

Abstract
Prokaryotic genomes acquire and eliminate blocks of DNA sequence by lateral gene transfer and spontaneous deletion, respectively. The basic parameters of spontaneous deletion, which are expected to influence the course of genome evolution, have not been determined for any hyperthermophilic archaeon. We therefore screened a number of independent pyrimidine auxotrophs of Sulfolobus acidocaldarius for deletions and sequenced those detected. Deletions accounted for only 0.4% of spontaneous pyrE mutations, corresponding to a frequency of about 10−8 per cell. Nucleotide sequence analysis of five independent deletions showed no significant association of the endpoints with short direct repeats, despite the fact that several such repeats occur within the pyrE gene and that duplication mutations in pyrE reverted at high frequencies. Endpoints of the spontaneous deletions did not coincide with short inverted repeats or potential stem-loop structures. No consensus sequence common to all the deletions could be identified, although two deletions showed the potential of being stabilized by octanucleotide sequences elsewhere in pyrE, and another pair of deletions shared an octanucleotide at their 3′ ends. The unusually low frequency and low sequence dependence of spontaneous deletions in the S. acidocaldarius pyrE gene compared to other genetic systems could not be explained in terms of possible constraints imposed by the 5-fluoroorotate selection.
Sequence analysis of entire genomes has made it clear that despite the gradual accumulation of nucleotide substitutions, much of the divergence among prokaryotic species occurs through acquisition and loss of blocks of DNA sequences. Although bacterial genomes carry significant amounts of sequences originating in other lineages, related species exhibit similar genome sizes (21, 22), implying a general balance of losses against gains. Notable exceptions to this size constancy occur in species that have adopted a parasitic life strategy; the genomes of these species are either small or rapidly shrinking, reflecting the elimination of many genes whose functions have become superfluous (1, 6, 24). The mechanistic basis of genome reduction appears to be a short-term capacity to delete DNA that exceeds the capacity to acquire it through lateral gene transfer. This kinetic bias in favor of deletion implies that the maintenance of genome size seen in most bacterial lineages reflects selection for function of the vast majority of genes in the genome (24). It also explains the genome features observed in most free-living prokaryotes, including compactness (i.e., high open reading frame [ORF] density) and a dearth of full-length but nonfunctional ORFs (1, 24).
Data from genomes of hyperthermophilic archaea suggest a similar interplay among DNA acquisition, DNA loss, and selection for function. For example, Pyrococcus species can acquire large blocks of DNA by lateral transfer (9), yet they have also maintained relatively constant genome sizes during speciation (35). However, these and other archaea from geothermal biotopes have extreme evolutionary divergence from the organisms in which most DNA transactions have been analyzed at the molecular level, and they grow optimally at temperatures which destabilize the primary and secondary structure of DNA. Thus, the frequency with which deletions occur in nonessential sequences, the average size of individual deletions, and the influence of DNA sequences on the positioning of endpoints cannot be assumed to correspond to those of the bacterial or eukaryotic systems in which the process of spontaneous deletion has been analyzed. Since these parameters should influence the rate and course of DNA removal, determining them in hyperthermophilic archaea represents an important step in understanding the evolutionary dynamics of their genomes.
In this study, we combined genetic selection and sequence analysis to determine the frequency and molecular nature of spontaneous deletions in the chromosome of Sulfolobus acidocaldarius, a crenarchaeote that populates acidic hot springs and grows optimally at about 80°C and pH 3. To our knowledge, this is the first quantitative molecular analysis of deletion formation in any archaeon or hyperthermophile. In contrast to the bacterial and eukaryotic target systems that have been similarly examined, spontaneous deletion events in the S. acidocaldarius pyrE gene were infrequent and not directed by repeated sequences at their ends.
MATERIALS AND METHODS
Recovery of deletions.
A total of 155 independent 5-fluoroorotic acid (FOA)-resistant (Foar) mutants of wild-type S. acidocaldarius (strain DG185) were selected, clonally purified, and preserved as previously described (15, 28). After the strains were screened to eliminate those exhibiting reversion, the pyrE coding region was amplified from 84 mutants by PCR (28). The PCR products were first screened by electrophoresis in 1.5% agarose gels; any that appeared to be smaller than normal were then digested with TaqI endonuclease and resolved on a nondenaturing 10% polyacrylamide gel. New product was amplified from each of the resulting candidates and subjected to dye terminator sequencing on both strands, yielding two confirmed deletions (one each in strains MR123 and MR311). Other deletions were previously identified in strains MR31 and MR103 following detection of unusual recombinational properties (28), and in strain JDS22 by direct (brute-force) sequencing of a collection of over 100 spontaneous mutants (15).
The frequency of deletion formation in the wild-type pyrE gene was calculated from the isolations that yielded strains MR123, MR311, and JDS22. The calculations had to account for the fact that about 60% of all spontaneous Foar mutants have a leaky phenotype. This class of mutants was included in a set of 79 strains that yielded JDS22 (15) but had been purged during assembly of the remaining strains (284 strains). Therefore, deletion mutants MR123, MR311, and JDS22 represent the results of screening a total of 79 + (284/0.40), or approximately 790, independent Foar mutants initially isolated.
Genetic assays.
The frequency of Foar cells was determined by plating aliquots of 113 small liquid cultures (less than 108 cells each) on selective medium and by plating appropriate dilutions of the cultures on nonselective (xylose-tryptone-uracil) medium to enumerate viable cells. The average frequency of mutants under these conditions was 2.5 × 10−6 per viable cell. Deletion of tandemly duplicated DNA was similarly measured by three independent determinations of phenotypic reversion under conditions previously described (34).
Sequence analysis.
Deletion endpoints were defined by aligning their pyrE sequences with the wild-type pyrE (orotate phosphoribosyltransferase) coding sequence of 594 nucleotides (nt) (GenBank accession number Y12822). This sequence, and for some analyses, a sequence of 2,251 nt encompassing pyrE and the adjacent aspartate carbamyltransferase (pyrB) and orotidine 5′-monophosphate decarboxylase (pyrF) coding regions, was analyzed using programs of the GCG package supported by the SeqWeb interface (Accelerys Inc., Madison, Wis.). Direct repeats (DRs) and inverted repeats (IRs) were found by COMPARE, and consensus sequences were found by PILEUP, using penalties of 5 for gap creation and 1 for gap extension. Sequences surrounding the deletion endpoints were randomized by SHUFFLE, and six potential stem-loop structures stable at 65°C were identified by M-FOLD.
Probabilities.
The expected numbers of short DRs or IRs near deletion endpoints were calculated from the number of N-mers that fit within a 20-nt window and the probability of matching any given N-mer at both ends of the deletion. Thus, for N = 3, there are 18 overlapping trimers in each 20-nt region and a 1/64 probability that a given trimer in the 5′ region will define a DR or complementary IR compared with a given trimer in the 3′ region. When the trimers at the ends are not paired in phase (i.e., when position relative to the deletion endpoint is ignored), all unordered combinations of trimers, i.e., 1/2(18)(19) = 171, are considered. This leads to an expected value per deletion of 171/64 for N = 3. Corresponding calculations yield values of 153/256 for N = 4 and 136/1,024 for N = 5. These expected yields were multiplied by the number of deletions available (five) to give the overall expected number of observed DRs or IRs.
The probability that n of 10 deletion endpoints would fall within duplication-prone regions comprising 104 bp (17.5%) of the pyrE gene is slightly greater than the Poisson term Pn = (xn/n!)e−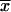 for 10 endpoints distributed randomly among six bins. In the latter case, x = 10/6, and the probability that three endpoints would fall into any one bin is P3 = [(10/6)3/3!]e−10/6 = 0.146.
for 10 endpoints distributed randomly among six bins. In the latter case, x = 10/6, and the probability that three endpoints would fall into any one bin is P3 = [(10/6)3/3!]e−10/6 = 0.146.
RESULTS
Frequency of spontaneous deletions in S. acidocaldarius.
Having observed pyrE deletion mutations in the course of other studies on S. acidocaldarius Foar mutants, we conducted a systematic search to recover additional deletions in this gene and measure their frequency. Screening additional mutants by PCR yielded two strains, MR123 and MR311, in which pyrE deletions were confirmed by DNA sequencing. When these results are combined with the results of directly sequencing a smaller set of mutants (15), deletions account for about 0.4% of spontaneous pyrE mutations. On the basis of the overall frequency of Foar mutants (Materials and Methods), pyrE deletions occur at a frequency of about 10−8 in S. acidocaldarius cultures. For comparison, deletions account for about 13% of all Escherichia coli lacI mutants, representing an overall frequency of about 2.5 × 10−7 per viable cell (30).
To our knowledge, S. acidocaldarius strains MR123 and MR311, together with strains MR31, MR103, and a strain which we provisionally designate JDS22 (Materials and Methods), comprise the only collection of mutants of a hyperthermophilic archaeon having spontaneous deletions. For simplicity, the alleles are here designated Δ123, Δ311, Δ31, Δ103, and Δ22, respectively. Figure 1 shows the relative sizes and positions of the deletions. Only two (Δ31 and Δ123) preserve the translational reading frame. Although overlaps occur among four of the deletions, the endpoints do not coincide, and the endpoints do not appear to be tightly clustered (Fig. 1).
FIG. 1.

Deletions and other sequence features of the S. acidocaldarius pyrE gene. Horizontal open bars on the genetic map depict coding sequences of the pyrB, pyrE, and pyrF genes of S. acidocaldarius; arrows show direction of transcription. Open bars above the map show the size and location of spontaneous deletions (designated in italics). All other numbers identify nucleotide positions in the pyrE coding sequence (GenBank accession number Y12822). Positions of the first and last nucleotides removed are given above the genetic map on either side of the open bars. Regions of potential stem-loop structures, numbered in order of decreasing thermodynamic stability, are indicated above the map. The first and last nucleotides of these regions (in roman numerals) are as follows: I, 200 to 217; II, 133 to 153; III, 67 to 88; IV, 528 to 541; V, 481 to 508, VI, 340 to 350. Regions which have been duplicated by various spontaneous mutations are represented by the trapezoidal symbols below the genetic map. Sites of frequent mutation are shown as partial sequences in parentheses below the map; numbers indicate the position of the first nucleotide in each region. More-detailed information on the gene sequence and mutations are provided in reference 15.
The deletion endpoints do not correspond to sites prone to other forms of mutation. The pyrE gene contains four mutational hot spots (Fig. 1), each of which accounts for 5% or more of Foar mutants sequenced (15). One of these regions includes a short trinucleotide repeat, whereas the others are short homopolymeric runs of purines in the nontranscribed (sense) strand (Fig. 1). None of the deletion endpoints occur within these four mutation-prone regions. The 3′ endpoint of Δ22 could be considered to be located as close as 2 bp from the first region, but this result is not significant at the 95% confidence level. (A total of 16 bp, or 2.7%, of the pyrE gene lies within 2 bp of one of these four mutation-prone regions, so the probability that at least 1 of 10 deletion endpoints randomly distributed within pyrE would fall within 2 bp of a hot spot is about 0.27.) Similarly, deletion endpoints were not significantly associated with sites promoting duplication. Duplications of pyrE regions that total 104 bp have been found by sequencing spontaneous mutants, but none of the duplication endpoints coincide with deletion endpoints (Fig. 1). Deletion Δ22 lies entirely within a duplication-prone region of 43 bp at the 5′ end of the coding region, and one endpoint of Δ103 falls within a region near the 3′ end of pyrE. However, the probability that 3 of 10 randomly distributed endpoints would fall within regions of pyrE totaling 104 bp is greater than 0.146 (see Materials and Methods), so this result is not considered significant.
Sequence analysis of deletion endpoints. (i) DRs.
In other genetic systems, most deletions occur between DRs of less than 15 nt and remove one of the repeats. Mechanisms proposed to explain this pattern include misalignment of a growing 3′ end between DRs, such that a stretch of template is bypassed during replication, or resection of the 5′ ends of a double-strand break to generate single-stranded 3′ ends which then anneal at DRs and support gap filling (3). In a previous study, Δ22 was found to have occurred between two GCT triplets in the 5′ region of the pyrE, leaving one triplet (15). This fits the pattern seen in other systems, but the DR is shorter than most of the other examples (8, 13, 25, 30). Only one other spontaneous deletion in the set, Δ123, has endpoints defined by a repeat, and this repeat is only 1 nt (Table 1).
TABLE 1.
Sequence contexts of deletion endpoints
| Deletion | No. of bp deleted | 5′ Regiona | 3′ Regiona |
|---|---|---|---|
| Δ31 | 18 | TGCAGTCGACATAGTAAAAG | AGGTATTAATTTTGATATGA |
| Δ22 | 20 | TTTCGTGAAAGCTCTACTTGATA | ATAAAAAATGCTTTTGATAGGA |
| Δ123 | 27 | GCTGGAGGTAAAGTTGAACAT | GTTATAGTCGATAGAGAAGAA |
| Δ311 | 322 | CTATTACCTAGATTTAAGGA | GTTGAACATGCTCTAGTTAT |
| Δ103 | 329 | GACATATTTTCATTCGTCGT | TGAAAAATTAGAAAGTGTAG |
Each region shown encompasses 10 bp on either side of the deletion endpoints. The sequences that remain after the deletion event are in bold type; the GCT triplet at the Δ22 endpoints and the A residues at the Δ123 endpoints are italic and underlined.
We assessed the significance of the Δ22 DR in two ways. The first was based on the observation that Δ22 could have arisen by removing 20 consecutive bp starting at any of four positions in the nontranscribed strand (Table 1): the first G, the first C, the first T, or the next nucleotide (C). This reflects the general property that a deletion involving two DRs N nucleotides long has N + 1 possible pairs of internucleotide breakpoints. As a result, a given deletion event will meet the criterion of involving a DR of N nucleotides if any of N + 1 pairs of windows N nucleotides wide coincides with a DR when scanned in phase across the actual breakpoints. Thus, any given deletion has a 4/64 probability of involving DRs of 3 bp, and the probability that at least one of the five deletions would exhibit this property by chance is 20/64, or 0.31. Our second assessment of significance was based on the proposed role of DRs as sites of strand annealing. Since the stability of annealing increases dramatically with longer complementary sequences, we searched the region for longer DRs that would anneal with correspondingly higher probability. The pyrE sequence itself contains one DR of 10 nt, nine DRs of 8 nt, and 13 DRs of 7 nt, and spacings between these DRs do not differ markedly from the size distribution of the pyrE deletions recovered (Table 2). Thus, the failure to use longer DRs for deletion does not reflect their scarcity in this region or a constraint on the size of deletions recovered in the selection. These considerations argue that the coincidence of deletion endpoints with the GCT repeat is largely fortuitous.
TABLE 2.
DRs in the wild-type pyrE sequence
| DR | Position (nt) of DR
|
No. of intervening bp | |
|---|---|---|---|
| 5′ end | 3′ end | ||
| 7-nt DRs | 12 | 387 | 367 |
| 13 | 549 | 528 | |
| 29 | 452 | 411 | |
| 33 | 223 | 182 | |
| 61 | 254 | 185 | |
| 181 | 578 | 389 | |
| 267 | 584 | 309 | |
| 310 | 334 | 16 | |
| 316 | 434 | 110 | |
| 319 | 490 | 163 | |
| 380 | 491 | 103 | |
| 447 | 543 | 88 | |
| 543 | 572 | 21 | |
| 8-nt DRs | 36 | 442 | 398 |
| 38 | 171 | 125 | |
| 95 | 383 | 280 | |
| 232 | 401 | 161 | |
| 262 | 535 | 265 | |
| 378 | 394 | 8 | |
| 447 | 572 | 116 | |
| 468 | 487 | 11 | |
| 547 | 585 | 30 | |
| 10-nt DR | 66 | 396 | 320 |
We also evaluated experimentally the potential of DRs to promote deletion, using a genetic selection provided by tandem duplications that inactivate the pyrE gene (Fig. 1). In these mutants, precise deletion of the duplication restores gene function, making this a selectable event. Five duplications were assayed for phenotypic reversion, and five pyrE frameshift mutants were assayed in parallel for comparison (Table 3). The frameshift mutations were stable, but most of the tandem duplications reverted, and the revertant frequency increased dramatically with length of the duplicated segment, up to 36 bp. This result showed that DRs can promote deletion formation in the S. acidocaldarius chromosome under appropriate conditions. Our ability to observe this only with tandem duplications could be explained, in part, by the fact that most of these duplications were much longer than the DRs occurring naturally in pyrE. The smallest and most stable duplication evaluated (the 6-bp duplication of strain JDS66) was nevertheless deleted more frequently than any of the native DRs. We compared the locations of the natural DRs and tandem duplications and found the natural DRs throughout pyrE, including in the regions where the duplications formed (Table 2 and Fig. 1). Thus, location per se could not be implicated as the main factor discouraging deletion between DRs in the wild-type pyrE sequence.
TABLE 3.
Reversion of tandem duplications
| Mutant | Mutationa | Mean no. of revertants/ 108 CFU (SD) |
|---|---|---|
| JDS66 | TD, 6 bp | 3.9 (5.2) |
| JDS178 | TD, 19 bp | 2,850 (990) |
| JDS12 | TD, 22 bp | 5,750 (6,800) |
| JDS18 | TD, 36 bp | 20,000 (6,700) |
| JDS64b | TD, 43 bp | <0.48 |
| JDS21 | −C | <0.35 |
| JDS36 | −G | 0.46 (0.68) |
| JDS10 | +G | <3.1 |
| JDS37 | +G | 0.40 (0.97) |
| JDS183 | +T | <0.24 |
TD, tandem duplication (the 6-bp duplication was of nt 346 to 351; the others were those diagrammed in Fig. 1); −C, C deleted; +G, G added.
It could not be ruled out that in the clone used for these experiments, the original duplication had mutated to a nonrevertable form.
(ii) IRs and secondary structure.
Another pattern often observed with spontaneous deletions is that their endpoints coincide with the outer boundaries of a short IR in the original sequence (12, 30). In other cases, one endpoint is associated with a region capable of forming more extensive secondary structure (26), or a nearby sequence is found that can anneal across the novel joint (12). As shown in Table 1, only Δ31 is defined by a self-complementary IR, removing 5′AT… .TA3′ from the nontranscribed strand. This minimal IR was not considered significant; the annealing product has negligible stability, and a 2-nt IR is expected at least once in five deletions by chance with a probability of 5/16, or 0.31. We reexamined the endpoint regions using a relaxed criterion in which self-complementary IRs of 3 nt or greater could occur anywhere within 10 bp of the deletion endpoint. This revealed eight trimeric IRs, two tetrameric IRs, and one pentameric IR. For comparison, the yields expected by chance for five deletions are 13.4 trimeric, 3.0 tetrameric, and 0.66 pentameric IRs (see Materials and Methods). We also confirmed that the region is not deficient in IRs of higher quality; pyrE contains one IR of 9 nt, one IR of 8 nt, and 10 IRs of 7 nt dispersed throughout the gene (not shown).
To evaluate possible placement of secondary structure at one end of a deleted sequence, we identified the six most stable stem-loop structures predicted in single-stranded DNA of the pyrE region (Fig. 1). These structures encompass a total of 124 nt, or 21% of the coding sequence, but no deletion endpoints fall within any of them. One deletion endpoint is adjacent to one of the stem-loop structures (the 3′ end of Δ31 and stem-loop II) (Fig. 1). We also searched for sequences in pyrE and the two adjacent genes that could anneal across both ends of any of the five deletions. The two best examples of such potentially “templating” sequences are shown in Fig. 2. Each represents an octanucleotide, the first half of which matches the 5′ flank of a deletion and the last half of which matches the 3′ flank. Annealing of these segments would have minimal base pairing for stabilization (4 bp for each segment) and would entail formation of large single-stranded regions (Fig. 2). Nevertheless, three observations make us hesitant to exclude a mechanistic role for these sequences in the formation of Δ22 and Δ103. (i) The two deletions have different sizes and positions, yet their potential templates occur at nearly the same location. (ii) The two potentially templating octanucleotides do not occur in the flanking pyrB or pyrF genes and are predicted to occur by chance once in every 20 to 50 kb of S. acidocaldarius DNA. (iii) In E. coli, one lacI deletion, S24, is known whose proposed intermediate closely resembles the structures depicted in Fig. 2 (12).
FIG. 2.

Octanucleotides corresponding to novel joints. As a convenient way to show the relative positions of the sequences, the upper strand of the wild-type pyrE gene is drawn annealed to the octanucleotide in the lower strand that represents the novel joint of the corresponding deletion. This method of depiction is that used in other deletion studies (11, 12, 16, 30) and is not a proposal of a specific mechanism. Sequences remaining after deletion are shown in bold type, as for Table. 1. Numbers with vertical lines indicate the nucleotide position in each strand with respect to the pyrE coding sequence; the remaining numbers indicate the sizes of the loops shown.
(iii) Consensus sequences.
Finally, we investigated the possibility that spontaneous deletion in S. acidocaldarius occurs predominantly at a consensus sequence which may not appear as a DR or IR due to interruptions of symmetry or lack of correspondence between the two ends of the deletion. For example, Δ31 and Δ22 share an octanucleotide sequence (TTTTGATA) at their 3′ ends (Table 1). In order to test the generality of shared sequences, we aligned the 5′-end regions of the deletions with each other, the 3′-end regions with each other, and the 5′-end regions with the reverse complements of the 3′-end regions. Alignments involving all five deletions revealed no base conserved at a level greater than 60% at any position fixed with respect to the endpoint. To test for a sequence whose position may vary, we aligned the 5′-end regions with each other and the 3′ end regions with each other, without constraint with respect to the position of the deletion endpoint. This yielded 100% consensus sequences TNNNT (where N is any nucleotide) (5′ ends) and A (3′ ends), with the deletion endpoints falling over a 10-nt range and a 9-nt range, respectively. To confirm that these consensus sequences are not significant, we separately randomized each of the end region sequences and repeated the alignments. This yielded 100% consensus sequences of ANT and ANA for the 5′- and 3′-end regions, respectively. Thus, the consensus sequences derived from randomized end regions were of equal or higher quality as those found near the actual deletion endpoints.
DISCUSSION
Comparison to other target genes.
Our review of the literature revealed few examples of chromosomal target genes in which unconstrained, spontaneous deletion has been analyzed both quantitatively and in terms of nucleotide sequence. Fortunately, however, the genetic systems meeting these criteria are phylogenetically diverse, representing animals, fungi, and gram-positive and gram-negative bacteria. Certain features of spontaneous deletion in the S. acidocaldarius pyrE gene become apparent when compared to these other systems (Table 4). One is the low frequency of spontaneous deletions in S. acidocaldarius. When the data are normalized with respect to the size of the target gene, only Caenorhabditis elegans exhibits a distinctly lower frequency, whereas the bacterial systems exhibit 13- and 65-fold-higher frequencies (Table 4). Alternatively, when deletions are measured as a proportion of all spontaneous mutants, S. acidocaldarius falls well outside the range of the bacterial, fungal, and animal systems. Another feature of S. acidocaldarius is the low correlation of deletion endpoints with DRs, even those as short at 3 nt. This property is not shared by any of the other systems (Table 4).
TABLE 4.
Properties of spontaneous deletion in diverse target genes
| Organisma | Target gene | Size (bp) | Deletion frequency
|
Proportion (%) of mutants | No. analyzed | % at DRs of > 2 nt | |
|---|---|---|---|---|---|---|---|
| Entire gene | Per kbp | ||||||
| S. acidocaldarius | pyrE | 594 | 1 × 10−8 | 1.7 × 10−8 | 0.4 | 5 | 20 |
| L. bulgaricus | lac | ∼3,100 | 3.3 × 10−6 | 1.1 × 10−6 | 9.3 | 8 | 88 |
| E. coli | lacIb | 1,080 | 2.5 × 10−7 | 2.3 × 10−7 | 12.6 | 22 | 68 |
| N. crassa | mtr | 1,472 | 1.0 × 10−7 | 6.8 × 10−8 | 35 | 11 | 100 |
| C. elegans | UNC-54 | ∼6,000c | 4.5 × 10−8 | 7.5 × 10−9 | 15 | 16 | 69 |
| Hamster | APRT | 670d | 1 × 10−8 | 1.5 × 10−8 | 10 | 6 | 67 |
Properties of the FOA selection.
Evaluating these results must account for the fact that analysis of spontaneous deletion generally requires a target gene whose inactivation can be selected, which has the potential drawback that properties of the selection can influence the types of deletions recovered. This study used FOA plus uracil to select mutations inactivating the pyrE or pyrF gene (14); this selection is widely used in yeast genetics and has been successfully applied to other hyperthermophilic archaea (33). More importantly, its possible influences on the types of deletions recovered can be evaluated on the basis of published data for S. acidocaldarius.
For example, the frequency of deletions in S. acidocaldarius pyrE would resemble that of E. coli lacI if about 90% of pyrE deletions went undetected (Table 4). Potential reasons for inefficient detection of a deletion include the following: (i) a leaky phenotype, (ii) inviability, or (iii) failure to be recognized as a deletion during screening. Interference from a leaky phenotype (possibility i) is excluded by the fact that FOA selects single-nucleotide frameshifts, small in-frame deletions, and small in-frame insertions throughout pyrE (15). Inviability of a large proportion of pyrE deletions (possibility ii) is inconsistent with recovery of deletions removing most of the gene and deletions that sharply decrease expression of the cotranscribed pyrF gene (15, 28). With regard to the failure to be recognized as a deletion (possibility iii), our screening of PCR products on agarose gels successfully identified an 18-bp deletion, and our screening probably has a detection threshold of 12 to 15 bp. Thus, a significant underestimation of the deletion frequency under our conditions would require the majority of pyrE deletions to be very short (i.e., less than about 15 bp) or to occur in pyrF. Neither of these alternatives agrees with the spectrum of spontaneous mutation in the pyrE and pyrF genes, in which only one deletion was observed among 108 independent, randomly chosen Foar mutants. This deletion (Δ22) removes 20 bp and lies in pyrE, which was the location of about 95% of all spontaneous mutations conferring the Foar phenotype (15).
Another property, the relatively low proportion of deletions for the S. acidocaldarius pyrE gene compared to the other genes in Table 4 could, in principle, result from the following: (i) production of other classes of mutation at a much higher rate than those in the other genetic systems or (ii) selection of leaky mutants at a much higher rate than those in the other systems. Possibility i is excluded by two sets of independent mutation rate measurements, which show that pyrE has a forward mutation rate very close to those of E. coli lacI and other bacterial genes (15, 18). Situation ii is more difficult to exclude, as about 60% of mutants selected by 50 μg of FOA per ml exhibit a leaky phenotype, and it is not known whether the other systems of Table 4 yield a significant proportion of leaky mutants. Therefore, the frequency of deletions among nonleaky S. acidocaldarius pyrE mutants (about 1%) may provide a more conservative value for comparison to the other systems, but this would still be only one-ninth of the closest value in Table 4.
Our failure to recover a deletion extending beyond the boundaries of the pyrE gene also raises the question of whether the FOA selection can recover such deletions in S. acidocaldarius. These would be precluded if DNA sequences flanking pyrE were essential, for example. The transcript beginning with pyrE includes pyrF and at least one other ORF, designated orf8 (32). However, two observations argue against essential roles for either pyrF or orf8: (i) frameshift mutations in pyrE which exert strong polar effects on pyrF expression are frequently isolated (28), and (ii) several pyrF frameshifts and one nonsense mutation, which inactivate pyrF and should exert similar polarity on orf8 expression, have also been isolated (15). An alternative explanation for the observed placement of deletions is that some position-dependent property of pyrE makes it more susceptible than pyrF to various forms of mutation, including deletion. This is consistent with the following facts: duplication mutations are relatively common in pyrE but have not been found in pyrF (15); point mutations are 20-fold more frequent in pyrE than pyrF (15); in S. solfataricus, IS elements insert threefold more frequently into pyrE than into pyrF (23); and diverse forms of DNA damage all induce mutation in S. acidocaldarius (29, 34).
Potential deletion mechanisms.
The low frequency of deletions in S. acidocaldarius impeded our efforts to collect many examples for analysis, yet sequences of the available alleles distinguish the dominant mode (or modes) of their formation from those commonly proposed for other organisms. No association of deletion endpoints with DRs, IRs, or stem-loop structures was supported at the 95% confidence level, despite the fact that these structures are about as abundant in the S. acidocaldarius pyrE gene as in the various target genes where they have been found to promote spontaneous deletions (3, 8, 12, 25, 26). However, genetic assays in S. acidocaldarius do provide evidence of strand slippage mechanisms of deletion under certain conditions. Tandem duplications reverted at high frequencies, which increased with increasing length of the repeated sequence (Table 3). Furthermore, homopolymeric runs and short repeats have been shown to promote frameshift mutations and triplet expansions, respectively, in the wild-type pyrE gene (15). Our observations that tandem duplications were deletion-prone, whereas natural DRs were not may reflect the fact that the natural DRs are shorter (on average) and interrupted by nonrepeated sequences.
In some systems, consensus sequences occur at the endpoints of spontaneous deletions and have been attributed to normal or aberrant processing by DNA breaking or joining enzymes such as topoisomerases (3, 5). The available set of S. acidocaldarius deletions included only one example of a common sequence found at the 3′ ends of two deletions (Δ22 and Δ31), and this remains difficult to interpret mechanistically. The lack of AA or TT dinucleotides at deletion endpoints argues that an activity resembling topoisomerase II of Sulfolobus shibatae, which cleaves DNA predominantly at AA or TT, yielding 2-nt, 5′ extensions (4), is not a significant source of small- and medium-sized deletions in S. acidocaldarius. However, it should be emphasized that DNA sequence specificity has not been established for other potentially relevant enzymes of S. acidocaldarius. For example, the reverse gyrase of S. acidocaldarius (a topoisomerase I) exhibits the same minimal sequence specificity of its counterpart in S. shibatae (19), so it is difficult to rule it out as a potential source of deletions. The possible role of nearby sequences in stabilizing nascent deletions (“templating” [12]) could be inferred in two cases and will require isolation of additional deletions to evaluate. Our observations also seem consistent with repair of double-strand breaks by some form of nonhomologous end joining (3), which, in turn, is consistent with the associations of MRE11 and RAD50 homologues in the genome of S. acidocaldarius and other hyperthermophilic archaea (7).
Implications for genome evolution.
According to mutational analysis of the pyrE and pyrF genes, S. acidocaldarius has one of the lowest genomic error rates yet measured, despite its extremely harsh growth conditions (15). The present study further shows that, within the low mutant frequency, deletions make up an unusually low proportion of spontaneous mutations. The relative frequencies of different classes of mutations now documented in S. acidocaldarius predict a pattern for the elimination of a gene following release of selective pressure. Initially, frameshift mutations should accumulate in homopolymeric runs, then elsewhere in the sequence, followed by slower accumulation of base pair substitutions and duplications, followed in turn by yet slower accumulation of deletions. It also seems significant that in S. acidocaldarius the more-frequent classes of mutations are reversible. This would serve to provide a window of time during which function of a mutated gene can be restored should moderate selection be reapplied. This idea finds experimental support in the observation that eight lineages of S. acidocaldarius subjected to multiple cycles of forward and reverse mutation failed to generate a functional pyrE gene with an altered sequence (2). Also, analysis of other genomes, such as that of Saccharomyces cerevisiae, has identified defective but recoverable genes which represent a reserve of potential functionality (17).
The slow, incremental nature of gene elimination predicted by the mutational processes in S. acidocaldarius seems to favor genome streamlining while minimizing concomitant inactivation of beneficial genes. It should be noted, however, that this situation may not typify all hyperthermophilic archaea. S. acidocaldarius has few, if any, active IS elements on the basis of genetic assays (15) and preliminary sequence analysis of the genome (R. Garrett, personal communication), but other Sulfolobus species have a number of them (20, 23, 31). In particular, transposition of several distinct families of IS elements causes a high frequency of spontaneous mutation at the pyrE and pyrF genes of S. solfataricus and may also generate frequent chromosomal rearrangements (23, 31). In such lineages, IS elements may assume the dominant role in inactivating nonessential genes (through insertion) and in removing them (through transposase-catalyzed imprecise excision).
Acknowledgments
We thank M. Reilly for providing strains; New England Biolabs, Inc., for providing D.W.G. with computing facilities; and J. Drake for communicating unpublished results and for comments on the manuscript.
This work was supported in part by grant MCB9733303 from the National Science Foundation.
REFERENCES
- 1.Andersson, J. O., and S. G. E. Andersson. 1999. Genome degradation is an ongoing process in Rickettsia. Mol. Biol. Evol. 16:1178-1191. [DOI] [PubMed] [Google Scholar]
- 2.Bell, G. D., and D. W. Grogan. 2002. Loss of genetic accuracy in mutants of the thermoacidophile Sulfolobus acidocaldarius. Archaea 1:45-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bierne, H., S. D. Ehrlich, and B. Michel. 1997. Deletions at stalled replication forks occur by two different pathways. EMBO J. 16:3332-3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buhler, C., J. H. G. Lebbink, C. Bocs, R. Ladenstein, and P. Forterre. 2001. DNA topoisomerase VI generates ATP-dependent double-strand breaks with two-nucleotide overhangs. J. Biol. Chem. 276:37215-37222. [DOI] [PubMed] [Google Scholar]
- 5.Bullock, P., J. J. Champoux, and M. Botchan. 1985. Association of crossover points with topoisomerase I cleavage sites: a model for nonhomologous recombination. Science 230:954-958. [DOI] [PubMed] [Google Scholar]
- 6.Cole, S. T., K. Eiglmeier, J. Parkhill, K. D. James, N. R. Thomson, P. R. Wheeler, N. Honore, T. Garnier, C. Churcher, K. Harris, K. Mungall, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. M. Davies, K. Devlin, S. Duthoy, T. Feltwell, A. Fraser, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, C. Lacroix, J. Maclean, S. Moule, L. Murphy, K. Oliver, M. A. Quall, M.-A. Rajandream, K. M. Rutherford, S. Rutter, K. Seeter, S. Simon, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, K. Taylor, S. Whitehead, J. R. Woodward, and B. G. Barrell. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007-1011. [DOI] [PubMed] [Google Scholar]
- 7.Constantinesco, F., P. Forterre, and C. Elie. 2002. NurA, a novel 5′-3′ nuclease gene linked to RAD50 and MRE11 homologs of thermophilic archaea. EMBO Rep. 3:537-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dillon, D., and D. Stadler. 1994. Spontaneous mutation at the mtr locus in Neurospora: the molecular spectrum in a wild-type and a mutator strain. Genetics 138:61-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiRuggiero, J., D. Dunn, D. L. Maeder, R. Holley-Shanks, J. Chatard, R. Horlacher, F. T. Robb, W. Boos, and R. B. Weiss. 2000. Evidence of recent lateral gene transfer among hyperthermophilic archaea. Mol. Microbiol. 38:684-693. [DOI] [PubMed] [Google Scholar]
- 10.Dush, M. K., J. M. Sikela, S. A. Khan, J. A. Tischfield, and P. J. Stambrook. 1985. Nucleotide sequence and organization of the mouse adenine phosphoribosyltransferase gene: presence of a coding region common to animal and bacterial phosphoribosyltransferases that has a variable intron/exon arrangement. Proc. Natl. Acad. Sci. USA 82:2731-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farabaugh, P. J., U. Schmeissner, M. Hofer, and J. H. Miller. 1978. Genetic studies of the lac repressor. VII. On the molecular nature of spontaneous hotspots in the lacI gene of Escherichia coli. J. Mol. Biol. 126:847-857. [DOI] [PubMed] [Google Scholar]
- 12.Glickman, B. W., and L. S. Ripley. 1984. Structural intermediates of deletion mutagenesis: a role for palindromic DNA. Proc. Natl. Acad. Sci. USA 81:512-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordenin, D. A., and M. A. Resnick. 1999. Yeast ARMs (at-risk motifs) can reveal sources of genome instability. Mutat. Res. 400:45-58. [DOI] [PubMed] [Google Scholar]
- 14.Grogan, D. W., and R. P. Gunsalus. 1993. Sulfolobus acidocaldarius synthesizes UMP via a standard de novo pathway: results of a biochemical-genetic study. J. Bacteriol. 175:1500-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grogan, D. W., G. T. Carver, and J. W. Drake. 2001. Genetic fidelity under harsh conditions: analysis of spontaneous mutation in the thermoacidophilic archaeon Sulfolobus acidocaldarius. Proc. Natl. Acad. Sci. USA 98:7928-7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halliday, J. A., and B. W. Glickman. 1991. Mechanisms of spontaneous mutation in DNA repair-proficient Escherichia coli. Mutat. Res. 250:55-71. [DOI] [PubMed] [Google Scholar]
- 17.Harrison, P., A. Kumar, N. Lan, N. Echols, M. Snyder, and M. Gerstein. 2002. A small reservoir of disabled ORFs in the yeast genome and its implications for the dynamics of proteome evolution. J. Mol. Biol. 316:409-419. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs, K. L., and D. W. Grogan. 1997. Rates of spontaneous mutation in an archaeon from geothermal environments. J. Bacteriol. 179:3298-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaxel, C., M. Duguet, and M. Nadal. 1999. Analysis of DNA cleavage by reverse gyrase from Sulfolobus shibatae B12. Eur. J. Biochem. 260:103-111. [DOI] [PubMed] [Google Scholar]
- 20.Kawarabayashi, Y., Y. Hino, H. Horikawa, K. Jin-So, M. Takahashi, M. Sekine, S. Baba, A. Ankai, H. Kosugi, A. Hosoyama, S. Fukui, Y. Nagai, K. Nishijima, R. Otsuka, H. Nakazawa, M. Takamiya, Y. Kato, T. Yoshizawa, T. Tanka, Y. Kudoh, J. Yamazaki, N. Kushida, A. Oguchi, K. Aoki, S. Masuda, M. Yanagii, M. Nishimura, A. Yamagishi, T. Oshima, and H. Kikuchi. 2001. Complete genome sequence of an aerobic thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain 7. DNA Res. 8:123-140. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence, J. G. 2001. Catalyzing bacterial speciation: correlating lateral transfer with genetic headroom. Syst. Biol. 50:479-498. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence, J. G., and H. Ochman. 1998. Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. USA 95:9413-9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martusewitsch, E., C. W. Sensen, and C. Schleper. 2000. High spontaneous mutation rate in the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by transposable elements. J. Bacteriol. 182:2574-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mira, A., H. Ochman, and N. A. Moran. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17:589-596. [DOI] [PubMed] [Google Scholar]
- 25.Mollet, B., and M. Delley. 1990. Spontaneous deletion formation within the β-galactosidase gene of Lactobacillus bulgaricus. J. Bacteriol. 172:5670-5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nalbantoglu, J., D. Hartley, G. Phear, G. Tear, and M. Meuth. 1986. Spontaneous deletion formation at the aprt locus of hamster cells: the presence of short sequence homologies and dyad symmetries at deletion termini. EMBO J. 5:1199-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pulak, R. A., and P. Anderson. 1988. Structures of spontaneous deletions in Caenorhabditis elegans. Mol. Cell. Biol. 8:3748-3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reilly, M. S., and D. W. Grogan. 2001. Characterization of intragenic recombination in a hyperthermophilic archaeon via conjugational DNA exchange. J. Bacteriol. 183:2943-2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reilly, M. S., and D. W. Grogan. 2002. Biological effects of DNA damage in the hyperthermophilic archaeon Sulfolobus acidocaldarius. FEMS Microbiol. Lett. 208:29-34. [DOI] [PubMed] [Google Scholar]
- 30.Schaaper, R. M., B. N. Danforth, and B. W. Glickman. 1986. Mechanisms of spontaneous mutagenesis: an analysis of the spectrum of spontaneous mutation in the Escherichia coli lacI gene. J. Mol. Biol. 189:273-284. [DOI] [PubMed] [Google Scholar]
- 31.She, Q., R. Singh, F. Confalonieri, Y. Zivanovic, G. Allard, J. Awayez, C. Chan-Weiher, I. Clausen, B. Curtis, A. DeMoors, G. Erauso, C. Fletcher, P. Gordon, I. Heilkamp-de Jong, A. Jeffries, C. Kozera, N. Medina, X. Peng, H. Thi-Ngoc, P. Redder, M. Shenk, C. Theriault, N. Tolstrup, R. Charlebois, W. Doolittle, M. Duguet, T. Gaasterland, R. Garrett, M. Ragan, C. Sensen, and J. Van de Oost. 2001. The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc. Natl. Acad. Sci. USA 98:7835-7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thia-Toong, T.-L., M. Roovers, V. Durbecq, D. Gigot, N. Glansdorff, and D. Charlier. 2002. Genes of de novo pyrimidine biosynthesis from the hyperthermophilic crenarchaeote Sulfolobus acidocaldarius: novel organization in a bipolar operon. J. Bacteriol. 184:4430-4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watrin, L., and D. Prieur. 1996. UV and ethyl methanesulfonate effects in hyperthermophilic archaea and isolation of auxotrophic mutants of Pyrococcus strains. Curr. Microbiol. 33:377-382. [DOI] [PubMed] [Google Scholar]
- 34.Wood, E. R., F. Ghane, and D. W. Grogan. 1997. Genetic responses of the thermophilic archaeon Sulfolobus acidocaldarius to short-wavelength UV light. J. Bacteriol. 179:5693-5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zivanovic, Y., P. Lopez, H. Philippe, and P. Forterre. 2002. Pyrococcus genome comparison evidences chromosome shuffling-driven evolution. Nucleic Acids Res. 30:1902-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]