

Abstract
Polar development and cell division in Caulobacter crescentus are controlled and coordinated by multiple signal transduction proteins. divJ encodes a histidine kinase. A null mutation in divJ results in a reduced growth rate, cell filamentation, and mislocalized stalks. Suppressor analysis of divJ identified mutations in genes encoding the tyrosine kinase (divL) and the histidine kinase (cckA). The divL and cckA suppressor alleles all have single amino acid substitutions, some of which confer a temperature-sensitive phenotype, particularly in a wild-type background. Analysis of transcription levels from several positively regulated CtrA-dependent promoters reveals high expression in the divJ mutant, suggesting that DivJ normally serves to reduce CtrA activity. The divL and cckA suppressors reduce the amount of transcription from promoters positively regulated by CtrA, indicating that the mutations in divL and cckA are suppressing the defects of the divJ mutant by reducing the abnormally high level of CtrA activity. Immunoblotting showed no major perturbations in the CtrA protein level in any of these strains, suggesting that the high amount of CtrA activity seen in the divJ mutant and the reduced amount of activity in the suppressors are regulated at the level of activation and not transcription, translation, or degradation. In vivo phosphorylation assays confirmed that divJ mutants have elevated levels of CtrA phosphorylation and that this level is reduced in the suppressors with mutations in divL.
The aquatic gram-negative bacterium Caulobacter crescentus has a dimorphic life cycle, beginning as a motile, piliated swarmer cell incapable of DNA replication. The swarmer cell differentiates into a stalked cell by ejecting its flagellum, retracting the pili, and synthesizing a stalk with an adhesive holdfast at the same pole that contained the flagellum. The stalked cell initiates DNA replication and cell division and synthesizes a new flagellum at the pole opposite the stalk, producing a new motile swarmer cell during each cell cycle. Multiple signal transduction proteins are involved in coordinating polar development and cell division in C. crescentus. The global response regulator CtrA controls the expression of at least 144 genes (25) involved in cell division, DNA methylation, holdfast synthesis, flagellum biogenesis, and pilus biogenesis (22, 25, 34, 43). CtrA prevents the initiation of DNA replication in swarmer cells by binding to the origin of replication (35) and inhibits cell division by repressing transcription of ftsZ, which encodes the first cell division protein to localize to the site of division (22). Late in the cell cycle, CtrA activates transcription from the PQA promoter, which cotranscribes ftsQ and ftsA, ensuring ordered expression of ftsZ, ftsQ, and ftsA (22, 38, 50).
CtrA activity is controlled by phosphorylation and proteolysis. CtrA is cleared from the swarmer cell during swarmer-to-stalked cell differentiation (9) in a ClpXP-dependent manner (21). Both CtrA and CtrA∼P can be degraded by ClpXP (37), so dephosphorylation of CtrA during swarmer-to-stalked cell differentiation is not required for, nor is it likely to be the cue for, cell cycle-dependent proteolysis (37). DivK, an essential single-domain response regulator, plays a role in signaling the degradation of CtrA (16).
DivK is regulated by two histidine kinases, DivJ and PleC (15, 19, 51). divJ has been shown to be epistatic to pleC in terms of morphological phenotype (49). DivJ and PleC have antagonistic effects on DivK activity. DivJ localizes to the stalked pole of the cell, where it acts as a kinase for DivK, whereas PleC localizes to the flagellar pole and acts as a phosphatase, controlling the release of DivK from the pole (18, 49). The phosphorylation state of DivK controls its localization (24), causing it to shuttle between the flagellar pole and stalked pole in predivisional cells (28). Upon completion of cytokinesis, the shuttling ceases, and DivK no longer localizes to the flagellated pole, allowing development of the swarmer cell to occur (28). Thus, DivK is able to signal the state of cell compartmentalization during division (28). DivJ and PleC also have an antagonistic role on the GGDEF diguanylate cyclase response regulator PleD, which is involved in turning off flagellar rotation and in stalk synthesis (45). There is in vivo evidence that DivJ phosphorylates PleD, whereas PleC dephosphorylates PleD∼P (3).
CtrA phosphorylation is controlled by DivL, an essential tyrosine kinase, and CckA, an essential hybrid kinase, which have the same localization pattern during the cell cycle (17, 18, 41, 52). DivL phosphorylates CtrA in vitro (52) and interacts with DivK, although the role of this interaction is not yet known (30). CckA is required for CtrA phosphorylation in vivo (17, 18), regulates the same subset of genes as CtrA, and is phosphorylated at the same time of the cell cycle as CtrA (17). CckA seems to have a stabilizing effect on the CtrA protein in addition to its putative role as a kinase for CtrA, as the half-life of CtrA decreases by over 50% in the absence of CckA (17). It remains to be seen whether both DivL and CckA are cognate kinases for CtrA or whether one or both are involved in a different stage of the pathway leading to the activation of CtrA.
CtrA activity controls so many cell cycle events, including cell division and DNA replication, that regulation of the level of CtrA activity is critical for optimal growth. In this paper, we show that divJ mutants have mislocalized holdfasts in addition to the mislocalized stalks, slow growth, and cell division defects previously described (40, 49). We use suppressor analysis to show that the defects of a divJ null mutant are due, at least in part, to increased CtrA activity. We provide in vivo evidence that DivJ normally acts to lower the activity of CtrA; therefore, mutations in divL and cckA suppress the divJ deletion mutant because they also lower the activity of CtrA.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are described in Table 1. Escherichia coli strains were grown in Luria-Bertani (LB) broth or on LB agar (39) supplemented with kanamycin (30 μg/ml in broth, 50 μg/ml in agar), streptomycin (5 μg/ml in broth and agar), spectinomycin (100 μg/ml in broth, 50 μg/ml in agar), gentamicin (15 μg/ml in broth, 20 μg/ml in agar), and tetracycline (12 μg/ml in broth and agar) as necessary. C. crescentus strains were grown in peptone-yeast extract (PYE) broth or on PYE agar (31) supplemented with kanamycin (5 μg/ml in broth, 20 μg/ml in agar), spectinomycin (25 μg/ml in broth, 100 μg/ml in agar), gentamicin (2.5 μg/ml in agar), nalidixic acid (20 μg/ml in agar), tetracycline (1 μg/ml in broth, 2 μg/ml in agar), and 3% sucrose (in agar) as needed. C. crescentus strains transduced with ΔdivJ::spec were grown on M2G agar (10) supplemented with spectinomycin (350 μg/ml) or kanamycin (100 μg/ml).
TABLE 1.
Strains and plasmids
| Strain or plasmid | Description or construction | Source or reference(s) |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5αF′ | φ80dlacZΔM15 Δ(lacZYA-argF)U169 endA1 recA1 hsdR17(r− m+) deoR thi-1 supE44 λ−gyrA96 relA1 | 26 |
| S17-1 | E. coli 294::RP4-2(Tc::Mu)(Km::Tn7) | 42 |
| BL21(λDE3) | F−ompT hsdSB(rB− mB−) gal dcm (λDE3) | Novagen |
| C. crescentus | ||
| NA1000 | syn-1000; previously called CB15N | 12 |
| CB15 | CB15; wild type | 31 |
| CMS1-CMS40 | Genomic marker strains; Kanr | 48 |
| UJ590 | CB15 ATCC 19089 ΔpilA | 1 |
| SU214 | CB15N rpoN::Tn5 | 6 |
| YB1382 | CB15N pleC::mini-Tn5 lacZ2 I29 | 13 |
| YB3059 | CB15 ΔdivJ::spec | A. Hinz, unpublished |
| YB3202 | NA1000 ΔdivJ::spec | This study |
| YB1388 | NA1000 divJ::mini-Tn5 lacZ2 | This study |
| YB3215 | NA1000 ΔdivJ::spec sdj-21 (original suppressor) | This study |
| YB3216 | NA1000 ΔdivJ::spec sdj-22 (original suppressor) | This study |
| YB3219 | NA1000 ΔdivJ::spec sdj-25 (original suppressor) | This study |
| YB3227 | NA1000 ΔdivJ::spec sdj-33 (original suppressor) | This study |
| YB3229 | NA1000 ΔdivJ::spec sdj-35 (original suppressor) | This study |
| YB3314 | NA1000 divL22 (exchanged suppressor) | This study |
| YB3315 | NA1000 ΔdivJ::spec divL22 (exchanged suppressor) | This study |
| YB3316 | NA1000 divL33 (exchanged suppressor) | This study |
| YB3317 | NA1000 ΔdivJ::spec divL33 (exchanged suppressor) | This study |
| YB3318 | NA1000 divL35 (exchanged suppressor) | This study |
| YB3319 | NA1000 ΔdivJ::spec divL35 (exchanged suppressor) | This study |
| YB3325 | NA1000 cckA21 (exchanged suppressor) | This study |
| YB3326 | NA1000 ΔdivJ::spec cckA21 (exchanged suppressor) | This study |
| YB3327 | NA1000 cckA25 (exchanged suppressor) | This study |
| YB3328 | NA1000 ΔdivJ::spec cckA25 (exchanged suppressor) | This study |
| Plasmids | ||
| pNPTS138 | Used for two-part selection for in-frame deletions | 4, 46 |
| pHP45Ω | Used to obtain Ω cassette for spectinomycin resistance | 32 |
| pBGST18 | Used to construct markers | 4 |
| pGSZ | Used to construct markers, a Gentr derivative of pGMTZ1 | 5 |
| pJS70 | Used to measure PpilA activity | 43 |
| plac290/HB2.0BP | Used to measure PftsZ activity | 22 |
| pMSP8LC | Used to measure PftsQA activity | 38 |
| pMR20 | Medium-copy Tetr plasmid | 36 |
| pJW375 | Used to overexpress DivL299 | 52 |
| pNPTS138UP, DN, Ω | Used to delete divJ | This study |
| pMR20divJ | Used to complement divJ null mutants | This study |
| pBGST18CMS12.3 | Used to generate genomic marker 3/10 of the way between CMS12 and CMS13 | This study |
| pBGST18CMS12.4 | Used to generate genomic marker 4/10 of the way between CMS12 and CMS13 | This study |
| pBGST18CMS12.49 | Used to generate genomic marker 49/100 of the way between CMS12 and CMS13 | This study |
| pBGST18CMS12.54 | Used to generate genomic marker 54/100 of the way between CMS12 and CMS13 | This study |
| pBGST18CMS12.59 | Used to generate genomic marker 59/100 of the way between CMS12 and CMS13 | This study |
| pBGST18CMS12.6 | Used to generate genomic marker 6/10 of the way between CMS12 and CMS13 | This study |
| pBGST18CMS37.65 | Used to generate genomic marker 65/100 of the way between CMS37 and CMS38 | This study |
| pBGST18CMS37.75 | Used to generate genomic marker 75/100 of the way between CMS37 and CMS38 | J. Wagner, unpublished |
| pGSZCMS12.3 | Used to generate genomic marker 3/10 of the way between CMS12 and CMS13 | This study |
| pGSZCMS37.65 | Used to generate genomic marker 65/100 of the way between CMS37 and CMS38 | This study |
Two-step gene replacement or deletion.
Two 450-bp fragments (UP and DN) on either side of divJ were amplified by PCR using oligonucleotides UPdivJN (5′-GTGGCGTATGCTAGCTACTGGGGG), UPdivJB (5′-GGGGAGGATGGATCCCGTCAAACC), DNdivJB (5′-TTTGCGCCGGGATCCGGCGCTCTG), and DNdivJH (5′-GCCTGTCGCAAAGCTTGAGCTTTGGG) and cloned into the vector pNPTS138 (4), along with a spectinomycin/streptomycin-resistant omega cassette (Ω) (32), to generate plasmid pNPTS138 UP, DN, Ω. The in-frame deletion was created by a two-step selection using the nptI gene to select for presence of the plasmid and the sacB and spectinomycin resistance genes to select against presence of the plasmid but for the presence of the Ω cassette, respectively (13, 47), to create YB3202 (ΔdivJ::spec). The same technique was used to exchange the mutant alleles from sdj-22, sdj-33, sdj-35, sdj-21, and sdj-25, except that the entire gene was amplified and the Ω cassette was not used. The exchanges were confirmed by sequencing.
Complementation of divJ mutants.
divJ and its presumptive promoter region were amplified by PCR using oligonucleotides divJUP and divJDN and cloned into the multicopy vector pMR20 (36), generating plasmid pMR20divJ.
Phage sensitivity assays.
Two hundred microliters of saturated culture grown overnight was mixed with 3 ml of molten PYE-0.5% top agar and plated onto plain PYE plates. Once the top agar had hardened, 5 μl of 1010 PFU/ml φCr30 (11) and φCbK (2) was spotted onto each plate and allowed to soak in. The plates were incubated at 30°C for 3 days. NA1000 was used as a positive (sensitive) control, UJ590 (CB15 ATCC 19089 ΔpilA), a pilus-minus mutant, was used as a negative (resistant) control for φCbK, and YB1382 (CB15N pleCI29) was used as a negative (resistant) control for both phages. For a more quantitative measure of phage sensitivity, approximately 170 PFU of φCr30 were mixed with 3 ml PYE-0.5% top agar and 200 μl of culture grown overnight and plated onto a plain PYE plate and incubated at room temperature for 3 to 5 days.
Swarm motility assay.
Five microliters of saturated cultures grown overnight or exponentially growing cells normalized to the same optical density at 600 nm (OD600) was injected into fresh PYE-0.3% swarm agar plates. The plates were incubated at room temperature in a humid chamber for 5 days. NA1000 was used as a positive control, and SU214 (CB15N rpoN::Tn5) (6), a flagellum-minus mutant, was used as a negative control.
Transductions.
Phage lysates were prepared as described previously (11). All transductions were done using a modification of the protocol described previously by West et al. (48). Briefly, 475 μl of culture grown overnight, 500 μl of PYE, and 25 μl of an irradiated phage lysate were mixed together. The mixture was incubated with shaking at 30°C for 1 to 4 h. Various amounts of the reaction mixture were plated onto PYE or M2G with appropriate antibiotics. φCr30 packages approximately 120 kb of DNA (L) (11), so if d is the distance between the gene and marker, and CTF is the cotransduction frequency, then CTF = [1 − (d/L)]3. For example, the distance between divL and CMS38 is about 30 kb, so the expected CTF is 42%. Markers with decimal numbers were generated in this study and located between markers previously described (48). For example, marker CMS37.65 lies 65/100 of the way between markers CMS37 and CMS38. For three-factor crosses, the phage was grown on a doubly marked donor strain.
Mapping the suppressor mutations.
We used transduction of markers spaced approximately 100 kb apart in the C. crescentus genome to map the locations of the suppressor mutations by transduction (48). The markers are numbered according to their location in the genome; for example, marker CMS37 is integrated at the 3.7-Mb position in the C. crescentus genome. We began by testing whether the suppressor mutations were linked to genes already known to be involved in developmental control, since it was likely that some of the suppressors of the divJ mutation would be in these genes. We identified markers linked to the wild-type allele of the suppressor mutation based on the ability of phage grown on a marked wild-type donor to transduce the suppressor strain back to the slow-growth phenotype of YB3202 (ΔdivJ::spec). The expected CTFs of each marker with each gene of interest were estimated based on the distance between the gene and the site at which the marker was integrated. By comparing experimental CTFs with expected CTFs, we identified candidate genes for sequencing to identify the suppressor mutants.
Three suppressor mutants, sdj-22, sdj-33, and sdj-35, were linked to CMS38 with an average CTF of 23%. These suppressors were also 14% linked to CMS37. The suppressors showed a very strong linkage (69%) to CMS37.75, which is placed at 75% of the distance between CMS37 and CMS38. We reasoned that possible sites for suppressors of divJ could be located in divL at 37.70, CC3474 (hk4) at 37.62, and CC3477 (rr4) at 37.64. hk4 (CC3474) and rr4 (CC3477) were identified as cell cycle-regulated signal transduction genes (25). Another marker, CMS37.65, was constructed (with gentamicin resistance) and used in combination with CMS38 for three-factor crosses for sdj-22, sdj-33, and sdj-35 to narrow the region to sequence for the suppressor mutation. The data for the three-factor cross indicated that the suppressor mutation mapped between CMS37.65 and CMS38, which ruled out hk4 and rr4 as possible sites for the mutation (data not shown).
Two suppressor mutants, sdj-21 and sdj-25, were cotransduced 23% and 20%, respectively, with marker CMS12. This marker is near divJ, cckN, and cckA. Because the CMS12 marker is within cotransduction distance of divJ, donor lysates for subsequent mapping within cotransduction distance of divJ were made from a ΔdivJ::spec background to avoid transducing the wild-type divJ allele along with the suppressor mutations. The sdj-21 and sdj-25 suppressor mutations were linked to CMS13 at 21% and 41%, respectively. This suggested that the mutation was between markers 12 and 13. Further mapping indicated that sdj-21 was 53% linked and sdj-25 was 58% linked to CMS12.6. A three-factor cross between a Gentr marker at position 12.3 and a Kanr marker at position 12.6 showed that the suppressor mutations in sdj-21 and in sdj-25 lay between CMS12.3 and 12.6. Kanr markers CMS12.4, 12.49, 12.54, and 12.59 were used to map the mutations more precisely. The suppressor mutants showed the greatest linkage to markers CMS12.49 and 12.54. sdj-21 was shown to be 80% and 87% linked to CMS12.49 and CMS12.54, respectively, while sdj-25 was 82% and 89% linked to the same markers, suggesting that the suppressor mutations were in cckA.
Sequencing.
divL and the presumptive promoter region (573 bp upstream from the start of divL) were amplified as two fragments by PCR using oligonucleotide pairs divLF1 and R4 and divLF3 and R1. Additional oligonucleotides (divLF2-6 and divLR2-6) were designed approximately 550 bp apart for sequencing of the resultant PCR product. Oligonucleotides cckAF1 and cckAR1 were used to amplify cckA and the presumptive promoter region (422 bp upstream from the start site of cckA). Oligonucleotides cckAF2-F5 and cckAR2-R6 were used to sequence the resultant PCR product with cckAF1 and cckAR1. Sequencing reactions were performed using the ABI Prism BigDye Terminator cycle sequencing ready reaction kit (Applied Biosystems). Sequencing was performed by the Institute for Molecular and Cellular Biology at Indiana University on an Applied Biosystems 3730 automated fluorescence sequencing system. Wisconsin Package version 10.3 (Accelrys Inc.) and Sequencher 4.2.2 (Gene Codes Corporation) were used to analyze sequence data. All oligonucleotide sequences are available upon request.
DivL antibody production, immunoblot analysis, and β-galactosidase assays.
Antibodies to DivL were generated by overexpressing the C-terminal 299 amino acids of DivL with a His tag from pJW375 (52) in BL21(λDE3) (Novagen) at 37°C in the presence of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The protein was purified from inclusion bodies as described previously (33). Polyclonal antibodies were raised in New Zealand White rabbits by Cocalico Biologicals (Reamstown, PA), and antibody was affinity purified as previously described (8).
Strains were grown to exponential phase (approximate OD600 of 0.6). From each culture, samples were removed for Lowry protein assay, Laemmli sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, and β-galactosidase assays (23, 27, 29). Equal amounts of total protein were resolved on SDS-polyacrylamide gels and transferred onto nitrocellulose. After staining with Ponceau-S to confirm even transfer (14), the blots were probed with affinity-purified anti-DivL antibody at a dilution of 1:500 and anti-CtrA crude serum at a dilution of 1:700 overnight and treated with goat anti-rabbit immunoglobulin G (heavy plus light chains)-horseradish peroxidase conjugate (Bio-Rad) at a dilution of 1:20,000. The DivL blots were developed with Pierce SuperSignal West Dura substrate, and the CtrA blots were developed with Pierce SuperSignal Pico substrate. All blots were analyzed using a Kodak Image Station 440CF and Kodak 1D software.
Temperature-sensitive (TS) assays.
Cultures of exponentially growing cells were diluted to an OD600 of 0.43, and 5 μl of 100, 10−2, and 10−5 dilutions was spotted onto three sets of plain PYE plates (only one set is shown). The plates were incubated at 30°C, 34°C, and 37°C for 2 days and scanned. For microscopy, cultures were grown overnight in PYE at 30°C, subcultured into two new cultures, and allowed to grow to exponential phase at 30°C, and one culture was then shifted to 37°C and one was left at 30°C for 4 h (approximately 2.5 doublings at 30°C).
Microscopy.
All images were captured using a Nikon Eclipse E800 with a Plan Apo ×100 oil immersion objective along with Princeton Instruments cooled charge-coupled-device digital camera model 1317 and MetaMorph imaging software (Universal Imaging Corporation). Lectin binding assays were performed as previously described (20), and fluorescence was observed using a Nikon FITC-HyQ filter cube (Chroma Technologies).
In vivo phosphorylation.
In vivo phosphorylation was carried out as described previously (19), with the following modifications. Cells were grown in M5GG medium to an OD600 of approximately 0.2. One milliliter of cells was removed for use in Western analysis. Five hundred microliters of cells was used for a Lowry protein assay. One milliliter of cells was washed with M5GG no-phosphate medium and labeled with 100 μCi [32P]H3PO4 for 10 min. The pellet of labeled cells was resuspended in 50 μl SDS lysis buffer and frozen on dry ice. Ten microliters of RNaseA (10 mg/ml) was added, and the pellet was thawed for 2 min at room temperature. Six hundred fifty microliters of K2 low-salt buffer was added to the pellets. Three 100-μl aliquots of the lysate were used for the rest of the assay to assess reproducibility among individual immunoprecipitations. Thirty microliters of protein A agarose was added to the lysate along with 650 μl K2 low-salt buffer and incubated on a rocker at 4°C for 10 min. The protein A agarose was pelleted, and the supernatant was transferred to a new tube. CtrA was immunoprecipitated with 2 μl anti-CtrA crude serum and 20 μl protein A agarose for 30 min by rocking at 4°C. Washes and gel analysis were performed as described previously (19).
RESULTS
Analysis of divJ mutants.
In a previous transposon mutagenesis screen for slow-growing mutants (13), we identified a mutant, called L35, that was deficient in cell division and exhibited a high frequency of stalk mislocalization. Sequencing of the C. crescentus DNA flanking the transposon revealed that the transposon was inserted after nucleotide 1018 of the divJ coding region, truncating the normally 597-amino-acid protein to 339 amino acids. This divJ::mini-Tn5 lacZ2 mutant (YB1388) had characteristics similar to those of the previously reported divJ partial deletion mutant (49), but they differed in some respects. For example, more than 30% of cells of the partial divJ deletion mutant had misplaced stalks (49), compared to only 9% in our divJ::mini-Tn5 lacZ2 mutant cells and 0.2% in wild-type NA1000. Because the previously reported divJ mutant was not a deletion of the entire gene, the differences between our divJ::mini-Tn5 lacZ2 mutant and the previous divJ mutant could be due to the remaining divJ sequences in either or both mutants. We therefore constructed a divJ null mutant, replacing the entire coding sequence with the Ω cassette carrying spectinomycin resistance, resulting in YB3202 (ΔdivJ::spec). The phenotype of the ΔdivJ::spec mutant was complemented by plasmid pMR20divJ containing divJ under the control of its own promoter.
We analyzed the phenotype of YB3202 (ΔdivJ::spec) by examining the length of the cells and stalks, the ability to swim in liquid and swarm in low-concentration agar, and sensitivity to phage (Table 2). YB3202 (ΔdivJ::spec) had an average of 8% mislocalized stalks, compared to 9% mislocalized stalks in YB1388 (divJ::mini-Tn5 lacZ2) and 0.2% mislocalized stalks in NA1000. YB3202 (ΔdivJ::spec) cells in a mixed population were approximately 4 μm long, compared to a 3-μm average length for NA1000 cells. The average length of stalks in YB3202 (ΔdivJ::spec) was 3 μm, twice as long as stalks of NA1000 cells (Table 2). The ΔdivJ::spec mutation was transduced into the CB15 background, creating YB3059 (CB15 ΔdivJ::spec), to assay for the presence and location of holdfasts. YB3059 (CB15 ΔdivJ::spec) produced holdfasts at the tips of polar stalks and at the tips of mislocalized side stalks (Fig. 1), suggesting that the ΔdivJ::spec mutant has a general defect in cell polarity and not simply a defect in stalk localization. We saw no obvious differences in the swimming ability of swarmer cells of YB3202 (ΔdivJ::spec) compared to that of cells of NA1000 by phase-contrast microscopy, but we found that YB3202 (ΔdivJ::spec) does not form as large a swarm in 0.3% swarm agar as NA1000, as previously reported (Table 2) (7, 44). Since the defect in swarming is not due to an obvious defect in swimming, it is likely that the filamentous cells and the slow growth of the mutant are responsible for the swarming defect. We also examined the sensitivity of YB3202 (ΔdivJ::spec) to the pilus-specific phage φCbK and the S-layer phage φCr30. YB3202 was as sensitive to both phages as NA1000 (Table 2). In summary, YB3202 (ΔdivJ::spec) is filamentous, has long, occasionally misplaced stalks, is deficient in swarming in 0.3% agar but not in swimming in liquid, and is sensitive to both S-layer and pilus-specific phage, and YB3059 (CB15 ΔdivJ::spec) makes holdfast material associated with polar and misplaced stalks.
TABLE 2.
Phenotypes of the divJ suppressors
| Strain | Locus | Avg cell length (μm) (no. of cells counted) | % of cells with stalks | Avg stalk length (μm) (no. of stalks counted) | φCbK | φCr30 | Swarm motility |
|---|---|---|---|---|---|---|---|
| NA1000 | NAa | 3.03 (200) | 67 | 1.48 (133) | Sb | S | wt |
| YB3202 (ΔdivJ::spec) | NA | 3.97 (200) | 64 | 3.00 (127) | S | S | divJe |
| YB3215 (ΔdivJ::spec sdj-21) | cckA | 4.02 (192) | 27 | 2.19 (51) | S | PSc | divJe |
| YB3219 (ΔdivJ::spec sdj-25) | cckA | 3.12 (240) | 13 | 1.81 (30) | S | PS | divJe |
| YB3216 (ΔdivJ::spec sdj-22) | divL | 3.25 (141) | 11 | 2.17 (16) | S | Rd | divJe |
| YB3227 (ΔdivJ::spec sdj-33) | divL | 3.47 (220) | 11 | 1.61 (23) | S | R | divJe |
| YB3229 (ΔdivJ::spec sdj-35) | divL | 3.10 (240) | 13 | 1.95 (31) | S | R | divJe |
NA, not applicable.
S, sensitive.
PS, partially sensitive.
R, resistant.
Less than the wild type (wt) and greater than a fla mutant.
FIG. 1.
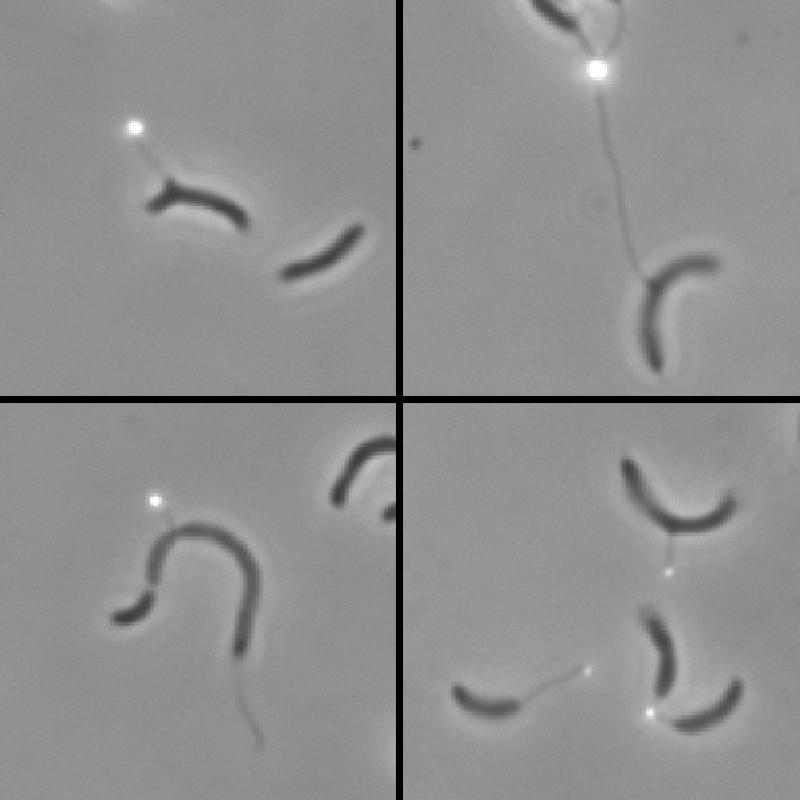
Presence of holdfasts on mislocalized side stalks of YB3059 (CB15 ΔdivJ::spec). Holdfasts were labeled with wheat germ agglutinin-fluorescein isothiocyanate and appear as white dots.
Isolation of fast-growing suppressors of a divJ null mutant.
To determine the genetic basis for slow growth in the divJ mutant, we isolated suppressors of the slow-growth phenotype. Suppressors of the divJ mutation were obtained by repeatedly subculturing YB1388 or YB3202 (divJ::mini-Tn5 lacZ2 or ΔdivJ::spec) and plating aliquots daily. Fast-growing suppressors of the divJ mutant formed larger colonies than the original mutant, allowing for their easy identification (Table 3). As soon as a suppressor colony was identified from each of the independent cultures, no additional suppressors were isolated from that culture to avoid accumulation of multiple mutations in the same strain. The suppressors were named sdj mutants for suppressor of divJ. Each of the suppressor mutants described in this study was isolated independently.
TABLE 3.
Doubling times of the divJ and sdj mutants
| Strain | Doubling time (min) ± SD |
|---|---|
| NA1000 | 94.1 ± 1.5 |
| YB3202 (ΔdivJ::spec) | 103.5 ± 3.1 |
| YB3326 (ΔdivJ::spec cckA21) | 96.3 ± 0 |
| YB3328 (ΔdivJ::spec cckA25) | 97.6 ± 1.4 |
| YB3315 (ΔdivJ::spec divL22) | 99.0 ± 0 |
| YB3317 (ΔdivJ::spec divL33) | 98.1 ± 0.8 |
| YB3319 (ΔdivJ::spec divL35) | 100.5 ± 0 |
The first group of suppressor mutants, YB2761 to YB2763 and YB2766 to YB2768, was isolated from YB1388 (divJ::mini-Tn5 lacZ2), and the second group of suppressors, YB3215 to YB3229, was isolated from YB3202 (ΔdivJ::spec). To eliminate any potential effects of the mini-Tn5 in the suppressors isolated from YB1388 (divJ::mini-Tn5 lacZ2) and to facilitate mapping, we replaced the original divJ::mini-Tn5 lacZ2 with ΔdivJ::spec in the appropriate mutants by transduction, resulting in YB3203, YB3235, YB3236, and YB3238 to YB3240. The phenotypes of the transductants were indistinguishable from those of the original suppressors (data not shown).
Mutations in divL suppress the divJ phenotype.
Mapping of the suppressor mutations in the sdj-22, sdj-33, and sdj-35 suppressors indicated that they were closely linked to divL (see Materials and Methods). Sequencing confirmed the presence of single-base-change mutations in divL in the sdj-22, sdj-33, and sdj-35 suppressors (Fig. 2A). In sdj-22, nucleotide 1259 of the coding sequence was changed from T to C, causing a V420A mutation in the DivL protein. In sdj-33, there was a T-to-G change at position 1560, corresponding to an F514V mutation in the protein. Finally, in sdj-35, nucleotide 1417 was changed from G to C, resulting in a G473R change in the protein. Each mutation was confirmed independently by PCR and sequencing. These mutations occur just upstream of the kinase domain of divL, possibly affecting phosphorylation activity.
FIG. 2.

Suppressor mutations mapping to divL and cckA. (A) Mutations in divL. Each mutation occurs only in the suppressor listed below it. The gray box represents the kinase domain, and Y is the catalytic tyrosine at position 550 in divL. (B) Mutations in cckA. Each mutation occurs only in the suppressor listed below it. The gray box represents the kinase domain, and H is the catalytic histidine at position 322 in cckA. The black box represents the cckA receiver domain, and D is the conserved aspartate at position 623.
We assayed each of the suppressors for several phenotypes (Table 2). sdj-22, sdj-33, and sdj-35 were all more filamentous than the wild type but less filamentous than YB3202 (ΔdivJ::spec). Fewer cells of sdj-22, sdj-33, and sdj-35 had stalks than either NA1000 or YB3202 (ΔdivJ::spec), and the length of the stalks was greater than NA1000 but less than YB3202 (ΔdivJ::spec) (Table 2). sdj-22, sdj-33, and sdj-35 were sensitive to φCbK but were resistant to φCr30. The resistance to φCr30 appeared to be a defect in supporting phage replication, as plaques were not formed, but cells were capable of being infected by phage, since they were able to be transduced. All three suppressors formed swarms similar in size to those of YB3202 (ΔdivJ::spec) (Table 2). Because the suppressors were not significantly filamentous, their swarming deficiency cannot be due to a cell division defect as was the case for the ΔdivJ::spec mutant. Microscopic examination of suppressor cultures revealed that the swarmer cells of these suppressors swam more slowly than the wild-type cells, which accounts for their small swarms in swarm agar.
We confirmed that each mutation in divL was solely responsible for the suppressor phenotype using allelic exchange. The divL alleles of each of the suppressors (sdj-22, sdj-33, and sdj-35) were exchanged in YB3202 (ΔdivJ::spec). In each case, the sdj allele suppressed the divJ phenotype. Therefore, the sdj-22, sdj-33, and sdj-35 alleles will hereafter be referred to as divL22, divL33, and divL35, respectively. The phenotypes of the allelic exchange mutants YB3315 (ΔdivJ::spec divL22), YB3317 (ΔdivJ::spec divL33), and YB3319 (ΔdivJ::spec divL35) were identical to those of the original suppressor mutants described above.
We performed allelic exchange of the divL suppressor alleles in wild-type strain NA1000. The NA1000 divL strains YB3314 (NA1000 divL22), YB3316 (NA1000 divL33), and YB3318 (NA1000 divL35) formed small colonies compared to the wild type. This is especially interesting since, when combined with the ΔdivJ::spec mutation, which also causes slow growth and small colonies, the double divJ divL mutants form large colonies and have growth rates similar to that of the wild type. The sdj divL alleles in the NA1000 background were also more filamentous than NA1000 and the ΔdivJ mutant. The cells had stalk lengths that were approximately the same as those of wild-type cells. Many cells of the strains with sdj divL alleles in the NA1000 background had multiple constrictions. Levels of DivL protein were lower in YB3202 (the ΔdivJ::spec mutant) but were at or slightly above wild-type levels in the suppressor mutants in both the NA1000 and YB3202 (ΔdivJ::spec) backgrounds, as determined by immunoblot (Table 4).
TABLE 4.
Relative levels of DivL and CtrA in the suppressors
| Strain | DivL level (% of wt ± SD)a | CtrA level (% of wt ± SD) |
|---|---|---|
| NA1000 | 100 | 100 |
| YB3202 (ΔdivJ::spec) | 64 ± 10 | 107 ± 10 |
| YB3314 (divJ+divL22) | 123 ± 40 | 100 ± 6 |
| YB3315 (ΔdivJ::spec divL22) | 118 ± 32 | 116 ± 25 |
| YB3316 (divJ+divL33) | 112 ± 25 | 102 ± 19 |
| YB3317 (ΔdivJ::spec divL33) | 110 ± 26 | 112 ± 23 |
| YB3318 (divJ+divL35) | 117 ± 26 | 87 ± 14 |
| YB3319 (ΔdivJ::spec divL35) | 89 ± 17 | 85 ± 17 |
wt, wild type.
The divL mutations confer TS lethality to wild-type cells and to a divJ null mutant.
Because mutations in essential genes often result in a TS phenotype, and a previous divL mutant was TS (52), we examined the effects of temperature on the divL sdj alleles. All three NA1000 sdj divL strains were sensitive to 35°C and 37°C, whereas NA1000 and YB3202 (ΔdivJ::spec) were not (Fig. 3). Two of the ΔdivJ::spec divL strains, YB3315 (ΔdivJ::spec divL22) and YB3317 (ΔdivJ::spec divL33), also showed a sensitivity to temperatures above 30°C. The TS phenotype of divL22 and divL35 was weaker in the YB3202 (ΔdivJ::spec) background than in the wild-type background (Fig. 3). YB3315 (ΔdivJ::spec divL22) exhibited reduced growth at 37°C but not at 35°C. YB3317 (ΔdivJ::spec divL33) was reduced for growth at both 35°C and 37°C, and YB3319 (ΔdivJ::spec divL35) was not sensitive to either temperature (Fig. 3). To examine the morphology of the cells at permissive and nonpermissive temperatures, the NA1000 divL and ΔdivJ::spec divL strains were grown to exponential phase in liquid culture at a permissive temperature (30°C) and shifted to a nonpermissive temperature (37°C) for 4 h. Five strains, YB3314 (NA1000 divL22), YB3315 (ΔdivJ::spec divL22), YB3316 (NA1000 divL33), YB3317 (ΔdivJ::spec divL33), and YB3318 (NA1000 divL35), became more filamentous after the incubation at 37°C, whereas NA1000, YB3202 (ΔdivJ::spec), and YB3319 (ΔdivJ::spec divL35) were not affected (Fig. 4). The most dramatic increase in cell length occurred in YB3316 (NA1000 divL33). The lack of increase in cell length in YB3319 (ΔdivJ::spec divL35) correlates with the robust growth of this strain on plates at 37°C compared to the other strains (Fig. 3 and 4). The fact that the filamentous cells containing the divL sdj alleles had extended areas without constrictions suggests that the early stages of cell division are delayed in these mutants. The cells were also wider than in the YB3202 (ΔdivJ::spec) background. Some cells of NA1000 divL35 were bumpy (Fig. 4).
FIG. 3.

Temperature sensitivity of divJ suppressors. Five microliters of exponential-phase cultures normalized to an OD600 of 0.43 and diluted as shown was spotted onto PYE plates in triplicate (only one set of plates is shown). Plates were incubated for 2 days at the indicated temperatures.
FIG. 4.

Temperature-sensitive phenotypes of divJ suppressors. Phase-contrast micrographs show cells from exponentially growing cultures that were incubated at 30°C or 37°C for 4 h.
Mutations in cckA suppress the divJ phenotype.
Mapping of the suppressor mutations in the sdj-21 and sdj-25 suppressors indicated that they were closely linked to cckA (see Materials and Methods). Sequencing confirmed the presence of single-base-change mutations in cckA in sdj-21 and sdj-25 (Fig. 2B). sdj-21 had a mutation at nucleotide 917 of the coding sequence, with a G-to-A mutation causing a Q306R substitution. In sdj-25, the C at nucleotide 1189 was changed to a T, resulting in an R397C amino acid substitution. Each mutation was confirmed independently by PCR and sequencing. The cckA suppressor alleles (sdj-21 and sdj-25) were exchanged with the wild-type allele in YB3202 (ΔdivJ::spec). In both cases, the sdj allele suppressed the divJ mutant phenotype. The sdj-21 and sdj-25 mutations will hereafter be referred to as cckA21 and cckA25, respectively. The phenotypes of the ΔdivJ::spec cckA allelic exchange mutants, YB3326 (ΔdivJ::spec cckA21) and YB3328 (ΔdivJ::spec cckA25), were identical to those of the original suppressor mutants.
The cckA21 and cckA25 suppressors were different with respect to cell and stalk length (Table 2). The average cell length of the cckA21 suppressor was similar to YB3202 (ΔdivJ::spec), whereas the length of the cckA25 suppressor was only slightly longer than NA1000. The cckA21 suppressor had more cells with stalks than the cckA25 strain, and the stalks were on average longer (Table 2). However, the cckA21 suppressor and the cckA25 suppressor were both sensitive to φCbK and partially sensitive to φCr30 and formed swarms similar in size to YB3202 (ΔdivJ::spec) (Table 2). As with the suppressor mutants in divL, the suppressors in cckA also swam more slowly than the wild type, accounting for their small swarms in 0.3% swarm agar.
The cckA21 and cckA25 alleles were exchanged in NA1000, as described above. YB3325 (NA1000 cckA21) and YB3327 (NA1000 cckA25) formed smaller colonies than the wild type. YB3325 (NA1000 cckA21) was more filamentous than NA1000 and had longer stalks than NA1000 but had fewer of them. YB3327 (NA1000 cckA25) was also more filamentous than NA1000 but had approximately wild-type-length stalks, although it had fewer stalks than NA1000. There were multiple sites of constriction along the cells for both YB3325 (NA1000 cckA21) and YB3327 (NA1000 cckA25), but unlike the NA1000 divL strains, these cells were not bumpy. The NA1000 cckA strains, like the NA1000 divL strains, both formed small colonies compared to those formed by NA1000. As with the divL suppressor alleles, when the cckA suppressor alleles were combined with ΔdivJ::spec, the two mutations, which independently cause small colonies and slow growth, restored wild-type colony size and growth.
The cckA mutations confer TS lethality to wild-type cells.
Because cckA is essential, and a previous cckA mutant was TS, we examined the effects of temperature on the strains with the cckA alleles (Fig. 3). YB3325 (NA1000 cckA21) and YB3327 (NA1000 cckA25) exhibited reduced growth at 35°C and 37°C. Both strains were sensitive to temperatures of 35°C and 37°C, as seen for the 10−5 dilutions. However, the alleles in the YB3202 (ΔdivJ::spec) background were virtually unaffected by temperatures above 30°C (Fig. 3). The growth of the spots of YB3326 (ΔdivJ::spec cckA21) and YB3328 (ΔdivJ::spec cckA25) was not reduced after incubation at 35°C and 37°C and was similar to the growth of spots grown at 30°C (Fig. 3). After exponentially growing cultures of YB3325 (NA1000 cckA21), YB3326 (ΔdivJ::spec cckA21), YB3327 (NA1000 cckA25), and YB3328 (ΔdivJ::spec cckA25) were shifted to 37°C for 4 h, YB3325 (NA1000 cckA21) had increased significantly in length, forming very long filaments with some areas of constriction, while YB3327 (NA1000 cckA25) had increased only slightly in length (Fig. 4). The filamentous cells of YB3325 (NA1000 cckA21) and YB3327 (NA1000 cckA25) with extended areas lacking constrictions suggest a delay in the early stages of cell division. Cells of the NA1000 cckA strains were also wider than wild-type cells. YB3326 (ΔdivJ::spec cckA21) and YB3328 (ΔdivJ::spec cckA25) only increased slightly in length, which correlates with the robust growth on plates at 35°C and 37°C (Fig. 3 and 4). NA1000 and YB3202 (ΔdivJ::spec) were not affected (Fig. 4). The late-cell-division phenotype of both the divL and cckA suppressors led us to examine CtrA activity in the ΔdivJ mutant and the suppressor mutants.
CtrA activity, but not protein level, is reduced in the sdj mutants.
CtrA phosphorylation occurs late in the cell cycle and is required to activate transcription of cell division genes ftsQ and ftsA, which are needed to complete the division process (38, 50). Therefore, we hypothesized that the suppression of the divJ phenotype was due to a lowered CtrA activity. The amount of CtrA, as determined by immunoblot, in the different suppressor mutants (in both the NA1000 and YB3202 [ΔdivJ::spec] backgrounds) did not differ significantly (Table 4). We used pilA-lacZ fusions as a measure of CtrA activity level since the transcription of pilA depends on CtrA (43) (Table 5). pilA-lacZ transcription was higher in YB3202 (ΔdivJ::spec) than in NA1000, suggesting that CtrA activity was higher in the ΔdivJ mutant, since the CtrA concentration was the same as that in the wild type (Table 4). pilA-lacZ transcription was reduced in the divL and cckA sdj mutants, more so in the NA1000 background than in the YB3202 (ΔdivJ::spec) background (Table 5). These results suggest that the phenotype of the ΔdivJ::spec mutant is due at least in part to increased CtrA activity and that the divL and cckA alleles suppress those defects by reducing the activity of CtrA. Since CtrA regulates cell division, and because cells of the suppressor alleles in NA1000 have a late-cell-division phenotype, we next examined the transcription of genes encoding proteins involved in cell division.
TABLE 5.
Relative activity of the pilA promoter in the presence of sdj mutations
| Strain | pilA transcription (% of wt ± SD)a |
|---|---|
| NA1000 | 100 ± 6.13 |
| YB3202 (ΔdivJ) | 128 ± 6.37 |
| YB3325 (divJ+cckA21) | 34 ± 7.73 |
| YB3326 (ΔdivJ::spec cckA21) | 66 ± 3.16 |
| YB3327 (divJ+cckA25) | 56 ± 4.80 |
| YB3328 (ΔdivJ::spec cckA25) | 88 ± 4.35 |
| YB3314 (divJ+divL22) | 44 ± 5.02 |
| YB3315 (ΔdivJ::spec divL22) | 52 ± 11.86 |
| YB3316 (divJ+divL33) | 46 ± 5.42 |
| YB3317 (ΔdivJ::spec divL33) | 47 ± 9.13 |
| YB3318 (divJ+divL35) | 49 ± 12.79 |
| YB3319 (ΔdivJ::spec divL35) | 64 ± 6.79 |
Activity was measured as Miller units and then expressed as a percentage of the wild type (wt), with wild-type activity equal to 100%.
ftsZ and ftsQA promoter activity is altered in the divJ mutant and in divJ suppressor strains.
The cell division phenotype of the divJ null mutant, with cells often containing extended regions without constrictions, suggested that the frequency of cell division initiation was reduced. This is consistent with our model that the divJ null mutant has an increased level of CtrA activity, since CtrA represses ftsZ transcription (22) and activates ftsQA transcription (50). ftsZ is required for the initiation of cell division, whereas ftsQ and ftsA, whose transcription is activated by CtrA, act later in cell division (38). We used β-galactosidase assays to examine the levels of transcription from the ftsZ and ftsQA promoters in the suppressor mutants along with NA1000 and YB3202 (ΔdivJ::spec) (Table 6). As expected, transcription from the ftsZ promoter was only about 60% of that of the wild type in YB3202 (ΔdivJ::spec). Conversely, ftsQA transcription was approximately 20% higher in YB3202 (ΔdivJ::spec) than in the wild type. All five sdj mutants (in both the NA1000 and the ΔdivJ::spec mutant backgrounds) had nearly wild-type levels of ftsZ transcription, while ftsQA transcription was at or below wild-type levels (Table 6). This was true for both the permissive temperature of 30°C and the nonpermissive temperature of 37°C. These results are consistent with the model that the divJ null mutant has an increased level of CtrA activity and that the divL and cckA alleles suppress the divJ phenotype by reducing CtrA activity.
TABLE 6.
ftsZ and ftsQA transcription in the presence of sdj mutations
| Strain | Transcription level (% of wt ± SD)a
|
|||
|---|---|---|---|---|
|
ftsZ
|
ftsQA
|
|||
| 30°C | 37°C | 30°C | 37°C | |
| NA1000 | 100 ± 17.46 | 100 ± 7.68 | 100 ± 2.95 | 100 ± 3.44 |
| YB3202 (ΔdivJ) | 64 ± 4.06 | 61 ± 9.48 | 127 ± 9.59 | 113 ± 5.03 |
| YB3325 (divJ+cckA21) | 96 ± 5.80 | 86 ± 6.03 | 67 ± 1.68 | 45 ± 1.17 |
| YB3326 (ΔdivJ::spec cckA21) | 103 ± 6.00 | 101 ± 2.85 | 112 ± 2.75 | 115 ± 6.13 |
| YB3327 (divJ+cckA25) | 88 ± 4.49 | 92 ± 8.01 | 70 ± 1.60 | 54 ± 7.71 |
| YB3328 (ΔdivJ::spec cckA25) | 84 ± 1.01 | 105 ± 2.80 | 101 ± 2.51 | 100 ± 5.12 |
| YB3314 (divJ+divL22) | 93 ± 2.05 | 85 ± 5.58 | 69 ± 4.14 | 41 ± 11.68 |
| YB3315 (ΔdivJ::spec divL22) | 106 ± 3.91 | 130 ± 5.99 | 102 ± 1.09 | 96 ± 1.95 |
| YB3316 (divJ+divL33) | 92 ± 8.17 | 91 ± 7.82 | 96 ± 6.41 | 55 ± 24.36 |
| YB3317 (ΔdivJ::spec divL33) | 108 ± 3.13 | 113 ± 4.75 | 97 ± 3.80 | 69 ± 19.14 |
| YB3318 (divJ+divL35) | 97 ± 5.49 | 93 ± 2.54 | 104 ± 7.50 | 91 ± 5.07 |
| YB3319 (ΔdivJ::spec divL35) | 105 ± 11.67 | 119 ± 4.02 | 113 ± 22.68 | 112 ± 3.18 |
Activity was measured as Miller units and then expressed as a percentage of the wild type (wt), with wild-type activity equal to 100%.
CtrA phosphorylation is increased in ΔdivJ::spec and reduced in an sdj mutant.
Results from promoter analysis suggested that the ΔdivJ::spec mutant has higher CtrA activity than wild-type cells and that the sdj mutations reduce CtrA activity. To directly test this possibility, we used in vivo phosphorylation assays. Cells were labeled with radioactive inorganic phosphate, followed by immunoprecipitation of CtrA and quantitation of the amount of CtrA∼P by phosphorimaging. The CtrA∼P values were normalized to the abundance of CtrA as determined by Western blot. The results in Fig. 5 show that the ΔdivJ::spec mutant has a higher level of CtrA∼P than wild-type cells. The presence of the divL35 allele in both the NA1000 background and the ΔdivJ::spec background (YB3318 and YB3319, respectively) reduced the amount of CtrA∼P to below that of the wild type (Fig. 5). These results provide direct support for the model that the divJ null mutant phenotype is due, at least in part, to increased CtrA∼P and that the divL alleles suppress the divJ null phenotype by reducing the amount of CtrA∼P. Since the cckA alleles had effects on gene expression similar to those of the divL alleles and had similar cell division phenotypes, we hypothesize that they also suppress the divJ phenotype by the same mechanism.
FIG. 5.

Levels of CtrA∼P relative to CtrA in the divJ null mutant and a divJ suppressor mutant (divL35). Data are averages of three independent experiments, each done in triplicate (nine samples total). The CtrA∼P band intensity was divided by the CtrA band intensity based on loading equal total protein for the in vivo phosphorylation and Western blot.
DISCUSSION
DivJ is involved in the pathway controlling CtrA activity by controlling the level of DivK∼P (15, 49), which in turn controls CtrA degradation (16). The kinase domain of DivJ can phosphorylate DivK in vitro (15), and the amount of DivK∼P in vivo is reduced in a divJ null mutant (49). Additional evidence for DivJ acting upstream of CtrA comes from the ability of the sokA allele of ctrA to suppress the cell division phenotype of a divJ cold-sensitive mutant (51). In this paper, we describe a screen for suppressors of a divJ null mutant that provides in vivo evidence for the role of DivJ in controlling the activity of CtrA. Suppression of a divJ null mutant by alleles of divL and cckA appears to reduce CtrA activity without reducing the cellular level of CtrA. This strongly suggests that at least part of the defect of a divJ null mutant is due to increased CtrA activity. Indeed, DivL can phosphorylate CtrA in vitro (52), and CckA is required for CtrA phosphorylation in vivo (17, 18).
Since DivJ is responsible for the phosphorylation of DivK, eventually leading to the degradation of CtrA∼P (16, 49), the excess active CtrA in the divJ mutant may be the result of inefficient degradation of CtrA∼P. However, this is unlikely, since the cellular level of CtrA is not significantly reduced in the divJ null mutant compared to the wild type. Therefore, it appears that the defect of the divJ null mutant is due in part to an imbalance of CtrA∼P compared to CtrA. The mutations in DivL and CckA are likely to be partial loss-of-function mutations, causing them to function less efficiently as kinases, leading to reduced CtrA∼P. This is supported by the fact that these mutations lead to filamentation of cells when present in an otherwise-wild-type background; this is the phenotype of DivL and CckA loss-of-function mutants (18, 52). Alternatively, the mutations may cause an increase in the phosphatase activity of DivL and CckA, again resulting in reduced CtrA∼P. Thus, the sdj mutations in DivL and CckA compensate for the lack of DivJ by causing a reduction in the levels of CtrA∼P but not a reduction in the overall amount of CtrA. It should be noted that the suppression of the divJ null phenotype is not perfect with respect to stalk presence and length. This could be due to faulty regulation of PleD, which is normally phosphorylated by DivJ (3). PleD controls loss of motility and stalk formation at the swarmer-to-stalked cell transition (45).
We believe that the divJ phenotype (filamentous cells with long, sometimes misplaced stalks) is due in part to an abnormally high level of active CtrA, CtrA∼P, in the cell. This phenotype is similar to that of divK(Cs) at nonpermissive temperatures (15), which results in an excess of CtrA activity as well, in this case by reducing CtrA degradation (16). The high levels of pilA transcription in the ΔdivJ mutant compared to that in the wild type support our hypothesis, since pilA transcription is directly activated by CtrA∼P (43). As we expected, the divL and cckA suppressor mutant alleles reduce pilA transcription in both the wild-type and YB3202 (ΔdivJ::spec) backgrounds. The high level of pilA-lacZ transcription in YB3202 (ΔdivJ::spec) and the reduced levels in the suppressor mutants suggest that the suppression occurs by reducing the amount of active CtrA (CtrA∼P) in the cells and not by altering the cellular level of the protein. Indeed, there is an increase in the amount of CtrA∼P in YB3202 (ΔdivJ::spec) and a reduction of CtrA∼P in YB3318 (NA1000 divL35) and YB3319 (NA1000 ΔdivJ::spec divL35) compared to the wild-type.
The overabundance of active CtrA can also explain the cell division phenotype seen in the divJ null mutant. Active CtrA represses transcription of ftsZ and activates transcription of ftsQ and ftsA from the PQA promoter (50). The ΔdivJ::spec mutant has reduced ftsZ transcription and increased transcription of ftsQA compared to the wild type. Since FtsZ is required for the initiation of cell division (33), and increased FtsA expression inhibits cell division (38), the effect of the divJ mutation on ftsZ and ftsQA transcription is to reduce the frequency of cell division. The divJ suppressors are not as filamentous as the ΔdivJ::spec mutant and grow at rate similar to that of the wild type, consistent with the fact that the sdj suppressors restore ftsZ and ftsQA expression to near-wild-type levels in the ΔdivJ::spec background.
Another explanation for the high level of active CtrA in the ΔdivJ::spec mutant is that DivJ may act as a phosphatase for CtrA∼P, in addition to its role as a kinase for DivK and PleD. In a divJ null mutant, levels of CtrA∼P would rise, resulting in the filamentous phenotype of the ΔdivJ::spec mutant. Mutations in DivL and CckA that reduce their effectiveness in phosphorylating CtrA would compensate for the loss of DivJ phosphatase activity by reducing the amount of CtrA∼P made in the first place.
In conclusion, our results support the hypothesis that the phenotype of divJ null mutants is the result of excess active CtrA. Mutations in DivL and CckA reduce the activity of these kinases in phosphorylating CtrA or increase their activity in dephosphorylating CtrA, thereby suppressing the loss of DivJ. Future work on the DivL mutant proteins will help determine the exact nature of this suppression.
Acknowledgments
This work was supported by NIH predoctoral training grant GM07757 to D.L.P. and NIH grant GM51986 to Y.V.B.
We thank A. Newton for the DivL overexpression construct for antibody production, C. Stephens for marker strains, K. Ryan and C. Jacobs-Wagner for advice on in vivo phosphorylation, and members of our laboratory for helpful discussions and critical reading of the manuscript.
REFERENCES
- 1.Ackermann, M., S. C. Stearns, and U. Jenal. 2003. Senescence in a bacterium with asymmetric division. Science 300:1920. [DOI] [PubMed] [Google Scholar]
- 2.Agabian-Keshishian, N., and L. Shapiro. 1970. Stalked bacteria: properties of deoxyribonucleic acid bacteriophage φCBK. J. Virol. 5:795-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldridge, P., R. Paul, P. Goymer, P. Rainey, and U. Jenal. 2003. Role of the GGDEF regulator PleD in polar development of Caulobacter crescentus. Mol. Microbiol. 47:1695-1708. [DOI] [PubMed] [Google Scholar]
- 4.Alley, M. R. K. Unpublished data.
- 5.Alley, M. R. K., S. L. Gomes, W. Alexander, and L. Shapiro. 1991. Genetic analysis of a temporally transcribed chemotaxis gene cluster in Caulobacter crescentus. Genetics 129:333-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brun, Y. V., and L. Shapiro. 1992. A temporally controlled sigma factor is required for cell-cycle dependent polar morphogenesis in Caulobacter. Genes Dev. 6:2395-2408. [DOI] [PubMed] [Google Scholar]
- 7.Burton, G. J., G. B. Hecht, and A. Newton. 1997. Roles of the histidine protein kinase PleC in Caulobacter crescentus motility and chemotaxis. J. Bacteriol. 179:5849-5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Din, N., E. M. Quardokus, M. J. Sackett, and Y. V. Brun. 1998. Dominant C-terminal deletions of FtsZ that affect its ability to localize in Caulobacter and its interaction with FtsA. Mol. Microbiol. 27:1051-1064. [DOI] [PubMed] [Google Scholar]
- 9.Domian, I. J., K. C. Quon, and L. Shapiro. 1997. Cell type-specific phosphorylation and proteolysis of a transcriptional regulator controls the G1-to-S transition in a bacterial cell cycle. Cell 90:415-424. [DOI] [PubMed] [Google Scholar]
- 10.Ely, B. 1991. Genetics of Caulobacter crescentus. Methods Enzymol. 204:372-384. [DOI] [PubMed] [Google Scholar]
- 11.Ely, B., and R. C. Johnson. 1977. Generalized transduction in Caulobacter crescentus. Genetics 87:391-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evinger, M., and N. Agabian. 1977. Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. J. Bacteriol. 132:294-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonin, M., E. M. Quardokus, D. O'Donnol, J. Maddock, and Y. V. Brun. 2000. Regulation of stalk elongation by phosphate in Caulobacter crescentus. J. Bacteriol. 182:337-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harlow, E., and D. Lane. 1988. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 15.Hecht, G. B., T. Lane, N. Ohta, J. M. Sommer, and A. Newton. 1995. An essential single domain response regulator required for normal cell division and differentiation in Caulobacter crescentus. EMBO J. 14:3915-3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hung, D. Y., and L. Shapiro. 2002. A signal transduction protein cues proteolytic events critical to Caulobacter cell cycle progression. Proc. Natl. Acad. Sci. USA 99:13160-13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobs, C., N. Ausmees, S. J. Cordwell, L. Shapiro, and M. Laub. 2003. Functions of the CckA histidine kinase in Caulobacter cell cycle control. Mol. Microbiol. 47:1279-1290. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs, C., I. J. Domian, J. R. Maddock, and L. Shapiro. 1999. Cell cycle-dependent polar localization of an essential bacterial histidine kinase that controls DNA replication and cell division. Cell 97:111-120. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs, C., D. Hung, and L. Shapiro. 2001. Dynamic localization of a cytoplasmic signal transduction response regulator controls morphogenesis during the Caulobacter cell cycle. Proc. Natl. Acad. Sci. USA 98:4095-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janakiraman, R. S., and Y. V. Brun. 1999. Cell cycle control of a holdfast attachment gene in Caulobacter. J. Bacteriol. 181:1118-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenal, U., and T. Fuchs. 1998. An essential protease involved in bacterial cell-cycle control. EMBO J. 17:5658-5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly, A. J., M. J. Sackett, N. Din, E. M. Quardokus, and Y. V. Brun. 1998. Cell cycle-dependent transcriptional and proteolytic regulation of FtsZ in Caulobacter. Genes Dev. 12:880-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 24.Lam, H., J. Y. Matroule, and C. Jacobs-Wagner. 2003. The asymmetric spatial distribution of bacterial signal transduction proteins coordinates cell cycle events. Dev. Cell 5:149-159. [DOI] [PubMed] [Google Scholar]
- 25.Laub, M. T., S. L. Chen, L. Shapiro, and H. H. McAdams. 2002. Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc. Natl. Acad. Sci. USA 99:4632-4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liss, L. R. 1987. New M13 host: DH5αF′ competent cells. Focus 9:3, 13. [Google Scholar]
- 27.Lowry, O. H., N. R. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement using the folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 28.Matroule, J. Y., H. Lam, D. T. Burnette, and C. Jacobs-Wagner. 2004. Cytokinesis monitoring during development; rapid pole-to-pole shuttling of a signaling protein by localized kinase and phosphatase in Caulobacter. Cell 118:579-590. [DOI] [PubMed] [Google Scholar]
- 29.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 30.Ohta, N., and A. Newton. 2003. The core dimerization domains of histidine kinases contain recognition specificity for the cognate response regulator. J. Bacteriol. 185:4424-4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poindexter, J. S. 1964. Biological properties and classification of the Caulobacter group. Bacteriol. Rev. 28:231-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prentki, P., and H. M. Krisch. 1984. In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303-313. [DOI] [PubMed] [Google Scholar]
- 33.Quardokus, E. M., N. Din, and Y. V. Brun. 2001. Cell cycle and positional constraints on FtsZ localization and the initiation of cell division in Caulobacter crescentus. Mol. Microbiol. 39:949-959. [DOI] [PubMed] [Google Scholar]
- 34.Quon, K. C., G. T. Marczynski, and L. Shapiro. 1996. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell 84:83-93. [DOI] [PubMed] [Google Scholar]
- 35.Quon, K. C., B. Yang, I. J. Domian, L. Shapiro, and G. T. Marczynski. 1998. Negative control of bacterial DNA replication by a cell cycle regulatory protein that binds at the chromosome origin. Proc. Natl. Acad. Sci. USA 95:120-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts, R. C., C. Toochinda, M. Avedissian, R. L. Baldini, S. L. Gomes, and L. Shapiro. 1996. Identification of a Caulobacter crescentus operon encoding hrcA, involved in negatively regulating heat-inducible transcription, and the chaperone gene grpE. J. Bacteriol. 178:1829-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryan, K. R., E. M. Judd, and L. Shapiro. 2002. The CtrA response regulator essential for Caulobacter crescentus cell-cycle progression requires a bipartite degradation signal for temporally controlled proteolysis. J. Mol. Biol. 324:443-455. [DOI] [PubMed] [Google Scholar]
- 38.Sackett, M. J., A. J. Kelly, and Y. V. Brun. 1998. Ordered expression of ftsQA and ftsZ during the Caulobacter crescentus cell cycle. Mol. Microbiol. 28:421-434. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 40.Sciochetti, S. A., T. Lane, N. Ohta, and A. Newton. 2002. Protein sequences and cellular factors required for polar localization of a histidine kinase in Caulobacter crescentus. J. Bacteriol. 184:6037-6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sciochetti, S. A., N. Ohta, and A. Newton. 2005. The role of polar localization in the function of an essential Caulobacter crescentus tyrosine kinase. Mol. Microbiol. 56:1467-1480. [DOI] [PubMed] [Google Scholar]
- 42.Simon, R., U. Prieffer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Biotechnology 1:784-790. [Google Scholar]
- 43.Skerker, J. M., and L. Shapiro. 2000. Identification and cell cycle control of a novel pilus system in Caulobacter crescentus. EMBO J. 19:3223-3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sommer, J. M., and A. Newton. 1991. Pseudoreversion analysis indicates a direct role of cell division genes in polar morphogenesis and differentiation in Caulobacter crescentus. Genetics 129:623-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sommer, J. M., and A. Newton. 1989. Turning off flagellum rotation requires the pleiotropic gene pleD: pleA, pleC, and pleD define two morphogenic pathways in Caulobacter crescentus. J. Bacteriol. 171:392-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spratt, B. G., P. J. Hedge, S. te Heesen, A. Edelman, and J. K. Broome-Smith. 1986. Kanamycin-resistant vectors that are analogues of plasmids pUC8, pUC9, pEMBL8 and pEMBL9. Gene 41:337-342. [DOI] [PubMed] [Google Scholar]
- 47.Stephens, C., A. Reisenauer, R. Wright, and L. Shapiro. 1996. A cell cycle-regulated bacterial DNA methyltransferase is essential for viability. Proc. Natl. Acad. Sci. USA 93:1210-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.West, L., D. Yang, and C. Stephens. 2002. Use of the Caulobacter crescentus genome sequence to develop a method for systematic genetic mapping. J. Bacteriol. 184:2155-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheeler, R. T., and L. Shapiro. 1999. Differential localization of two histidine kinases controlling bacterial cell differentiation. Mol. Cell 4:683-694. [DOI] [PubMed] [Google Scholar]
- 50.Wortinger, M., M. Sackett, and Y. Brun. 2000. CtrA mediates a DNA replication checkpoint that prevents cell division in Caulobacter crescentus. EMBO J. 19:4503-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu, J., N. Ohta, and A. Newton. 1998. An essential, multicomponent signal transduction pathway required for cell cycle regulation in Caulobacter. Proc. Natl. Acad. Sci. USA 95:1443-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu, J., N. Ohta, J. L. Zhao, and A. Newton. 1999. A novel bacterial tyrosine kinase essential for cell division and differentiation. Proc. Natl. Acad. Sci. USA 96:13068-13073. [DOI] [PMC free article] [PubMed] [Google Scholar]