

Abstract
Antibiotics that interfere with DNA replication and cell viability activate the SOS response. In Staphylococcus aureus, the antibiotic-induced SOS response promotes replication and high-frequency horizontal transfer of pathogenicity island-encoded virulence factors. Here we report that β-lactams induce a bona fide SOS response in S. aureus, characterized by the activation of the RecA and LexA proteins, the two master regulators of the SOS response. Moreover, we show that β-lactams are capable of triggering staphylococcal prophage induction in S. aureus lysogens. Consequently, and as previously described for SOS induction by commonly used fluoroquinolone antibiotics, β-lactam-mediated phage induction also resulted in replication and high-frequency transfer of the staphylococcal pathogenicity islands, showing that such antibiotics may have the unintended consequence of promoting the spread of bacterial virulence factors.
Previous studies have revealed that certain genes of Staphylococcus aureus, including those encoding proteins involved in cell wall metabolism and stress responses, are upregulated after treatment with β-lactam antibiotics (5, 15, 18), suggesting the existence of a cell wall stimulon induced in response to cell wall-active agents. In addition, it is well documented that in Escherichia coli, the SOS response is induced by antibiotics that interfere with cell wall synthesis (8, 12) as well as DNA replication (13). The SOS system represents a global response to DNA damage that upregulates genes involved in DNA repair and cell survival (2, 4). The SOS response is governed by the LexA and RecA proteins. The LexA protein binds to operator sites of SOS-regulated genes, effectively repressing their expression. Conversely, the presence of DNA lesions activates RecA, which promotes the autocatalytic cleavage of LexA at a specific Ala-Gly bond (7). Cleaved LexA is unable to bind DNA, leading to the derepression of SOS genes. Once DNA damage has been addressed, newly synthesized RecA and LexA restore repression to the system. Furthermore, the SOS response has been shown to induce the lateral transfer of antibiotic resistance encoded by the Vibrio cholerae integrating conjugative element SXT (1), of pathogenicity island-encoded virulence factors in staphylococci (16), and of prophage-encoded Shiga toxin in E. coli (19).
In this study we investigated the influence of subinhibitory concentrations of different antibiotics, including β-lactams (ampicillin, penicillin, ceftriaxone, and cloxacillin), macrolide-lincosamide-streptogramin B antibiotics (erythromycin), aminoglycosides (kanamycin), chloramphenicol, and tetracycline, on the replication and transfer of superantigen-carrying staphylococcal pathogenicity islands (SaPIs). In S. aureus, several related pathogenicity islands have been described, including SaPI1 to SaPI4, SaPbov1, SaPIbov2, and SaPIn1 to SaPIn3 (for reviews see references 10 and 11). These elements most commonly encode TSST-1 plus two or more other superantigen toxins, with the exception of SaPIbov2, which encodes the biofilm-associated protein Bap (17). Phage-assisted replication, transduction, and site-specific integration in a recA mutant strain demonstrated the mobility of SaPI1 (6, 14) and SaPIbov1 (16). Additionally, we demonstrated that the fluoroquinolone-induced SOS response is fully effective for the mobilization of SaPIbov1 and SaPI1 and, by implication, for that of all other SOS-induced SaPIs (16). In view of these results and of the results of Cohen and coworkers (8), we analyzed the possibility that other antibiotics used in clinical practice could also induce the SOS response, resulting in the dissemination of virulence factors in staphylococci. The results of our study show that β-lactams induce SaPI replication and transfer in an SOS-dependent manner and imply that they, as well as other SOS-inducing antibiotics, could thus increase microbial strain diversification and promote the spread of temperate phages and phage-inducible pathogenicity islands, resulting in the dissemination of superantigens and other virulence factors.
In the experiments presented here, we employed S. aureus strains RN27 and RN451, carrying SOS-inducible prophages 80α and φ11, respectively (9). Induction of the SOS response by any of the antibiotics analyzed would be expected to induce phage replication in these strains. Bacteria grown in Trypticase soy broth to an optical density at 540 nm of 0.4 were tested for prophage induction by the addition of subinhibitory concentrations (ranging from 0.05 μg/ml to 10 μg/ml) of ampicillin, penicillin, erythromycin, chloramphenicol, tetracycline, or kanamycin. Cultures were grown at 32°C with slow shaking (80 rpm). After 16 h, phage titers were determined by plating suitable dilutions on RN4220. Phage replication was stimulated by exposure of bacteria to ampicillin and penicillin (Table 1), although the phage titers were lower than those with mitomycin C induction (Table 1). In contrast, none of the non-β-lactam antibiotics tested induced phage replication (data not shown). Since the β-lactam antibiotics (ampicillin and penicillin) utilized have low clinical relevance in the treatment of staphylococcal infections, we repeated the phage inductions with the β-lactam antibiotics ceftriaxone and cloxacillin, which are extensively used in the treatment of staphylococcal infections. As shown in Table 1, exposure of bacteria to ceftriaxone and cloxacillin also increased the phage titers.
TABLE 1.
Phage titers of β-lactam-induced lysogenic staphylococcal strainsa
| Donor strain | Phage | Inducerb | Phage titerc |
|---|---|---|---|
| RN27 | φ80α | MC | 3.4 × 1010 |
| A | 2.4 × 108 | ||
| P | 7.3 × 108 | ||
| CL | 2.4 × 108 | ||
| CE | 3.2 × 108 | ||
| NI | 8.0 × 105 | ||
| RN451 | φ11 | MC | 3.6 × 109 |
| A | 3.6 × 106 | ||
| P | 2.8 × 106 | ||
| CL | 2.3 × 106 | ||
| CE | 3.0 × 106 | ||
| NI | 1.4 × 105 | ||
| RN1030 (recA mutant) | φ11 | MC | <10 |
| A | <10 | ||
| P | <10 | ||
| NI | <10 | ||
| JP83 [RN27 LexA (G94E)] | φ80α | A | 4.5 × 106 |
| NI | 4.0 × 105 | ||
| JP84 [RN451 LexA (G94E)] | φ11 | A | 1.3 × 106 |
| NI | 3.0 × 104 |
Results from a representative experiment are shown.
MC, mitomycin C (2 μg/ml); A, ampicillin (10 μg/ml); P, penicillin G (10 μg/ml); CL, cloxacillin (10 μg/ml); CE, ceftriaxone (10 μg/ml); NI, not induced.
Number of plaque-forming phages per milliliter of induced culture, using RN4220 as the indicator strain.
To determine whether ampicillin-mediated phage induction was SOS dependent, we measured the phage titers after antibiotic treatment of strain RN1030, a recA-defective strain lysogenic for φ11 (9). As is evident in Table 1, no induction was observed in the presence of the antibiotic.
To resolve the basis of ampicillin-mediated SOS induction further, we investigated the effect of ampicillin on expression of lexA. Thus, reverse transciption-PCR analysis using internal oligonucleotides specific to the S. aureus lexA gene were carried out as previously reported (3). As shown in Fig. 1, our experiments showed that the presence of ampicillin produces an increase of approximately 19-fold in S. aureus lexA expression from strain RN450 (9), while expression of the gene in its derivative recA mutant strain RN981 (9) was not affected, indicating that this β-lactam induces the SOS response.
FIG. 1.
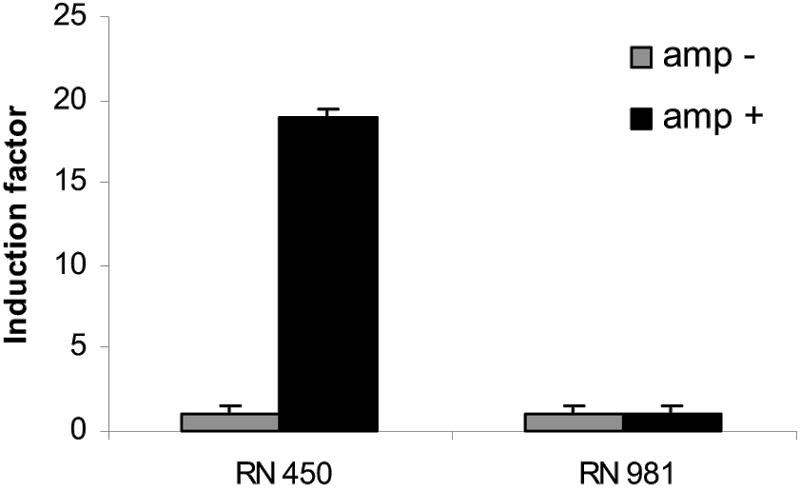
Ampicillin-mediated induction factor of the lexA gene in S. aureus RN450 or its recA-mutant strain RN981. The induction factor was measured by quantitative reverse transciption-PCR, and in all cases, it is the ratio of the relative lexA mRNA concentration in ampicillin-treated cells to that in untreated cells. The relative lexA mRNA concentration was calculated as described previously (3). Values were calculated 5 h after ampicillin addition. In each case, the mean value from three independent experiments (each in triplicate) is shown. Amp+, addition of ampicillin at 0.2 μg/ml; Amp−, no ampicillin added.
This conclusion was additionally confirmed by mutational inactivation of the LexA protein and testing for phage induction. The substitution of glutamate for glycine at the Ala-Gly RecA cleavage site in the LexA protein results in a noncleavable repressor that was predicted to be less sensitive to SOS induction (7). The noncleavability mutation was introduced into lexA in strains RN27 and RN451, using oligonucleotides lexA-1mB (5′-CGCGGATCCGGCTGTTTGCTCCTTTGCTTCTTC-3′), lexA-2c (5′-CTCAGCCATTAATGAATTCTATTGGTC-3′), lexA-3m (5′-GGTAAAGTCACAGCTGAGGTTCCTATTACCGC-3′), and lexA-4c (5′-GCGGTAATAGGAACCTCAGCTGTGACTTTACC-3′), as previously described (16). The resulting strains, JP83 and JP84, respectively, each encoded noncleavable LexA [LexA (G94E)]. Strain JP83 [RN27 LexA (G94E)] showed diminished phage titers upon ampicillin treatment in comparison to those induced by ampicillin in strain RN27, confirming the role of LexA in the ampicillin-mediated SOS response. Surprisingly, although ampicillin induced φ11, similar phage titers were obtained upon ampicillin treatment of strains JP84 [RN451 LexA (G94E)] and RN451 (Table 1), suggesting the existence of a LexA-independent pathway in ampicillin-mediated SOS induction of φ11.
We have previously shown that mitomycin C induction of the SOS response in lysogenic SaPI1- and SaPIbov1-containing S. aureus induced replication and high-frequency transduction of the island (6, 14, 16). Similarly, growth of lysogenic SaPI1 or SaPIbov1 donor cells in ciprofloxacin, a widely used fluoroquinolone antibiotic that activates the SOS response (13), also increased SaPI1 and SaPIbov1 replication and transfer (16). To determine whether the β-lactam-mediated SOS response could induce SaPIbov1, strains JP44 (RN27 SaPIbov1 tst::tetM [16]) and JP47 (RN451 SaPIbov1 tst::tetM [16]) were grown with ampicillin, penicillin, ceftriaxone, or cloxacillin (10 μg/ml); as shown in Table 2, all the β-lactam antibiotics analyzed also increased SaPIbov1 replication and transfer.
TABLE 2.
Transduction frequencies of β-lactam-induced SaPI-containing lysogenic strainsa
| Donor strain | Phage | SaPI | Inducerb | Transduction titerc |
|---|---|---|---|---|
| JP44 | φ80α | SaPIbov1 | MC | 7.2 × 109 |
| A | 2.7 × 107 | |||
| P | 3.0 × 107 | |||
| CL | 1.6 × 107 | |||
| CE | 3.2 × 107 | |||
| NI | 8.1 × 104 | |||
| JP47 | φ11 | SaPIbov1 | MC | 2.1 × 106 |
| A | 6.0 × 104 | |||
| P | 5.8 × 104 | |||
| CL | 4.4 × 104 | |||
| CE | 5.6 × 104 | |||
| NI | 1.7 × 103 | |||
| JP50 (recA mutant) | φ11 | SaPIbov1 | MC | <10 |
| A | <10 | |||
| NI | <10 | |||
| JP85 [JP83 LexA (G94E)] | φ80α | SaPIbov1 | MC | 5 × 105 |
| A | 4 × 104 | |||
| NI | 2.3 × 103 | |||
| JP52 | φ11 cIG131E | SaPIbov1 | MC | <10 |
| A | <10 | |||
| NI | <10 | |||
| RN8667 | φ80α | SaPI1 | A | 2.1 × 105 |
| P | 1.6 × 105 | |||
| CL | 2.2 × 105 | |||
| CE | 2.9 × 105 | |||
| NI | 6.0 × 103 |
Results from a representative experiment are shown.
MC, mitomycin C (2 μg/ml); A, ampicillin (10 μg/ml); P, penicillin G (10 μg/ml); CL, cloxacillin (10 μg/ml); CE, ceftriaxone (10 μg/ml); NI, not induced.
Number of transductants per milliliter of induced culture, using RN4220 as the recipient strain.
To confirm that the stimulation of SaPIbov1 transfer by ampicillin was a consequence of SOS induction, we induced JP50 (a recA mutant S. aureus strain, a derivative of RN1030, lysogenic for φ11, that carries SaPIbov1 tst::tetM [16]) and JP85 (a derivative of JP83 that carries SaPIbov1 tst::tetM). As expected, neither replication nor significant transfer was observed upon ampicillin induction of strain JP50 (Table 2), while a significant reduction in SaPIbov1 transfer was observed after ampicillin induction of strain JP85. Additionally, the role of ampicillin in the SOS-mediated transfer of SaPIbov1 was confirmed by analysis of strain JP52, a SaPIbov1-positive strain, lysogenic for φ11, that carries a mutation eliminating the φ11 phage repressor (cI) cleavage site (16). Ampicillin did not increase SaPIbov1 transfer in this strain (Table 2), suggesting that cI cleavage mediates the SOS enhancement of SaPIbov1 transfer.
Finally, we addressed the question of whether SaPI1, the prototypical S. aureus pathogenicity island, could also be excised, replicated, and transferred at a high frequency after antibiotic treatment. For that purpose, RN8667 (SaPI1 positive, lysogenic for φ80α [6]) was grown with ampicillin, penicillin, ceftriaxone, or cloxacillin, as described above. As expected, the β-lactam-activated SOS response increased SaPI1 replication and transfer (Table 2). Thus, activation of the SOS response by β-lactam antibiotics greatly stimulates the transfer of S. aureus pathogenicity islands.
Recently, it has been reported that β-lactam antibiotics induce the SOS response in E. coli through the DpiBA two-component signal transduction system (8). This event, which requires the SOS-promoted cleavage of RecA and LexA as well as dpiA, transiently halts bacterial cell division, enabling the organisms to survive otherwise lethal antibiotic exposure. In addition, it has been reported that transcription of the SOS-regulated dinB gene, encoding DNA polymerase IV, is induced by inhibition of cell wall synthesis by β-lactam antibiotics (12). Whether corresponding genes are involved in SOS induction by β-lactams in S. aureus remains to be determined. Nevertheless, our findings indicate that β-lactam antibiotics are extracellular stimuli of the SOS response in S. aureus as well as in E. coli and demonstrate a novel mechanism for horizontal dissemination of staphylococcal virulence factors. Thus, our results, even though anticipated, reinforce the need for great caution in the use of SOS response-inducing antibiotics. Such antibiotics not only promote the dissemination of antibiotic resistance genes and the production of toxins regulated by repressors sensitive to RecA cleavage but also promote the induction of prophages and SaPIs, staphylococcal elements that frequently encode virulence factors.
Acknowledgments
This work was supported by grants BIO2002-04542-C02-01 and BIO2005-08399-C02-02 from the Comisión Interministerial de Ciencia y Tecnología (C.I.C.Y.T.), grants from the Cardenal Herrera-CEU University, the Conselleria de Agricultura, Pesca i Alimentació (CAPiA), and the Generalitat Valenciana (CTIDIA/2002/62, CTESPP/2003/027, and AE04-8) to J.R.P., and a grant from the MEC (AGL2005-03574/GAN) to J.B. Fellowship support for C.U. from CAPiA and for E.M. from the Cardenal Herrera-CEU University is gratefully acknowledged.
REFERENCES
- 1.Beaber, J. W., B. Hochhut, and M. K. Waldor. 2004. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427:72-74. [DOI] [PubMed] [Google Scholar]
- 2.Courcelle, J., A. Khodursky, B. Peter, P. O. Brown, and P. C. Hanawalt. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dinamarca, M. A., I. Aranda-Olmedo, A. Puyet, and F. Rojo. 2003. Expression of the Pseudomonas putida OCT plasmid alkane degradation pathway is modulated by two different global control signals: evidence from continuous cultures. J. Bacteriol. 185:4772-4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandez De Henestrosa, A. R., T. Ogi, S. Aoyagi, D. Chafin, J. J. Hayes, H. Ohmori, and R. Woodgate. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 35:1560-1572. [DOI] [PubMed] [Google Scholar]
- 5.Kuroda, M., H. Kuroda, T. Oshima, F. Takeuchi, H. Mori, and K. Hiramatsu. 2003. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49:807-821. [DOI] [PubMed] [Google Scholar]
- 6.Lindsay, J. A., A. Ruzin, H. F. Ross, N. Kurepina, and R. P. Novick. 1998. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 29:527-543. [DOI] [PubMed] [Google Scholar]
- 7.Little, J. W. 1993. LexA cleavage and other self-processing reactions. J. Bacteriol. 175:4943-4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller, C., L. E. Thomsen, C. Gaggero, R. Mosseri, H. Ingmer, and S. N. Cohen. 2004. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 305:1629-1631. [DOI] [PubMed] [Google Scholar]
- 9.Novick, R. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155-166. [DOI] [PubMed] [Google Scholar]
- 10.Novick, R. P. 2003. Mobile genetic elements and bacterial toxinoses: the superantigen-encoding pathogenicity islands of Staphylococcus aureus. Plasmid 49:93-105. [DOI] [PubMed] [Google Scholar]
- 11.Novick, R. P., P. Schlievert, and A. Ruzin. 2001. Pathogenicity and resistance islands of staphylococci. Microbes Infect. 3:585-594. [DOI] [PubMed] [Google Scholar]
- 12.Perez-Capilla, T., M. R. Baquero, J. M. Gomez-Gomez, A. Ionel, S. Martin, and J. Blazquez. 2005. SOS-independent induction of dinB transcription by β-lactam-mediated inhibition of cell wall synthesis in Escherichia coli. J. Bacteriol. 187:1515-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillips, I., E. Culebras, F. Moreno, and F. Baquero. 1987. Induction of the SOS response by new 4-quinolones. J. Antimicrob. Chemother. 20:631-638. [DOI] [PubMed] [Google Scholar]
- 14.Ruzin, A., J. Lindsay, and R. P. Novick. 2001. Molecular genetics of SaPI1—a mobile pathogenicity island in Staphylococcus aureus. Mol. Microbiol. 41:365-377. [DOI] [PubMed] [Google Scholar]
- 15.Singh, V. K., R. K. Jayaswal, and B. J. Wilkinson. 2001. Cell wall-active antibiotic induced proteins of Staphylococcus aureus identified using a proteomic approach. FEMS Microbiol. Lett. 199:79-84. [DOI] [PubMed] [Google Scholar]
- 16.Ubeda, C., E. Maiques, E. Knecht, I. Lasa, R. P. Novick, and J. R. Penades. 2005. Antibiotic-induced SOS response promotes horizontal dissemination of pathogenicity island-encoded virulence factors in staphylococci. Mol. Microbiol. 56:836-844. [DOI] [PubMed] [Google Scholar]
- 17.Ubeda, C., M. A. Tormo, C. Cucarella, P. Trotonda, T. J. Foster, I. Lasa, and J. R. Penades. 2003. Sip, an integrase protein with excision, circularization and integration activities, defines a new family of mobile Staphylococcus aureus pathogenicity islands. Mol. Microbiol. 49:193-210. [DOI] [PubMed] [Google Scholar]
- 18.Utaida, S., P. M. Dunman, D. Macapagal, E. Murphy, S. J. Projan, V. K. Singh, R. K. Jayaswal, and B. J. Wilkinson. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149:2719-2732. [DOI] [PubMed] [Google Scholar]
- 19.Zhang, X., A. D. McDaniel, L. E. Wolf, G. T. Keusch, M. K. Waldor, and D. W. Acheson. 2000. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J. Infect. Dis. 181:664-670. [DOI] [PubMed] [Google Scholar]