transfer functions, orf13-orf24, in its tra region (30). It is a highly promiscuous element able to transfer between different species and genera of gram-positive organisms as well as gram-negative bacteria, such as Escherichia coli and Pseudomonas fluorescens (6, 24). Conjugative transposons have been shown to mediate transfer of antibiotic resistance in complex microbial communities, such as the intestinal flora (31).
Transposition of Tn916 begins with an excision event catalyzed by the transposon-encoded integrase (Int) and Xis proteins (17, 25), resulting in the formation of a circular, nonreplicative intermediate. Genetic evidence suggests that a single strand of the intermediate is then transferred to the recipient (28), where it undergoes replication and subsequent integration into the new host's genome (33). The segment of Tn916 designated the origin of conjugal transfer (oriT) lies in the noncoding region between orf20 and orf21 (14). This 500-bp DNA segment can catalyze in cis the mobilization of a nonconjugal plasmid by Tn916. The sequence of oriT possesses a number of inverted repeats and sequences that are similar to origins of transfer in IncP and F-like plasmids. Certain regions within oriT were postulated to be nic sites, where transfer is initiated by cleavage by a putative endonuclease/relaxase and the formation of a protein-DNA complex that serves as a substrate for DNA transfer (the relaxosome) (14). However, neither the exact location of nic nor the protein responsible for cleavage within oriT has been identified.
The broad-host-range nature of Tn916 and comparisons with other bacterial conjugation systems (40) suggest that the protein(s) responsible for formation of the relaxosome is carried on the element. The only Tn916 protein that has been shown to cleave DNA is the integrase, Int (34), which binds and produces staggered cleavages at the transposon ends (3). Hinerfeld and Churchward (12) showed that Int binds specifically within the minimal required region for oriT; however, no cleavage of oriT by Int was observed. This protein is unlikely to act as the nicking endonuclease that initiates DNA transfer due to its DNA cleavage polarity. At the ends of Tn916, Int leaves a 5′ OH terminus and remains covalently attached to the 3′ phosphate (34). This is in contrast to the 3′ OH left by relaxation enzymes (2), which can serve as a primer for rolling-circle DNA synthesis.
Two gene products of Tn916 could potentially serve as oriT-nicking enzymes. The gene product of orf23 is similar to MbeA, the mobilization protein of the E. coli plasmid ColE1 (6). This 12-kDa protein is considerably smaller than the 57.8-kDa MbeA protein, and its region of similarity to MbeA lies outside the residues of MbeA thought to be involved in relaxation (36). A second possible candidate is Orf20, which is similar to the 41.3-kDa RstA protein of Vibrio cholerae CTXφ. RstA is the only CTXφ protein required for rolling-circle replication of extrachromosomal pCTX (37). Both the size, 40 kDa, and location, immediately downstream of oriT, of Orf20 make it likely to act as the relaxase for Tn916. Here, we describe the purification and characterization of Tn916 Orf20 and show that Int confers endonucleolytic cleavage specificity on Orf20.
MATERIALS AND METHODS
Construction of pMAL-Orf20.
To produce maltose binding protein (MBP)-Orf20, orf20 (991 bp) was amplified from 150 ng Bacillus subtilis CKS102 (29) chromosomal DNA using the primers Orf20-1 (5′-CTGTTTGATTATGTAAGGATTC-3′) and Orf20-2 Eco (5′-GAATTTGAATTCGAGTTATTTTTTTG-3′). The PCR was performed with 2.5 U PfuTurbo (Stratagene) in a 50-μl reaction mixture (95°C for 2 min; 30 cycles of 95°C for 30 seconds, 43°C for 30 seconds, 72°C for 1 min, and 72°C for 10 min). The Orf20-2 Eco primer introduces an EcoRI site (underlined) following the TAA stop codon (in bold) for orf20 and includes 6 bases after the restriction site to aid in cleavage. The PCR was purified with the QIAquick PCR cleanup kit (QIAGEN) and digested with EcoRI in a 20-μl reaction mixture containing 1× MULTI-CORE buffer (Promega) at 37°C for 1.5 h. The pMAL-c2x vector (New England Biolabs) was digested with XmnI in a 20-μl reaction mixture containing 1× MULTI-CORE buffer (Promega) for 30 min at 37°C. EcoRI was added, and incubation was continued for 1.5 h. Both reactions were heat inactivated at 70°C for 13 min and purified as described above. A 10-μl T4 DNA ligase reaction mixture was created with 1× ligase buffer (Promega) with a 3:1 insert-to-vector ratio for 2 h at 25°C and heat inactivated at 70°C for 5 min. The entire ligation reaction was used to transform chemically competent E. coli TOP10 cells (Invitrogen) via heat shock at 42°C for 2 min. Transformants were selected overnight at 37°C on LB plates supplemented with 100 μg ampicillin and then screened by picking onto the LB-ampicillin plates containing 80 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside; Sigma). Positive clones (white colonies) were screened for the presence of orf20 via colony PCR using Taq PCR Master Mix (QIAGEN), the above-described cycling parameters, and primers Orf20-1 and Orf20-2. Plasmid from a positive clone was prepared using the QIAprep plasmid miniprep kit (QIAGEN) and used to transform electrocompetent E. coli SG22094 (clpP lon) (10). The colony PCR screening procedure was repeated. Three clones were tested for production of the 80-kDa MBP-Orf20 fusion protein by inducing 10-ml cultures with 0.3 mM IPTG at 37°C and performing sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis on the whole-cell lysates. Plasmid from these clones was prepared as described above and sequenced. One clone, clone T, was chosen to use for large-scale preparation of MBP-Orf20.
Purification of MBP-Orf20.
Purification was undertaken essentially as suggested by the pMAL vector supplier (New England Biolabs). In short, 1 liter of LB containing 1% d-glucose and 100 μg/ml ampicillin was inoculated with 10 ml of an E. coli SG22094/pMAL-Orf20 culture grown at 37°C. The culture was allowed to shake at 37°C until the A600 reached ∼0.5. Cultures continued to grow for 3 hours after the addition of 0.3 mM IPTG. Cells were harvested by centrifugation in a Sorvall GSA rotor at 4,000 × g and 4°C for 20 min. Cells were resupended in 50 ml column buffer (20 mM Tris-Cl [pH 7.4], 200 mM NaCl, and 1.0 mM EDTA) and stored overnight at −20°C. The cells were thawed in cold water and kept on ice throughout the remainder of the lysis procedure. Cells were lysed via one pass through a French pressure cell (at 10,000 lb/in2) and then centrifuged in a Sorvall SS-34 rotor at 9,000 × g for 30 min at 4°C. The supernatant was diluted 1:5 with cold column buffer. To prepare the column, 15 ml of amylose resin (New England Biolabs) was applied to a 2.5- by 10-cm Kontes FlexColumn and washed with eight column volumes of column buffer at 4°C. The diluted lysate was applied to the column and then washed with 12 column volumes of column buffer and eluted with 30 ml of column buffer plus 10 mM maltose. The presence of MBP-Orf20 was determined via SDS-polyacrylamide gel electrophoresis, where protein of the appropriate size appeared in the eluate. Protein concentration was determined using the Bradford assay (1a).
Preparation of radiolabeled oriT200-2 DNA.
Two primers were employed to differentially label the transferred (T) (200-2 F, 5′-AAGCGGAAGTCGCAGGTGTG-3′) or nontransferred (NT) (200-2 R, 5′-AAATCCCTCCAATCAAAAAGGC-3′) strand. Primer (12 pmol) was labeled with 17 pmol [γ-32P]ATP via T4 polynucleotide kinase (Epicentre) in a 25-μl reaction mixture. Labeled primer (100 nM) was paired with unlabeled primer (800 nM) and used with Taq PCR Master Mix (QIAGEN) in a PCR (94°C for 5 min; 30 cycles of 95°C for 30 seconds, 48°C for 30 seconds, 72°C for 30 seconds, and 72°C for 10 min) with 150 ng (per 50 μl) B. subtilis CKS102 (29) chromosomal DNA as a template. The PCR product was purified on a 5% polyacrylamide Tris-borate-EDTA gel. The fragment was cut out of the gel, crushed via centrifugation, soaked in QIAGEN PB buffer at 37°C overnight, and spun down at 12,000 rpm every 5 min; the supernatant was applied to a QIAGEN miniprep column and eluted as described in the QIAGEN miniprep protocol. DNA was stored at −20°C.
Cleavage assays.
Radiolabeled PCR product (150 pM) was incubated with MBP-Orf20 (37.5 nM, 125 nM, 375 nM, or 1.25 μM) in a 100-μl reaction mixture containing 1× binding buffer (13) at room temperature for 1.5 h. DNA was precipitated by the addition of 400 μl stop solution B (80% ethanol, 570 mM ammonium acetate) and 2 μl Pellet Paint (Novagen). Samples were washed twice with cold 70% ethanol and then briefly dried. Pellets were resuspended in 5 μl formamide dye, boiled 5 minutes, and loaded alongside GATC dideoxy (dd) sequencing reactions (Epicentre kit) on an 8% sequencing gel (26) prerun for 30 min. The same labeled primer used to prepare PCR product was used for the sequencing reactions, which were performed via the isothermic method using 4 μg pAM5160 DNA (14) as a template (1 cycle; 95°C for 5 min, 25°C for 75 min, and 65°C for 75 min). The gel was run approximately 2 hours at 75 W. The gel was fixed for 10 min in 10% acetic acid, vacuum dried for 40 min, and exposed to a PhosphorScreen (Molecular Dynamics) overnight.
To assay the effect of Int on cleavage, reactions were set up and processed as described above with 125 nM MBP-Orf20 and Int at 40, 80, 160, and 320 nM. The Int was prepared as described previously using the “cloning and expression of Int in E. coli” method (34).
Testing for generation of 3′ OH.
The template DNA was prepared via PCR with unlabeled primers (200-2 F and 200-2 R) and then purified via electrophoresis as described above. The binding reaction mixtures were set up as described above (125 nM MBP-Orf20 and 160 nM Int) and incubated at room temperature for 1.5 h. After incubation, 100 μg/ml proteinase K and 0.5% SDS were added to each reaction, and the samples were incubated at 37°C for 2 hours. The reactions were each extracted with 100 μl phenol-chloroform-isoamyl alcohol (25:24:1) (Sigma). The aqueous phase was removed to a fresh tube, precipitated as described above, and washed once with cold 70% ethanol. The pellets were air dried and stored overnight at 4°C. The pellets were resuspended in 50 μl sterile, deionized water and denatured by the addition of 0.2 M sodium hydroxide and incubation at 37°C for 10 min (27). The DNA was precipitated as described above, and the pellets were allowed to air dry. The samples were resuspended in sterile, deionized water and labeled with [α-32P]ddATP via terminal deoxynucleotidyl transferase (Promega) at 37°C for 1.75 h. The reactions were precipitated as described above using 300 μl stop solution B, washed twice with cold 70% ethanol, and allowed to air dry. The pellets were resuspended in 5 μl formamide dye, run out on an 8% sequencing gel, and dried as described above. The gel was exposed to the PhosphorScreen (Molecular Dynamics) for approximately 4 days before development.
Mutation of nic from TGGT to GTTG.
Two primer sets were used to introduce the GTTG mutation into the nic site by preparing fragments GTTG-1 and GTTG-2, which were subsequently used in overlapping PCRs to generate the full-length oriT fragment containing GTTG. GTTG-1 was prepared using primers oriTF2-SalI (5′-GTCGACATGATTTCTTGGAGGAAATTAAAAAG-3′) and oriT-GTTG1R (5′-CCCTTGTACAACAAGGATTTTCTAATGTTTTTTTG-3′). The incorporated SalI site is underlined, and the introduced mutation is inbold.GTTG-2 was prepared with primers oriT-GTTG2F (5′-ATCCTTGTTGTACAAGGGATTTACAAAATTTCA-3′) and oriTR-BamHI (5′-CTTGGATCCAGGGACTGCTGTAAATC-3′). Again, the mutation is in bold, and the BamHI site is underlined. Both PCRs used B. subtilis CKS102 (29) chromosomal DNA as a template in a 50-μl reaction mixture with 2.5 U PfuTurbo (Stratagene) in the provided buffer and 200 μM deoxynucleoside triphosphate mix. The following cycling parameters were used: 95°C for 2 min; 30 cycles of 95°C for 30 seconds, 51.8°C for 30 seconds, 72°C for 1 min, and 72°C for 10 min. The full-length PCR was generated using equimolar amounts of GTTG-1 and GTTG-2 as templates with the above-described reaction conditions and cycling parameters. Primers oriTF2-SalI and oriTR-BamHI were used to amplify the full-length product. The resulting PCR was ligated into pCR-BluntII-TOPO (Invitrogen) following the manufacturer's instructions and sequenced to verify the change from TGGT to GTTG.
To prepare the template for the cleavage reaction, primer 200-2 F was labeled with [γ-32P]ATP as described above and used in conjunction with cold 200-2 R to amplify a GTTG-containing oriT200-2 fragment from the GTTG template using the same cycling parameters outlined above for preparation of oriT200-2. This fragment was purified and stored as described above. Cleavage reactions with the GTTG-containing fragment were carried out as for oriT200-2.
RESULTS
Cleavage of oriT by Orf20.
Relaxase proteins interact with their target DNAs and cleave one of the two strands, leaving a 3′ OH and remaining covalently attached to the 5′ phosphate (2, 12). In order to observe the endonuclease activity of MBP-Orf20, 5′-end-labeled oriT200-2 DNA was generated by PCR employing one labeled primer and one unlabeled primer. This differential labeling allowed us to assay the protein's action separately on the transferred (T) and nontransferred (NT) strands. Cleavage assays were expected to reveal a single cleavage product when MBP-Orf20 was allowed to interact with the labeled T strand, an indication of the single-strand nick. No cleavage products were expected when the NT strand was labeled. After incubation for 1.5 h at room temperature, a reproducible pattern of multiple endonucleolytic cleavages by MBP-Orf20 was observed with both DNA templates (Fig. 1). The banding pattern did not change with increasing amounts of protein. Comparison of the cleavage sites on the T strand and NT strand showed that the cleavages did not align in a manner that would suggest that MBP-Orf20 causes double-strand breaks. Thus, it appears that MBP-Orf20 possesses endonucleolytic activity but that cleavage of Tn916 oriT does not occur in the expected strand- and sequence-specific manner. As shown in Fig. 2, of 19 nonspecific cleavages by Orf20 on oriT200-2, 9 come before a T (47%), 8 come before an A (42%), and 1 each before a G or C (5%). Ten of the 19 cleavages occur predominantly after G (53%), 4 each after A or C (21%), and none after T. The most frequent nucleotide couplet cleaved is GT (7 of 19 or 37%). No other couplet appears more than twice. Cleavage tends to occur after a G or C and before an A or T (13 of 19 or 68%).
FIG. 1.

Cleavage of Tn916 oriT200-2 by MBP-Orf20. The left panel shows cleavage of the T strand. Lanes 1 to 4: DNA-sequencing reactions. Lane 5: DNA probe alone. Lanes 6 to 9: MBP-Orf20 increasing from 37.5 nM to 1.25 μM. The right panel shows cleavage of the NT strand. Lane 1: DNA probe alone. Lanes 2 to 5: MBP-Orf20 increasing from 37.5 nM to 1.25 μM. Lanes 6 to 9: DNA-sequencing reactions.
FIG. 2.

Map of oriT200-2. The TGGT motif identified as nic is underlined, with the cleavage site indicated by a black downward pointing arrow. MBP-Orf20 nonspecific cleavage sites are identified by thin upward pointing arrows. The Int binding site is indicated by a dashed line above the sequence. Inverted arrow pairs indicate inverted repeats identified by Jaworski and Clewell (14). Instances of GT at cleavage sites are boldface.
Effect of Int on oriT cleavage by Orf20.
Since interaction of the relaxase with its cognate oriT is often mediated by an accessory protein (40) and MBP-Orf20 alone was unable to generate site- and strand-specific cleavage at oriT, it seemed likely that another protein was involved. The only other Tn916 protein known to interact with oriT is the tyrosine recombinase, Int. Therefore, we determined the effect of Int on MBP-Orf20 cleavage within oriT. The cleavage assays described in the legend to Fig. 1 were repeated with a constant concentration of MBP-Orf20 in the presence of increasing concentrations of Int (Fig. 3). No cleavage of oriT200-2 DNA was observed in samples containing Int alone (Fig. 3, both panels, lane 1). When the labeled NT strand was used as a template, increasing amounts of Int suppressed all cleavage products (Fig. 3, right panel). With the labeled T strand as a template, increasing the concentration of Int suppressed the nonspecific cleavage products and led to the generation of a new, specific cleavage product (Fig. 3, left panel). The specific cleavage occurs at one of two TGGT sequences within oriT. This sequence lies immediately adjacent to the segment of oriT DNA that is protected by Int (Fig. 2) (12).
FIG. 3.

Effect of Int on cleavage by MBP-Orf20. Left panel: T strand. Right panel: NT strand. Lane 1: DNA probe in the presence of 320 nM Int. Lane 2: 125 nM MBP-Orf20. Lanes 3 to 6: 125 nM MBP-Orf20 with increasing amounts of Int from 40 nM to 320 nM. Lanes 7 to 10: DNA-sequencing reactions.
Polarity of DNA cleavage by Orf20.
Upon cleavage, relaxases are known to leave a 3′ OH group and remain covalently attached to the 5′ phosphate (2). A biologically relevant cleavage event at oriT by MBP-Orf20 should generate a 3′ OH, which can be demonstrated by the addition of [α-32P]ddATP using terminal transferase. To test for generation of a 3′ OH, the cleavage reaction was repeated using unlabeled oriT200-2 DNA and concentrations of Int and MBP-Orf20 that produced site-specific cleavage. After incubation for 1.5 h at room temperature, the proteins were digested with proteinase K and removed by phenol extraction. The DNA was then washed, denatured, and labeled with [α-32P]ddATP using terminal transferase. DNA alone and DNA plus Int controls (Fig. 4, lanes 1 and 2) showed that in the absence of MBP-Orf20, only the full-length fragment was labeled. In the sample with MBP-Orf20 alone, the same patterns of cleavage products as those in Fig. 2 were visible (Fig. 4, lane 3), indicating that endonucleolytic cleavage by MBP-Orf20 generates a 3′ OH. The addition of Int to the reaction resulted in a single labeled cleavage product (Fig. 4, lane 4) that corresponds to the specific product generated in the cleavage reactions using 5′-end-labeled oriT200-2 DNA (Fig. 3, left panel, lanes 3 to 6). Thus, Int-associated specific cleavage of Tn916 oriT by MBP-Orf20 produces a 3′ OH group that could serve as a primer during conjugal DNA replication.
FIG. 4.
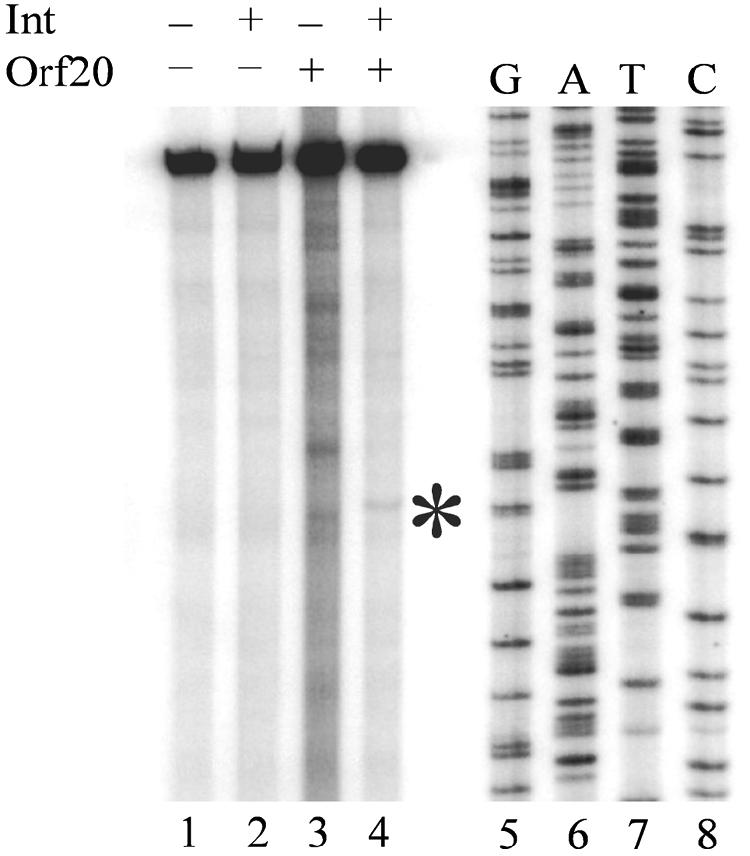
Cleavage by MBP-Orf 20 leaves a 3′ OH group. Lane 1: DNA alone. Lane 2: DNA plus 160 nM Int. Lane 3: DNA plus 125 nM MBP-Orf20. Lane 4: DNA plus 160 nM Int plus 125 nM MBP-Orf20. Lanes 5 to 8: DNA sequence ladder. The asterisk indicates the specific cleavage product produced in the presence of MBP-Orf20 and Int.
Substrate specificity of MBP-Orf20 cleavage.
To determine whether MBP-Orf20 recognizes a specific sequence for cleavage, as opposed to cleaving any sequence that might be adjacent to the Int binding site in oriT, we constructed a template where the TGGT motif on the transferred strand was converted to GTTG. As shown in Fig. 5, this substrate was cleaved by MBP-Orf20 alone in the same positions as those of the wild-type template (compare Fig. 1 and 5). However, in the presence of Int, no cleavage was observed at the site of the substitution, indicating that MBP-Orf20 must recognize the TGGT motif for cleavage to occur.
FIG. 5.
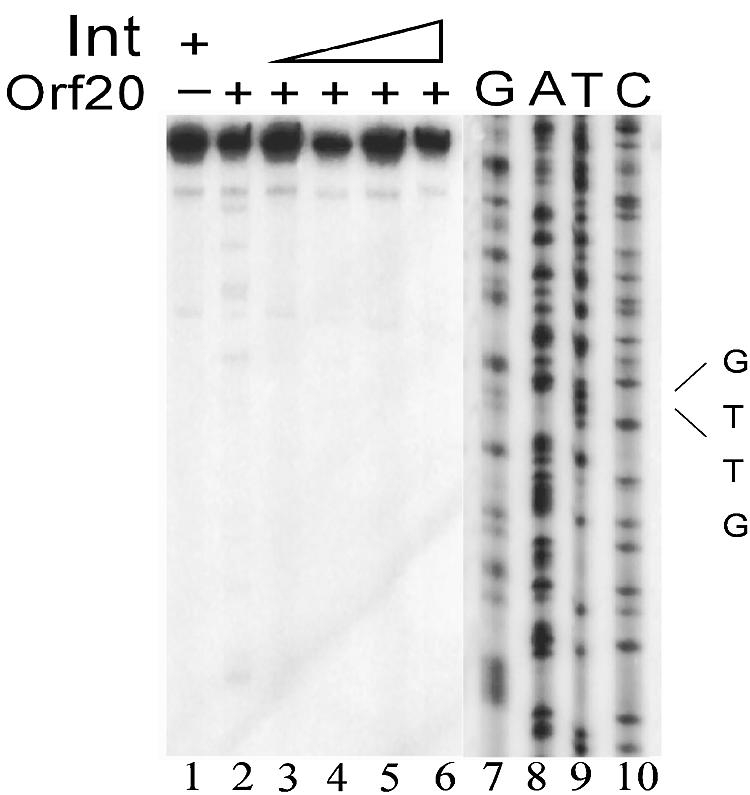
Absence of cleavage of a mutant DNA template by MBP-Orf20. Lane 1: DNA plus 320 nM Int. Lane 2: 125 nM MBP-Orf20. Lanes 3 to 6: 125 nM MBP-Orf20 plus increasing amounts of Int from 40 nM to 320 nM. Lanes 7 to 10: DNA sequence ladder. The position of the sequence alteration from TGGT to GTTG is indicated.
DISCUSSION
We have demonstrated that Orf20, encoded by Tn916, is an endonuclease that cleaves one strand of a DNA duplex, leaving a 3′ OH group. In the presence of Int, MBP-Orf20 cleaves oriT in a strand- and sequence-specific manner. We suggest that this specific cleavage site, TGG↓T, is the nic site in the origin of transfer of Tn916 and that Orf20 and Int act in concert to carry out the initial steps in the conjugal transfer of Tn916. We propose a model in which Int correctly positions Orf20, the relaxase, for cleavage at nic, an unprecedented role for a tyrosine recombinase.
Orf20 is the Tn916 relaxase protein.
Orf20 has several properties characteristic of plasmid relaxases. Typically, the open reading frame for a plasmid-encoded relaxase lies downstream of its cognate oriT; yet, no more than 700 nucleotides generally separate the oriT site from the coding sequence (15). The Tn916 oriT sequence lies in the 395-bp intergenic region between orf21 and orf20, with the orf20 sequence on the downstream side. Second, Orf20 cleaves DNA, leaving a free 3′ OH group that can be labeled with terminal transferase and ddATP. Third, it appears that Orf20 remains in close contact with the site of the nick. It has been shown that after the initial transesterification reaction, the relaxase remains closely associated with the DNA (2). When we tried to determine the presence of a 3′ OH group after cleavage of Tn916 oriT, the nic site was available for labeling only after the proteins in the reaction were digested with proteinase K (data not shown). Fourth, if Orf20 is the Tn916 relaxase and remains covalently attached to the 5′ phosphate group at the site of cleavage, one would expect that it would be transferred from the donor to the recipient. In other plasmids that employ type IV secretion systems for DNA transfer, protein substrates are characterized by the presence of large numbers of basic residues at the C-terminal end of the protein (5). Of all the Tn916-encoded proteins, Orf20 has the most basic residues (12 of 50) at its C terminus.
While it is possible that the site- and strand-specific cleavage product observed was due to action by Int rather than MBP-Orf20, this scenario is extremely unlikely. First, the int gene is located upstream of oriT, at the other end of the transposon. Second, Int cleavage at the transposon ends leaves a 5′ OH and not a 3′ OH. In order for Int to be responsible for the oriT cleavage, it would have to change its cleavage polarity, which has never been described for a tyrosine recombinase. MBP-Orf20, however, leaves a 3′ OH regardless of the presence of Int (Fig. 4, lane 3). In addition, the oriT cleavage site lies immediately downstream of where Int binds; however, cleavage at the transposon ends occurs in the middle of the region protected from DNase I digestion by Int (16). Thus, in order for Int to be responsible for oriT cleavage, it would have to change both its cleavage polarity and the positioning of its active site with respect to the substrate DNA.
Int acts as an accessory protein.
The requirement for accessory proteins involved in determining specificity and providing stability for relaxase-DNA complexes is not unprecedented. The IncP family RP4 relaxase, TraI, requires three accessory proteins for maximum efficiency. TraJ acts as a specificity determinant to allow TraI access to the nic site by binding to the nic-proximal portion of the inverted repeat (21). TraK plays a similar role by interacting with the nic-distal region to adjust the DNA topology (41), and TraH interacts with the other accessory proteins to stabilize the relaxosome (21). The IncF and IncW groups require accessory proteins TraY and IHF or TrwA, respectively, to cleave relaxed or linear templates (18, 20), while the IncQ MobA protein shows enhanced cleavage in vitro when MobB and MobC are present (27). The unique property of Tn916 is that Int apparently plays a dual role in the movement of the transposon from the donor to the recipient. No other instances of a tyrosine recombinase acting as an accessory protein for a separate DNA relaxation reaction are known. However, it has previously been demonstrated that excision of the Bacteroides mobilizable element NBU1 requires a segment of DNA that includes, in addition to the end of the element and the integrase gene, the origin of conjugal transfer (32). Thus, an association between recombination and conjugation functions is not unique to Tn916.
Properties of Orf20.
Tn916 Orf20 shows general similarity to the V. cholerae CTXφ RstA protein, which is responsible for extrachromosomal replication of phage DNA. Proteins involved in rolling-circle replication fall into two classes: one consisting of those proteins involved in replication (Rep) and the other consisting of those proteins involved in conjugal DNA transfer. These proteins have three characteristic motifs (15, 23, 36, 37). We have compared the sequences of Tn916 Orf20 and its close orthologues (Fig. 6). The proteins are divided into two groups: those showing greater than 63% identity and 83% similarity to Orf20 (group 1) and those showing less than 40% identity and 59% similarity to Orf20 (group 2). Both of these groups contain motifs that are recognizably similar to those in the rolling-circle replication proteins (15). However, these Tn916-related proteins are more similar to the superfamily 1 replication proteins (of which the A protein of the bacteriophage φX174 is the prototype) than to the relaxase proteins.
FIG. 6.

Alignment of Tn916 Orf20 and related proteins. The proteins are separated into two groups based on their similarity to Tn916 Orf20. The first group (group I) includes those proteins scored as at least 63% identical and 82% similar to Orf20 by BLASTP (1) analysis. The second group (group II) is comprised of proteins scoring less than 40% identical and 59% similar by BLASTP. Alignments were generated by CLUSTAL W (35). Markings below the alignments indicate degrees of similarity between the sequences: *, single fully conserved residue (group I); :, conservation of strong groups (group I); · , conservation of weak groups (group I); !, single fully conserved residue (all sequences); ‡, conservation of strong groups (all sequences); †, conservation of weak groups (all sequences); no mark, no conservation. Highly conserved tyrosines among all proteins are shaded light gray. The one completely conserved tyrosine is indicated by white letters on a black background. Residues implicated in the modified 3-H motif are shaded dark gray. The proposed motifs (based on Rep protein motifs assigned by Koonin and Ilyina [15]) are indicated above the sequences. The protein sequences shown and their accession numbers are as follows: Orf20 of E. faecalis Tn916, AAB60013; Orf20 of C. difficile Tn5397, AAO24811; Enterococcus faecium DO putative replication initiation factor protein EfaeDRAFT_2437, ZP_00603105; E. faecalis V583 Cro/CI family protein EF1886, AAO81639; Listeria monocytogenes EDG-e Orf20 homolog lmo1111, AG1213; Staphylococcus aureus Mu50 putative phage replication protein SAV0408, BAB56570; S. aureus COL replication initiation factor family protein SACOL1583, AAW38199; S. aureus MRSA252 conserved hypothetical protein SAR1297, CAG40299; B. subtilis 168 transposon-like protein YdcR, CAB12294; Geobacillus stearothermophilus cryptic plasmid pSTK1 Orf3, 2102242C; Streptococcus thermophilus putative transfer protein OrfJ, CAE52362; and Streptococcus mutans UA159 putative transposon protein SMU.207c, AAN57979.
The sequence identified in Fig. 6 as motif III contains the only completely conserved tyrosine, so we suggest that this is the active-site tyrosine of Orf20. We have identified a sequence, HEDUXUYSYXE, similar to motif II of the rolling-circle replication proteins (15). Analysis of Mg2+ coordinating proteins from the Protein Data Bank showed that aspartic acid, glutamic acid, serine, threonine, asparagine, and glutamine residues act more frequently in Mg2+ coordination than does histidine (11). Therefore, we suggest that the HSE residues of this sequence act in the same way as the motif II of other rolling-circle replication proteins.
Given the motif differences combined with the demonstrated endonucleolytic activity of Orf20, it seems likely that Orf20 and its closely related proteins share a progenitor distinct from that of the Rep and relaxase proteins but convergent in function. Due to this likely departure in origin, we propose a new identifier for Tn916 proteins indicated in conjugal transfer of the element, the transposon-encoded conjugation region (tec). Orf13 becomes TecA, and Orf20 becomes TecH.
Significance of the functional interaction between Orf20 and Int.
The results presented here provide a rationale for our previous observation that Int binds specifically to oriT (12). The functional interaction between these two proteins may also reflect other aspects of the regulation of conjugative transposition by Tn916. Int is encoded at the opposite end of the transposon from the transfer genes and is potentially expressed by several constitutive promoters. Thus, Int is likely to be present in cells that have not undergone transposon excision and may bind to oriT. The transfer genes are expressed only upon excision and circularization of the transposon from a promoter upstream of Int (4). Thus, under normal circumstances, transfer can occur only subsequent to excision and can result in transfer of the entire element. However, depending upon the site of integration of Tn916, the transfer genes could be expressed from an appropriately positioned chromosomal promoter, resulting in Hfr-type transfer of part of the element and adjacent chromosomal sequences. Binding of Int to oriT could prevent such premature transfer from occurring and could contribute to a multistage regulatory system that ensures that only intact copies of Tn916 are transferred from the donor to the recipient. This notion is supported by observations that Tn916 cannot mobilize flanking plasmid markers upon transfer from the donor to the recipient (7).
Our results raise the question of whether a similar interaction between integrase and relaxase occurs in elements that are similar to Tn916. In particular, the element Tn5397 discovered in Clostridium difficile is very similar in sequence to Tn916, except that the int gene and part of the xis gene have been replaced by a substitution that encodes a serine recombinase (38). It would be unlikely that an interaction at oriT between the Tn5397 Orf20 relaxase and the recombinase is maintained in the face of replacement with a structurally completely different recombinase enzyme. The loss of such a functional interaction might explain the low transfer frequencies reported for Tn5397 compared to those for Tn916 (19, 39).
Acknowledgments
This work was supported by grant number MCB-0131471 from the National Science Foundation and award number 2002111 from the University Research Committee of Emory University.
We thank Huiping Ling for technical assistance.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1a.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 2.Byrd, D. R., and S. W. Matson. 1997. Nicking by transesterification: the reaction catalysed by a relaxase. Mol. Microbiol. 25:1011-1022. [DOI] [PubMed] [Google Scholar]
- 3.Caperon, M. G., and J. R. Scott. 1989. Excision and insertion of the conjugative transposon Tn916 involves a novel recombination mechanism. Cell 59:1027-1034. [DOI] [PubMed] [Google Scholar]
- 4.Celli, J., and P. Trieu-Cuot. 1998. Circularization of Tn916 is required for expression of the transposon-encoded transfer functions: characterization of long tetracycline-inducible transcripts reading through the attachment site. Mol. Microbiol. 28:103-117. [DOI] [PubMed] [Google Scholar]
- 5.Christie, P. J. 2004. Type IV secretion: the Agrobacterium VirB/D4 and related conjugation systems. Biochim. Biophys. Acta 1694:219-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clewell, D. B., S. E. Flannagan, and D. D. Jaworski. 1995. Unconstrained bacterial promiscuity: the Tn916-Tn1545 family of conjugative transposons. Trends Microbiol. 3:229-236. [DOI] [PubMed] [Google Scholar]
- 7.Clewell, D. B., and C. Gawron-Burke. 1986. Conjugative transposons and the dissemination of antibiotic resistance in Streptococci. Annu. Rev. Microbiol. 40:635-659. [DOI] [PubMed] [Google Scholar]
- 8.Flannagan, S. E., L. A. Zitzow, Y. A. Su, and D. B. Clewell. 1994. Nucleotide sequence of the 18-kb conjugative transposon Tn916 from Enterococcus faecalis. Plasmid 32:350-354. [DOI] [PubMed] [Google Scholar]
- 9.Franke, A. E., and D. B. Clewell. 1981. Evidence for a chromosome-borne resistance transposon (Tn916) in Streptococcus faecalis that is capable of “conjugal” transfer in the absence of a conjugative plasmid. J. Bacteriol. 145:494-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottesman, S. 1990. Minimizing proteolysis in Escherichia coli: genetic solutions. Methods Enzymol. 185:119-129. [DOI] [PubMed] [Google Scholar]
- 11.Harding, M. M. 2004. The architecture of metal coordination groups in proteins. Acta Crystallogr. Sect. D. 60:849-859. [DOI] [PubMed] [Google Scholar]
- 12.Hinerfeld, D., and G. Churchward. 2001. Specific binding of integrase to the origin of transfer (oriT) of the conjugative transposon Tn916. J. Bacteriol. 183:2947-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinerfeld, D., and G. Churchward. 2001. Xis protein of the conjugative transposon Tn916 plays dual opposing roles in transposon excision. Mol. Microbiol. 41:1459-1467. [DOI] [PubMed] [Google Scholar]
- 14.Jaworski, D. D., and D. B. Clewell. 1995. A functional origin of transfer (oriT) on the conjugative transposon Tn916. J. Bacteriol. 177:6644-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koonin, E. V., and T. V. Ilyina. 1993. Computer-assisted dissection of rolling circle DNA replication. BioSystems 30:241-268. [DOI] [PubMed] [Google Scholar]
- 16.Lu, F., and G. Churchward. 1995. Tn916 target DNA sequences bind the C-terminal domain of integrase protein with different affinities that correlate with transposon insertion frequency. J. Bacteriol. 177:1938-1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marra, D., and J. R. Scott. 1999. Regulation of excision of the conjugative transposon Tn916. Mol. Microbiol. 31:609-621. [DOI] [PubMed] [Google Scholar]
- 18.Moncalian, G., G. Grandoso, M. Llosa, and F. de la Cruz. 1997. oriT-processing and regulatory roles of TrwA protein in plasmid R388 conjugation. J. Mol. Biol. 270:188-200. [DOI] [PubMed] [Google Scholar]
- 19.Mullany, P., M. Wilks, I. Lamb, C. Clayton, B. Wren, and S. Tabaqchali. 1990. Genetic analysis of a tetracycline resistance element from Clostridium difficile and its conjugal transfer to and from Bacillus subtilis. J. Gen. Microbiol. 136:1343-1349. [DOI] [PubMed] [Google Scholar]
- 20.Nelson, W. C., M. T. Howard, J. A. Sherman, and S. W. Matson. 1995. The traY gene product and integration host factor stimulate Escherichia coli DNA helicase I-catalyzed nicking at the F plasmid oriT. J. Biol. Chem. 270:28374-28380. [PubMed] [Google Scholar]
- 21.Pansegrau, W., D. Balzer, V. Kruft, R. Lurz, and E. Lanka. 1990. In vitro assembly of relaxosomes at the transferred origin of plasmid RP4. Proc. Natl. Acad. Sci. USA 87:6555-6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pansegrau, W., and E. Lanka. 1996. Enzymology of DNA transfer by conjugative mechanisms. Prog. Nucleic Acid Res. Mol. Biol. 54:197-251. [DOI] [PubMed] [Google Scholar]
- 23.Pansegrau, W., W. Schroder, and E. Lanka. 1994. Concerted action of three distinct domains in the DNA cleaving-joining reaction catalyzed by relaxase (TraI) of conjugative plasmid RP4. J. Biol. Chem. 269:2782-2789. [PubMed] [Google Scholar]
- 24.Poyart, C., J. Celli, and P. Trieu-Cuot. 1995. Conjugative transposition of Tn916-related elements from Enterococcus faecalis to Escherichia coli and Pseudomonas fluorescens. Antimicrob. Agents Chemother. 39:500-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudy, C., K. L. Taylor, D. Hinerfeld, J. R. Scott, and G. Churchward. 1997. Excision of a conjugative transposon in vitro by the Int and Xis proteins of Tn916. Nucleic Acids Res. 25:4061-4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 27.Scherzinger, E., R. Lurz, S. Otto, and B. Dobrinski. 1992. In vitro cleavage of double- and single-stranded DNA by plasmid RSF1010-encoded mobilization proteins. Nucleic Acids Res. 20:41-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott, J. R., F. Bringel, D. Marra, G. Van Alstine, and C. K. Rudy. 1994. Conjugative transposition of Tn916: preferred targets and evidence for conjugative transfer of a single strand and for a double-stranded circular intermediate. Mol. Microbiol. 11:1099-1108. [DOI] [PubMed] [Google Scholar]
- 29.Scott, J. R., P. A. Kirchman, and M. G. Caparon. 1988. An intermediate in the transposition of the conjugative transposon Tn916. Proc. Natl. Acad. Sci. USA 85:4809-4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Senghas, E., J. M. Jones, M. Yamamoto, C. Gawron-Burke, and D. B. Clewell. 1988. Genetic organization of the bacterial conjugative transposon Tn916. J. Bacteriol. 170:245-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoemaker, N. B., H. Vlamakis, K. Hayes, and A. A. Salyers. 2001. Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl. Environ. Microbiol. 67:561-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoemaker, N. B., G.-R. Wang, and A. A. Salyers. 2000. Multiple gene products and sequences required for excision of the mobilizable integrated Bacteroides element NBU1. J. Bacteriol. 182:928-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storrs, M. J., C. Poyart-Salmeron, P. Trieu-Cuot, and P. Courvalin. 1991. Conjugative transposition of Tn916 requires the excisive and integrative activities of the transposon-encoded integrase. J. Bacteriol. 173:4347-4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor, K., and G. Churchward. 1997. Specific DNA cleavage mediated by the integrase of conjugative transposon Tn916. J. Bacteriol. 179:1117-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varsaki, A., M. Lucas, A. S. Afendra, C. Drainas, and F. de la Cruz. 2003. Genetic and biochemical characterization of MbeA, the relaxase involved in plasmid ColE1 conjugative mobilization. Mol. Microbiol. 48:481-493. [DOI] [PubMed] [Google Scholar]
- 37.Waldor, M. K., E. J. Rubin, G. D. Pearson, H. Kimsey, and J. J. Mekalanos. 1997. Regulation, replication, and integration functions of the Vibrio cholerae CTXphi are encoded by region RS2. Mol. Microbiol. 24:917-926. [DOI] [PubMed] [Google Scholar]
- 38.Wang, H., A. P. Roberts, D. Lyras, J. I. Rood, M. Wilks, and P. Mullany. 2000. Characterization of the ends and target sites of the novel conjugative transposon Tn5397 from Clostridium difficile: excision and circularization is mediated by the large resolvase, TndX. J. Bacteriol. 182:3775-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, H., A. P. Roberts, and P. Mullany. 2000. DNA sequence of the insertional hot spot of Tn916 in the Clostridium difficile genome and discovery of a Tn916-like element in an environmental isolate integrated in the same hot spot. FEMS Microbiol. Lett. 192:15-20. [DOI] [PubMed] [Google Scholar]
- 40.Zechner, E. L., F. de la Cruz, R. Eisenbrandt, A. M. Grahn, G. Koriamann, E. Lanka, G. Muth, W. Pansegrau, C. M. Thomas, B. M. Wilkins, and M. Zatyka. 2000. Conjugative-DNA transfer processes, p. 87-174. In C. M. Thomas (ed.), The horizontal gene pool, 1st ed. Harwood Academic Publishers, Amsterdam, The Netherlands.
- 41.Ziegelin, G., W. Pansegrau, R. Lurz, and E. Lanka. 1992. TraK protein of conjugative plasmid RP4 forms a specialized nucleoprotein complex with the transfer origin. J. Biol. Chem. 267:17279-17286. [PubMed] [Google Scholar]