

Abstract
The nonhomologous end-joining (NHEJ) pathway is responsible for rejoining the majority of double-strand breaks in mammalian cells, including the programmed breaks introduced by V(D)J recombination. The regulation of the enzymatic activities associated with this recombination pathway is still largely unknown. Here we report that human XRCC4 (for X-ray cross-complementation group 4), a protein essential for NHEJ, is subject to posttranslational protein modification. The modifier peptide, SUMO, can be added to XRCC4 both in vitro and in vivo. The site of modification is mapped to lysine 210 by using specific mutagenesis. A protein mutated such that it cannot be SUMOylated remains localized in the cytoplasm rather than accumulating in the nucleus. Cells expressing only the mutated protein are radiation sensitive and fail to complete V(D)J recombination. Genetic fusion of the SUMO sequence to the C terminus of the mutant restores nuclear localization and radiation resistance. The modification may serve a regulatory role. Our finding fits with an emerging literature associating SUMO modification with the control of the repair and recombination associated with DNA breaks.
Double-strand DNA breaks (DSBs) arise naturally by means of a variety of mechanisms including direct breakage by ionizing radiation, replication of a nicked template, or enzymatic cleavage. Such events are likely to be lethal to a cell if left unresolved, so mechanisms to repair these lesions are quite important. The method that is used most commonly in mammalian cells is the nonhomologous end-joining (NHEJ) pathway (reviewed in references 24, 27, and 48). This pathway is of particular interest to immunologists because it is also essential for the completion of V(D)J recombination, the programmed DNA rearrangement that assembles the antigen receptors of B and T cells (reviewed in references 28 and 44).
One of the indispensable proteins in the NHEJ pathway is XRCC4. This protein forms higher-order complexes with itself (35) and DNA ligase IV (5, 10) and is necessary for NHEJ activity in vivo. Since ligase IV alone is capable of joining DNA as a purified protein (42), the role of XRCC4 appears to be regulatory, perhaps through a structural contribution to the repair complex.
Our previous finding of a ubiquitin ligase activity in RAG1 suggested that posttranslational peptide modifications may contribute to the regulation of V(D)J recombination (45, 62). The recognition of covalent modification of a protein by the addition of a peptide modifier was first recognized for ubiquitylation (reviewed in references 39 and 60). Subsequently, other peptide modifiers have been found (more than 15 to date), the addition of which leads to diverse consequences for the target proteins. Among these modifiers are the SUMO proteins, whose name derives from the phrase “small ubiquitin-related modifier.” The biochemistry and physiologic significance of this modification pathway have been recently reviewed (8, 13, 19, 36). SUMO modification is detected in several proteins concerned with DNA repair, including topoisomerases (2, 18), the base excision glycosylase TDG (12, 52), a complex including the yeast Ku70 (64), Ku80 (9), and the BLM helicase (7).
In examining the protein sequence for human XRCC4, we recognized the presence of conserved motifs that could function as targets for modification by SUMO. Here we report that XRCC4 is subject to posttranslational protein modification by the SUMO pathway both in vitro and in vivo and found that the modification controls intracellular localization. We then explored some of the physiologic consequences of interfering with the SUMOylation of XRCC4.
MATERIALS AND METHODS
Antibodies and reagents.
Monoclonal mouse antibodies (conjugated with horseradish peroxidase [HRP] or fluorescein isothiocyanate [FITC]) to the FLAG epitope were from Sigma-Aldrich (St. Louis, MO). HRP-conjugated monoclonal antibody to hemagglutinin (HA), His, and Myc tags were from Roche Applied Science (Indianapolis, IN). Antibody directed against histone H3 was from Cell Signaling Technology, Beverly, MA.
Size markers for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from Crystalgen (Huntington Station, NY). Markers for immunoblots were from Invitrogen (Carlsbad, CA).
DNA manipulations.
The GST-XRCC4 isoform 1 plasmid (pGEX4T-1) of the human gene was kindly provided by S. P. Lees-Miller (University of Calgary, Alberta, Canada). All point mutants (K140A, K140R, K210A, and K210R) were made by using a QuikChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and verified by sequencing.
For transient eukaryotic expression experiments, FLAG-XRCC4 or point mutants were cloned into the p3xFLAG-CMV7 vector (Sigma-Aldrich, St. Louis, MO). His-Xpress-XRCC4 or point mutants were cloned into the pcDNA3.1-His(C) vector (Invitrogen). GST-XRCC4 or point mutants were cloned into the pCMV-GST vector (58), kindly provided by R. R. Reed (Johns Hopkins University, Baltimore, MD) and Myc-XRCC4 or point mutants were cloned into the pCMV-Tag3B vector (Stratagene). For establishing stable cell lines XRCC4 wild type (wt) or XRCC4 K210R were cloned into p3xFLAG-CMV10 vector (Sigma-Aldrich).
SUMO fusions to XRCC4 and the K210R mutant were assembled by using PCR mutagenesis to add the SUMO-1 coding region in frame to the C terminus of XRCC4 in the vector above.
GST-PKA-SUMO1 was made from the GST-SUMO1 plasmid provided by R. Hay (University of St. Andrews, St. Andrews, Scotland) by cloning the coding region of SUMO-1 into the pGEX-2TK vector (Amersham Biosciences, Piscataway, NJ). The HA3-SUMO1 construct was provided by N. Agus-Schreiber (Albert Einstein College of Medicine, Bronx, NY). The SAE1/2-, Ubc9-, and GST-PML-expressing plasmids were provided by R. Hay. Myc-PIAS3 and Myc-PIAS Xβ (Miz1) plasmids were provided by M. Lechner (Drexel University, Philadelphia, PA). Myc-PIAS1 and Myc-PIAS1 C345,350,355S (the dominant-negative version) plasmids were made from the Escherichia coli GST-PIAS1 expression constructs provided by X.-H. Feng (Baylor College, Houston, TX) by cloning into the pCMV-Tag3B vector (Stratagene). HA-ubiquitin plasmid was provided by D. Bohmann (University of Rochester, Rochester, NY).
Mouse RAG1 cDNA encoding residues 1 to 1008, followed by the triple Myc tag (pMS119C) (46) was corrected to match the genomic sequence. Full-length T7 epitope-tagged RAG2 in pCAT7-Neo, kindly provided by R. Mizuta (Tokyo University of Science, Tokyo, Japan) (33) was used. The pJH200 and pJH290 extrachromosomal substrates for V(D)J recombination (14) were kindly provided by the lab of M. Gellert (National Institutes of Health, Bethesda, MD).
Protein purification.
E. coli produced glutathione S-transferase (GST)-tagged proteins were purified from the cleared lysates by affinity chromatography on glutathione-Sepharose (Amersham Biosciences, Piscataway, NJ) according to the manufacturer's protocol. Purification of the SAE1/SAE2 fusion protein expressed in E. coli was performed according to the recommendations of R. Hay.
Proteins from eukaryotic cells were harvested into radioimmunoprecipitation assay buffer (150 mM NaCl; 50 mM Tris-HCl, pH 8.0; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS) with protease inhibitor cocktail (Roche Applied Sciences, Indianapolis, IN). To preserve SUMOylation, cells were lysed in the presence of 5 mM N-ethylmaleimide (Fisher Scientific, Pittsburgh, PA).
Extraction into nuclear and cytoplasmic fractions was performed by Triton X-100 extraction in 3 mM MgCl2 as previously described (46) and by means of the NE-PER extraction kit (Pierce, Rockland, IL).
In vitro SUMOylation.
Radioactively labeled SUMO-1 was obtained by expressing the GST-PKA fusion protein in E. coli, binding the protein to glutathione-Sepharose beads and phosphorylating it on the beads by using cAMP-dependent protein kinase A catalytic subunit (New England Biolabs, Beverly, MA) and [32P]ATP (Amersham Biosciences). The resulting protein was cleaved from the GST partner by using biotinylated thrombin, which was subsequently removed by binding to streptavidin agarose (EMD Bioscience, Madison, WI). A total of 105 cpm of 32P-labeled SUMO-1 was used and assembled as described previously (56) with 0.5 μM SAE1/SAE2 and 1.5 μM Ubc9 enzymes (prepared in our laboratory or purchased from Boston Biochem, Cambridge, MA) in a 20-μl reaction. SUMOylation was performed by using target proteins bound to glutathione-Sepharose beads for 90 min at 30°C. The modified proteins were washed three times with phosphate-buffered saline, eluted in SDS sample buffer, separated by SDS-PAGE, detected by autoradiography, and quantified by using the PhosphorImager and ImageQuant software (Amersham Biosciences).
Cell culture and stable cell lines.
CHO.XR-1 (25, 51) and CHO-K1 (ATCC, Manassas, VA) cells were grown in F-12/DMEM medium (CellGro, Herndon, VA) supplemented with 10% fetal bovine serum, 100 U of penicillin/ml, and 100 μg of streptomycin/ml. Cells were transfected with plasmids by using Fugene-6 (Roche Applied Bioscience, Indianapolis, IN) according to the manufacturer's protocol. For stable cell line selection, 0.5 mg of Geneticin (Invitrogen)/ml was added to the culture medium 24 h after transfection. Cells were incubated in Geneticin-containing medium for 15 days. The medium was refreshed twice a week. Cell lines were generated from single cells cloned by dilution and maintained in 0.25 mg of Geneticin/ml.
Immunoprecipitation and Western blotting.
Anti-FLAG immunoprecipitation was performed by using EZview Red Anti-FLAG M2 affinity gel (Sigma-Aldrich). Typically, 100 to 500 μg of total protein was used for immunoprecipitation in radioimmunoprecipitation assay buffer containing a final concentration of 0.5 to 1.0 mM N-ethylmaleimide. HRP-labeled antibodies against FLAG (Sigma-Aldrich) and HA (Roche Applied Science) were used to avoid cross-reaction with immunoglobulin heavy chains when we visualized the blots. ECL and ECL Plus Western blotting reagents (Amersham Biosciences) were used for detection. Anti-HA immunoprecipitation was performed similarly using Mono-HA Affinity Matrix (Covance, Berkeley, CA).
Immunofluorescence.
For immunofluorescence microscopy, cells were grown on coverslips at 2 × 104 cells per slip, fixed with paraformaldehyde, and visualized by using FITC-conjugated anti-FLAG M2 monoclonal antibodies (Sigma-Aldrich) after counterstaining with DAPI (4′,6′-diamidino-2-phenylindole).
Images were acquired with a Zeiss Axioskop 2 microscope equipped with a digital camera. The resulting image files were processed and assigned false colors by using Photoshop software (Adobe, San Jose, CA).
V(D)J extrachromosomal recombination assay.
The extrachromosomal V(D)J assay was performed largely as described previously (14). CHO.XR-1 cells transfected with Myc-XRCC4 or its point mutants, or derived lines stably expressing a form of XRCC4, were cotransfected with RAG1-Myc, T7-RAG2, and a substrate plasmid. DNA was extracted at 48 h posttransfection by the method of Hirt (15) and digested with DpnI (New England Biolabs, Beverly, MA) to select for molecules that had replicated. E. coli DH5α was transformed by electroporation using a BTX ECM399 electroporator (Inovid Biomedical Corp., San Diego, CA). The efficiency of recombination is the ratio of colonies that grow on Amp+ Cam+ selective plates to the number of colonies that grow on Amp+ selective plates alone, corrected by confirming recombination events for a representative sampling by direct sequencing. The junctions were sequenced with the primer 5′-TGAGCGCAACGCAATTAATGTGAG-3′.
Radiosensitivity assay.
Cells (103) were plated and exposed to a range of ionizing radiation doses using a 137Cs source. Incubation under growth conditions was continued for 8 days, at which time cell colonies were fixed with 70% ethanol, stained with crystal violet, and counted as described previously (61).
RESULTS
Human XRCC4 is a 336-amino-acid protein composed of three major structural domains. There is an N-terminal globular domain (residues 1 to 119) with DNA-binding activity (34), a long helical stem that is responsible for multimerization and interaction with ligase IV (residues 119 to 203) (21, 50), and a C-terminal tail for which no enzymatic function is currently defined but may harbor a nuclear localization signal. Substantial portions of the C-terminal tail are dispensable for V(D)J recombination and NHEJ in cells (10).
Posttranslational peptide modifiers can be joined to a target protein at free amino groups, commonly lysine residues. For SUMO modification, additional local sequence preferences are often detectable. We examined the sequence of XRCC4 for the consensus motif common at SUMO targets, ΨKXE (43, 47), where Ψ represents a large hydrophobic residue, using the online tool SUMOplot provided by Abgent. Although SUMOylation is not always restricted to such sites, the data presented below show that the analysis provided a useful match in this case. The two candidate sequences with the best match to the consensus were IKQE surrounding K210 and AKNE at K140.
SUMO is conjugated to its target by means of a series of enzymatic steps requiring an E1 and an E2 enzyme analogous to ubiquitylation. In contrast to ubiquitylation, an E3 enzyme often is not required in vitro although the presence of an E3 can promote the reaction and is required in vivo. We first tested the XRCC4 protein as a potential substrate for SUMOylation in an in vitro SUMOylation assay (54, 56) using as a target GST or GST fusion proteins bound to glutathione-Sepharose beads. The reaction was assembled with the SUMO E1 (SAE1 and -2 either as a heterodimer or coexpressed as a single protein) and E2 (Ubc9) enzymes, SUMO-1, and ATP. In the experiment presented in Fig. 1, we used a 32P-labeled form of SUMO-1. Lane 1 shows the labeling of a known SUMO target, an 11-residue PML peptide expressed as a GST fusion (54). The band at 45 kDa is close to the anticipated mobility for the fusion protein (28 kDa) plus the addition of the SUMO peptide (13 kDa). The XRCC4 fusion protein (64 kDa) generates a prominent signal at 85 kDa (lane 2). GST protein alone (lane 3) is not a good substrate for the reaction and shows only a background level of signal that may correspond to contaminating proteins. As similar quantities of protein were reacted in each of lanes 1 to 3, SUMOylation of the XRCC4-fusion appears comparably efficient to the PML fusion in this system. Lane 4 represents the reaction mixture with no additional target protein added. Some of the weakly labeled bands that appear throughout seem to represent SUMOylation of the E1 and E2 enzymes (lane 4). The molecular masses of the unmodified enzyme components are 71 and 38 kDa for SAE1 and -2 and 18 kDa for Ubc9.
FIG. 1.

SUMOylation of XRCC4 in vitro. The transfer of 32P-labeled SUMO-1 to a target protein is visualized by autoradiography. The reactions are performed on glutathione beads using purified components then eluted and analyzed by SDS-PAGE. Lane 1 is the known SUMO target peptide from PML expressed as a GST fusion (56). Lane 2 is XRCC4 as a GST fusion. Lane 3 is GST alone. Lane 4 is the reaction mix without any added target protein. The molecular masses are shown in kilodaltons.
We used the same assay to determine whether the site of SUMOylation corresponded to either of the two predicted sites by mutating the two lysines K140 and K210 individually to the charge-conservative arginine or to alanine and then testing the resulting proteins. Figure 2A is the gel, stained with Coomassie brilliant blue, which shows comparable loading of the purified proteins appearing as the band in each lane at 70 kDa. Figure 2B shows that mutation of K210 to either alanine or arginine (lanes 3 and 4, respectively) eliminates the specific labeled product that appears at 85 kDa in lanes 1, 2, and 5. In contrast, mutation of K140 had no effect. This indicates that K210 is essential for SUMOylation and appears to be the only site of modification in this assay. Since similar behavior was seen with both K210 mutants, further experiments were confined to the K210R version since it is the more conservative change.
FIG. 2.
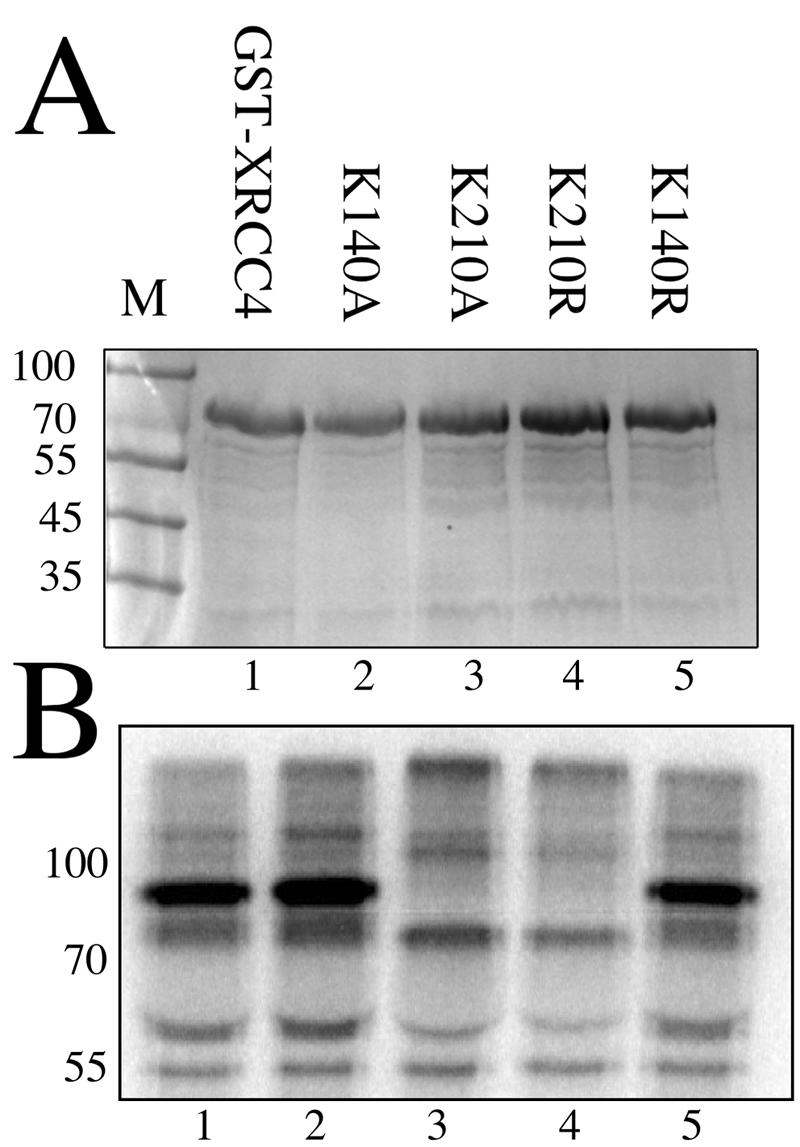
Mapping the SUMOylation site of XRCC4 to K210. (A) Coomassie brilliant blue stained SDS-polyacrylamide gel of the five GST-fusion proteins tested by the same SUMOylation assay as in Fig. 1. Marker lane is labeled M with molecular masses in kilodaltons. Each protein in lanes 1 to 5 is identical except for the mutation of a single lysine as listed. (B) Autoradiogram of the SUMOylation assay performed on the proteins from panel A. Parental XRCC4 (lane 1) and mutants at lysine 140 (lanes 2 and 5) are efficiently SUMOylated. Mutation of lysine 210 to alanine or arginine (lanes 3 and 4, respectively) prevents SUMOylation.
Next, we confirmed that a similar behavior could be obtained in mammalian cells. We expressed a FLAG epitope-tagged version of human XRCC4 along with an HA-tagged version of SUMO-1 by transient transfection of CHO.XR-1 cells that do not express endogenous XRCC4 (25, 51). XRCC4 protein was purified from the cell extract by immunoprecipitation with agarose-immobilized anti-FLAG antibody and then blotted and visualized with either anti-FLAG or anti-HA antibodies. Figure 3A shows an intense band in all lanes representing the unmodified XRCC4 protein at an apparent mobility of 65 kDa. A weaker second band is observed at 95 kDa in lane 1, representing the wild-type protein, and lane 3, the mutant at K140. That band is substantially reduced in lane 2 representing the K210R mutant. The identity of this second band as the SUMO modified form is confirmed by Fig. 3B, in which a parallel blot of the same immunoprecipitated proteins is developed with antibody directed against the HA epitope tag on the SUMO peptide. Again, the K210R lane is strikingly reduced, indicating that this mutation is sufficient to prevent SUMOylation. We note a minor band at 90 kDa, the identity of which is uncertain. This might represent a SUMOylated protein that coprecipitates with XRCC4. A low level of residual SUMOylated protein may be present in lane 2 that may indicate a small degree of SUMOylation at another site in the protein. Nevertheless, the majority of SUMOylation is prevented by mutation of K210. We conclude that SUMOylation of XRCC4 occurs in these cells under conditions where no effort was made to induce or overexpress the enzymes associated with this reaction. We note that the intensity of the upper band implies that the steady-state level of the SUMOylated form is only a few percent. This is also true for other SUMO-modified proteins (for examples, see references 29 and 57) and, in part, represents the dynamic and reversible nature of this modification owing to the continuous activity of SUMO-isopeptidases (30).
FIG. 3.

SUMOylation of XRCC4 in CHO.XR-1 cells. (A) Immunoblot directed against the FLAG epitope. CHO.XR-1 cells were cotransfected with plasmids for HA-SUMO-1 (all lanes) and a plasmid encoding FLAG-XRCC4 wild-type (lane 1, labeled WT) or equivalent constructs producing XRCC4 mutants K210R (lane 2) or K140R (lane 3). Three tandem FLAG peptides are incorporated in the XRCC4 constructs. Lysates were immunoprecipitated with anti-FLAG beads and analyzed by SDS-PAGE. The blot was visualized with HRP-conjugated anti-FLAG antibody. The unmodified protein is seen at 65 kDa. A modified form is present at 95 kDa in lanes 1 and 3. The molecular mass in kilodaltons is given to the left. (B) Confirmation that the modified form is SUMOylated. A parallel blot, representing proteins immunoprecipitated by anti-FLAG antibody, is visualized by anti-HA antibody directed against the triple HA tag on the SUMO-1 peptide. The predominant signal is seen at 95 kDa in lanes 1 and 3. This band is greatly reduced in intensity by the K210R mutation. A second band at 90 kDa may represent another SUMOylated protein that coprecipitates with XRCC4. The molecular masses of markers (lane M) are given in kilodaltons.
We determined whether coexpressing other enzymes known to contribute to this modification pathway would influence the degree of SUMOylation at steady state. Figure 4 shows that the abundance of the SUMOylated form can be raised in this way. As in the previous experiment, each lane represents FLAG-tagged XRCC4 protein expressed transiently along with HA-tagged SUMO-1 in CHO.XR-1 cells. The resulting proteins were immunoprecipitated using anti-FLAG antibodies, analyzed by SDS-PAGE, and visualized after blotting by antibody directed against the FLAG epitope. The major product is the unmodified protein at 65 kDa. The mono-SUMOylated form is visible at 95 kDa, and higher forms representing multiple SUMO modifications can also be recognized. Although poly-SUMOylation has been reported before, the significance is not clear (4, 54). The identity of these bands as SUMO-containing proteins was confirmed by parallel blots visualized with anti-HA antibody directed against the tag on the SUMO-1 moiety (not shown). Lane 1 shows again that the fraction SUMOylated in the absence of other transfected factors (lane designated “no add”) is only a small percentage of the unmodified protein. In contrast, overexpressing Ubc9, the unique E2 for the SUMO pathway, raises the SUMOylated fraction to about one-third of the total. Raising Ubc9 levels is expected to increase the degree of SUMOylation nonspecifically, but raising the levels of a SUMO E3 protein targets particular substrates. Lanes 3 to 5 represent the products of cells transfected with one of the following E3 proteins per lane: PIASxβ (also known as Miz1 or one isoform of PIAS2) (31), PIAS3 (37), and PIAS1 (26, 38), respectively. Both PIAS1 and PIASxβ raised the fraction of the SUMOylated form, the latter by a substantial degree to ca. 15% of the total. The form of PIASxβ we used was truncated to begin at amino acid residue 138, thereby removing the N-terminal DNA-binding domain of this protein. Full-length PIASxβ is known to activate and repress transcription in response to DNA damage (59). Our result indicates it may have a direct role in regulating DNA repair as well purely as a SUMO E3 ligase. Lane 6 represents cells transfected with a dominant-negative form of PIAS1 containing the three point mutations C345, 350, and 355S in the conserved RING motif (26). The SUMOylated form is slightly reduced from that in lane 1 in this and other repetitions of this experiment (not shown). This may indicate that PIAS1 contributes to the low level of SUMOylation seen when no additional enzymes are added.
FIG. 4.

SUMOylation of XRCC4 controlled by other enzymes. This immunoblot is directed against the FLAG epitope on XRCC4. Each lane represents protein derived from CHO.XR-1 cells cotransfected with plasmids encoding FLAG-XRCC4, HA-SUMO-1 and, for lanes 2 to 6, one additional plasmid encoding an enzyme of the SUMO pathway. Lane 1 was cotransfected with the empty expression vector. The unmodified protein is the major band at 65 kDa. Mono-SUMOylated XRCC4 is seen at 95 kDa, and higher-order adducts may be seen as well. The effects of the following enzymes were tested: Ubc9 (lane 2), PIASxβ (lane 3), PIAS3 (lane 4), PIAS1 (lane 5), and a dominant-negative form of PIAS1 (lane 6). The molecular masses in kilodaltons are listed on the left.
We investigated whether the SUMOylated form of XRCC4 differed in biochemical behavior from the unmodified form. Human XRCC4 residues 180 to 213 are known to be critical for dimerization and higher-order structure (21, 50). We sought to determine whether prevention of SUMOylation by mutation of K210 altered this behavior. Using protein produced by transient expression of individually tagged forms of the protein in CHO.XR-1 cells, we found that the non-SUMOylated K210R protein coprecipitated with the SUMOylated form of the normal protein (data not shown). In a DNA-binding assay, both the SUMOylated and the non-SUMOylated forms copurified (see Fig. S1 in the supplemental material). We detected no difference in the extent of ubiquitylation (see Fig. S2 in the supplemental material) or the kinetics of protein breakdown in the K210R mutant compared to the parental XRCC4 protein (data not shown). Although there are situations in which SUMOylation and ubiquitylation compete for the same lysine target (e.g., PCNA [16]), this does not seem to be the case in this instance. We also compared the physiologic properties of the K210R mutant with the parental XRCC4 protein by using the same transient-transfection approach. Under these conditions, V(D)J recombination of extrachromosomal substrates was almost equal in efficiency and fidelity. However, we now appreciate that the transient expression of XRCC4 does not recapitulate the normal regulation of the protein (discussed below).
We grew concerned that transient expression of XRCC4 might not adequately mimic the normal behavior of the protein with regard to the regulation of protein modification or intracellular localization. These activities could be sensitive to the high protein levels per cell achieved by transient expression or to other effects on cell physiology by the process of transfection itself. For example, transfection alone is sufficient to raise p53 levels (41). We therefore established cell lines with stable expression of FLAG epitope-tagged human XRCC4 protein or the K210R mutant integrated into the CHO.XR-1 parent. Individual clones were screened for the level of protein produced and qualitatively divided into high, moderate, or relatively low levels of XRCC4 on the basis of immunoblots directed against the FLAG epitope. A pair of cell lines expressing equal levels in the moderate range was analyzed by immunofluorescence microscopy to determine whether the wild-type XRCC4 protein and the K210R mutant showed the same subcellular localization. Cells in mid-log-phase growth were fixed and visualized by means of FITC-conjugated anti-FLAG antibody. Figure 5A shows the wild-type protein with diffuse nuclear localization and exclusion from the nucleoli. This is comparable to images made by using green fluorescent protein fusions of the normal protein (32). Figure 5B shows the DAPI-stained nuclei, and Fig. 5C shows panel A again, overexposed to reveal some residual cytoplasmic staining. A dramatically different behavior was obtained using the matched K210R mutant cell line in Fig. 5D. The non-SUMOylatable mutated protein now localized almost entirely to the cytoplasm. Figure 5E and F are the DAPI-stained and merged views of the same cells. Even though the steady-state level of SUMOylation was only a few percent, these cells revealed a substantial difference in the distribution of the total protein associated with the inability to SUMOylate it. It appears that SUMOylation is a transient process associated, at least in part, with nuclear transport. This is further explored below.
FIG. 5.

Intracellular localization of XRCC4 and mutant K210R. Cell lines stably expressing FLAG-XRCC4 were treated with FITC-conjugated anti-FLAG antibody to visualize the XRCC4 protein by immunofluorescence microscopy. Panels A to C show the distribution wild-type human XRCC4 to be largely nuclear. (A) FITC. (B) DAPI staining of nuclei. (C) Overexposure of panel A to show a small fraction of signal in the cytoplasm. Panels D to F show the cytoplasmic localization of XRCC4 in the K210R mutant cell line. The parallel cell line expressing the FLAG-XRCC4 mutant K210R was treated as described above. (D) FITC. (E) DAPI staining. (F) Merged image. Panels G to I depict SUMO-fusion of the wild-type XRCC4 that localizes to the nucleus. (G) FITC. (H) DAPI. (I) Overexposure of panel G to show residual cytoplasmic staining. Panels J to L show SUMO-fusion of the K210R mutant that now localizes to the nucleus. (J) FITC. (K) DAPI. (L) Overexposure of panel J to show residual cytoplasmic staining comparable to panel I.
The absence of XRCC4 from the nucleus in the K210R mutant cell lines might be expected to result in an equivalent absence of DNA repair activity. This was first tested by using a cell survival assay after exposure to graded doses of gamma radiation. Figure 6A represents the number of colonies, expressed as percentages, that grew after irradiation of 1,000 cells from each of three cell lines. The same cell lines were used in this analysis as presented in Fig. 5A and D, along with the control line generated by using the empty expression vector that does not encode XRCC4. This radiation-sensitive control cell line (labeled “empty vector”) showed 6.5% survival at 143 rads and was entirely killed with a dose of 610 rads, In contrast, the resistant cell line carrying the wild-type XRCC4 showed 35% survival at 143 rads and 1.7% survival at 610 rads. Consistent with the immunofluorescence data, the cell line expressing the XRCC4 K210R mutated protein demonstrated radiation sensitivity that was very similar to that of the negative control. This cell line displayed 8% survival at 143 rads and complete killing at 610 rads.
FIG. 6.

Radiation sensitivity of XRCC4 cell lines. Colony formation after exposure of cells to ionizing radiation is used as a measure of survival and is presented as a percentage of the initial number of cells. The cell line derived from CHO.XR-1 by stable incorporation of the empty expression plasmid serves as the radiation-sensitive control (labeled empty vector; squares). (A) The same cell lines used in Fig. 5A to F, stably expressing either unmutated XRCC4 (labeled wt XRCC4; diamonds) or the K210R mutated XRCC4 (circles), show radiation sensitivity that corresponds to the nuclear import of XRCC4. Error bars represent the standard error of the mean of duplicate experiments. (B) The cell lines used in Fig. 5G to L, representing SUMO-fusions to the wild-type XRCC4 (ovals) and K210R mutant (triangles), respectively, now show similar degrees of radiation resistance.
The V(D)J extrachromosomal recombination substrates pJH200 and pJH290 were used to demonstrate the ability of these cell lines to complete the recombination reaction. As in the radiation sensitivity assay, Table 1 shows a difference of >5-fold in recombination efficiency between the wild-type XRCC4 and cell lines expressing the K210R mutated protein. Note that the data for one of the recombination substrate plasmids, pJH200, have not been corrected by sequencing recombinant products. Hence, the factor of 5.3 difference between wild-type XRCC4 and the K210R mutant observed represents a conservative interpretation of the data and could represent an even greater difference if some of the rare recombinants are not legitimate V(D)J products.
TABLE 1.
Recombination efficiencies in cell lines expressing various versions of XRCC4 proteina
| CHO.XR-1 cell line | Recombination substrate | Recombination efficiency (%) | Fold reduction (mutant vs wt) |
|---|---|---|---|
| Vector only | pJH200 (signal joints) | 0 | |
| XRCC4 wt | pJH200 | 0.211 | |
| XRCC4 K210R | pJH200 | 0.040 | 5.3 (uncorrected by sequencing) |
| Vector only | pJH290 (coding joints) | 0.0009 | |
| XRCC4 wt | pJH290 | 0.188 | |
| XRCC4 K210R | pJH290 | 0.014 | 13.4 |
wt, wild type.
We pursued the role of SUMOylation by examining the behavior of the fusion protein formed by incorporating the SUMO peptide at the C terminus of the XRCC4 protein. This produces a protein that carries SUMO permanently, albeit not at the same location as when added naturally. A similar approach has previously been shown to redirect the localization of the NEMO protein to the nucleus, bypassing the requirement for regulated SUMOylation (17). We obtained an analogous result. Returning to Fig. 5, the control wild-type XRCC4 expressed as a SUMO fusion localizes to the nucleus (panels G to I). This is not surprising and serves to indicate that the fusion itself did not interfere with the protein stability or function (below). The interesting result was obtained by fusing SUMO to the C terminus of the K210R mutant (panels J to L). Now, the K210R mutation no longer prevents import, and the protein appears to be substantially nuclear. The SUMO fusion also restores XRCC4 function in the assay for radiation sensitivity. Figure 6B illustrates the activity in DNA repair of the same cell lines expressing the two SUMO fusion proteins in comparison to the wild-type XRCC4 and the cell line selected with the empty vector (therefore not expressing XRCC4). The SUMO-fusion of the K210R mutant now displays survival equivalent to the wild-type protein. Notably, the cell line expressing the empty vector showed only 11.7 and 2.4% viability at 200 and 400 rads, respectively, whereas the K210R mutant expressed as a SUMO fusion now yielded 53 and 18% viability under the same condition.
The final experiment explored the distribution of the SUMOylated form of XRCC4 (Fig. 7). The cell line expressing the FLAG-tagged wild-type protein was solubilized into cytoplasmic and nuclear fractions. Figure 7A shows three lanes, representing the abundance of the SUMOylated form of XRCC4 protein in total (T), nuclear (Nu), and cytoplasmic (Cy) fractions, as visualized by immunoblot. Despite the localization of the bulk protein to the nucleus (as in Fig. 5A), the SUMOylated form is almost entirely cytoplasmic. Figure 7B serves as a control for the fractionation, showing that histone H3 correctly distributes to the nuclear sample. The implications are discussed below.
FIG. 7.
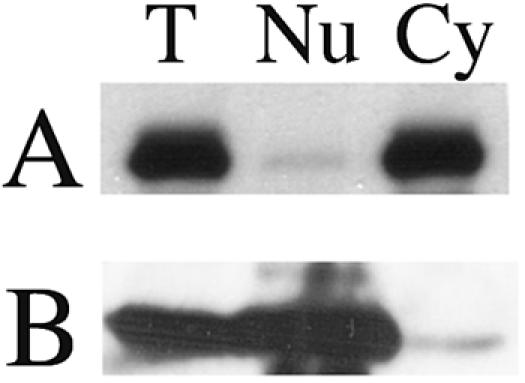
Fractionation of SUMOylated XRCC4. FLAG-tagged XRCC4 was harvested from the cell line and analyzed by immunoblot against the epitope tag. The band corresponding to the SUMO-modified form of the protein is shown in panel A. Lanes: T, total protein; Nu, nuclear fraction; Cy, cytoplasmic fraction. (B) Parallel blot directed against histone H3 to validate the fractionation.
DISCUSSION
The modification of proteins by the reversible covalent addition of a peptide is a flexible and powerful method of regulation. A large family of modifiers is recognized, and this is likely to continue to grow (49). Among these are the SUMO proteins, which exist as a four-gene family in humans: SUMO-1, SUMO-2, and SUMO-3 (reviewed in references 13, 19, and 36) and SUMO-4 (3). SUMOylation contributes to diverse physiologic functions, including the regulation of signaling pathways, transcription, and significantly for this report, the regulation of several components of DNA repair pathways. For example, p53 is regulated by a network of modifications that cause ubiquitin-mediated degradation during the uninduced state, and SUMOylation of p53 and several of its regulators (MDM2, p300, and HIPF2) upon induction (36). SUMOylation appears to be responsible for the subcellular localization of some proteins. One example is topoisomerase I, which appears to require SUMOylation to exit the nucleoli after treatment with camptothecin (40). Many other SUMOylated proteins localize to intranuclear structures called the PML nuclear bodies (reviewed in reference 19), and the BLM helicase appears to move between PML bodies and DNA damage-induced foci under the regulation of SUMO (7).
We were intrigued by the observation that human XRCC4 contained potential SUMOylation motifs and found that human XRCC4 is SUMOylated both in vitro and in vivo. We found that mutation of a single lysine (K210) prevents the modification, making this the likely target for SUMO addition. We note that these studies were performed based on the human protein, and the sites we manipulated are not precisely conserved in the mouse gene. Experiments are ongoing to compare phylogenetic differences. Our interest in SUMOylation originated with the recognition that the E3 enzymes for this pathway frequently contain the RING motif. This protein structure is also common among the ubiquitin E3 enzymes and is found in the RAG1 protein important for V(D)J recombination (20, 45, 62). However, we do not have evidence of a functional link between this aspect of RAG1 and the SUMOylation of XRCC4 at this time.
We found that only a small fraction of the XRCC4 pool, under 5% at steady state, appears to be SUMOylated. These data are derived from immunoblots of the protein expressed either transiently or in stably integrated expression constructs. Although the proportion of SUMOylated protein was similar on blots derived from these two methods of expressing the protein, we found a striking difference in the physiologic behavior between transient expression and expression at a lower level per cell obtained from integrated genes. When expressed transiently, the K210R mutant of XRCC4 was capable of complementing the defect in CHO.XR-1 cells and completed V(D)J recombination as effectively as its unmutated counterpart. In contrast, when expressed as an integrated transgene at a lower level of protein expression per cell, we found a considerable difference in behavior between cell lines expressing the K210R mutant versus the parental gene. In stable lines, the great majority of the XRCC4 K210R protein remained cytoplasmic. This change in localization was accompanied by a sensitivity to DNA DSBs and a sharp decrease in the ability to complete V(D)J recombination of test substrates. In retrospect, it appears that the failure to localize in the nucleus is not absolute. One explanation may be that a high level of expression of the mutant results in sufficient protein in the nucleus to complement the defect of CHO.XR-1 cells. We cannot exclude the possibility that the act of transient transfection itself could alter the fate of a DNA repair protein. Such a behavior has been noted with regard to the p53 protein (41). In other studies, transient transfection did not lead to detectable levels of SUMOylated PML-RAR fusion protein (6, 22), although this protein can be SUMOylated when expressed naturally. The authors of the second reference suggest that appropriate complex formation with other factors and subsequent localization to nuclear PML bodies must precede the modification. These concerns prompted our decision to use stable cell lines rather than transient expression for the later experiments.
SUMO modification has been associated with nuclear import previously. It is already appreciated that Ubc9, the SUMO E2 ligase, modifies RanGAP1 and the two proteins remain associated while bound to the cytoplasmic filaments of the nuclear pore complex (63). The SUMO-specific isopeptidase SENP2 is also associated with the nuclear pore and is located primarily on the nucleoplasmic side (11, 63). This configuration could easily allow SUMOylation to be a transient feature of transport into the nucleus. We cannot say whether a defect in nuclear localization is the only activity affected by preventing SUMOylation of XRCC4. Other aspects of intranuclear regulation by SUMOylation are known. For example, the mismatch excision repair enzyme thymine DNA glycosylase (12) binds the T-G mismatch and cleaves the thymine to generate an abasic site. The unmodified form of the enzyme has a high affinity for that site and appears to remain there until SUMOylated by another enzyme later in the repair pathway. This reduces the affinity of the glycosylase for DNA and returns it into solution until the SUMO is removed, thereby restoring the enzyme to its initial state. DNA topoisomerases I and II (2, 18) and the BLM helicase (7) show changes in localization under control of SUMO as well. It might appear that the result presented in Fig. 7 suggests that there is no role of SUMOylation of XRCC4 within the nucleus. It is certainly the case that, in this experiment, the majority of the SUMOylated form was retained in the cytoplasm. We note, however, that this experiment was performed under normal growth conditions. We are interested in whether conditions of DNA damage may lead to changes in XRCC4 modification, analogous to the induction of SUMOylation of the XPC protein by UV damage (53). It has also been reported that the degree of SUMO modification can be influenced by global stresses (23).
Although the characterization of the present study has been performed using the SUMO-1 member of the SUMO family, the other SUMO family members are known to have behaviors distinct from that of SUMO-1 (1, 55). It is therefore possible that additional complexity involving SUMOylation of XRCC4 will contribute to the regulation of this important DNA repair enzyme beyond its effect on cytoplasmic versus nuclear localization, perhaps through effects on the complexes it forms in the nucleus with other members of the DNA repair pathway.
In summary, we find that human XRCC4 can be SUMOylated in cells and that nuclear localization is itself regulated by the normally transient modification of the protein. The requirement for SUMOylation at K210 can be bypassed by incorporating SUMO at the C terminus of the protein.
Supplementary Material
Acknowledgments
This study was made possible by support from the National Institute of Allergy and Infectious Disease and institutional funds from the Albert Einstein College of Medicine. We are grateful for the use of facilities of the Cancer Center and of microscopy in the lab of R. Hazan. V.Y. is supported by NCI fellowship T32 CA09173 through an immuno-oncology training grant at AECOM.
We are grateful to N. Agus-Schreiber, D. Bohmann, X.-H. Feng, M. Gellert, M. Lechner, S. P. Lees-Miller, M. R. Lieber, R. Mizuta, and R. R. Reed who generously provided materials used in this study. We are especially appreciative of R. Hay and A. Ivanov for stimulating discussions and providing reagents. We are grateful to the anonymous reviewer for the suggestion to test the behavior of SUMO-fusion proteins.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Ayaydin, F., and M. Dasso. 2004. Distinct in vivo dynamics of vertebrate SUMO paralogues. Mol. Biol. Cell 15:5208-5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azuma, Y., A. Arnaoutov, and M. Dasso. 2003. SUMO-2/3 regulates topoisomerase II in mitosis. J. Cell Biol. 163:477-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bohren, K. M., V. Nadkarni, J. H. Song, K. H. Gabbay, and D. Owerbach. 2004. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 279:27233-27238. [DOI] [PubMed] [Google Scholar]
- 4.Bylebyl, G. R., I. Belichenko, and E. S. Johnson. 2003. The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J. Biol. Chem. 278:44113-44120. [DOI] [PubMed] [Google Scholar]
- 5.Critchlow, S. E., R. P. Bowater, and S. P. Jackson. 1997. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol. 7:588-598. [DOI] [PubMed] [Google Scholar]
- 6.Duprez, E., A. J. Saurin, J. M. Desterro, V. Lallemand-Breitenbach, K. Howe, M. N. Boddy, E. Solomon, H. de The, R. T. Hay, and P. S. Freemont. 1999. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localization. J. Cell Sci. 112(Pt. 3):381-393. [DOI] [PubMed] [Google Scholar]
- 7.Eladad, S., T. Z. Ye, P. Hu, M. Leversha, S. Beresten, M. J. Matunis, and N. A. Ellis. 2005. Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification. Hum. Mol. Genet. [DOI] [PubMed]
- 8.Gill, G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18:2046-2059. [DOI] [PubMed] [Google Scholar]
- 9.Gocke, C. B., H. Yu, and J. Kang. 2005. Systematic identification and analysis of mammalian small ubiquitin-like modifier substrates. J. Biol. Chem. 280:5004-5012. [DOI] [PubMed] [Google Scholar]
- 10.Grawunder, U., D. Zimmer, P. Kulesza, and M. R. Lieber. 1998. Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J: recombination and DNA double-strand break repair in vivo. J. Biol. Chem. 273:24708-24714. [DOI] [PubMed] [Google Scholar]
- 11.Hang, J., and M. Dasso. 2002. Association of the human SUMO-1 protease SENP2 with the nuclear pore. J. Biol. Chem. 277:19961-19966. [DOI] [PubMed] [Google Scholar]
- 12.Hardeland, U., R. Steinacher, J. Jiricny, and P. Schar. 2002. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 21:1456-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hay, R. T. 2005. SUMO: a history of modification. Mol. Cell 18:1-12. [DOI] [PubMed] [Google Scholar]
- 14.Hesse, J. E., M. R. Lieber, M. Gellert, and K. Mizuuchi. 1987. Extrachromosomal DNA substrates in pre-B cells undergo inversion or deletion at immunoglobulin V-(D)-J. joining signals. Cell 49:775-783. [DOI] [PubMed] [Google Scholar]
- 15.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 16.Hoege, C., B. Pfander, G. L. Moldovan, G. Pyrowolakis, and S. Jentsch. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419:135-141. [DOI] [PubMed] [Google Scholar]
- 17.Huang, T. T., S. M. Wuerzberger-Davis, Z. H. Wu, and S. Miyamoto. 2003. Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 115:565-576. [DOI] [PubMed] [Google Scholar]
- 18.Jacquiau, H. R., R. C. van Waardenburg, R. J. Reid, M. H. Woo, H. Guo, E. S. Johnson, and M. A. Bjornsti. 2005. Defects in SUMO conjugation and deconjugation alter cell sensitivity to DNA topoisomerase I-induced DNA damage. J. Biol. Chem. [DOI] [PubMed]
- 19.Johnson, E. S. 2004. Protein modification by SUMO. Annu. Rev. Biochem. 73:355-382. [DOI] [PubMed] [Google Scholar]
- 20.Jones, J. M., and M. Gellert. 2003. Autoubiquitylation of the V(D)J. recombinase protein RAG1. Proc. Natl. Acad. Sci. USA 100:15446-15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Junop, M. S., M. Modesti, A. Guarne, R. Ghirlando, M. Gellert, and W. Yang. 2000. Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. EMBO J. 19:5962-5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamitani, T., H. P. Nguyen, K. Kito, T. Fukuda-Kamitani, and E. T. Yeh. 1998. Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 273:3117-3120. [DOI] [PubMed] [Google Scholar]
- 23.Kurepa, J., J. M. Walker, J. Smalle, M. M. Gosink, S. J. Davis, T. L. Durham, D. Y. Sung, and R. D. Vierstra. 2003. The small ubiquitin-like modifier (SUMO) protein modification system in Arabidopsis. Accumulation of SUMO1 and -2 conjugates is increased by stress. J. Biol. Chem. 278:6862-6872. [DOI] [PubMed] [Google Scholar]
- 24.Lees-Miller, S. P., and K. Meek. 2003. Repair of DNA double strand breaks by non-homologous end joining. Biochimie 85:1161-1173. [DOI] [PubMed] [Google Scholar]
- 25.Li, Z., T. Otevrel, Y. Gao, H. L. Cheng, B. Seed, T. D. Stamato, G. E. Taccioli, and F. W. Alt. 1995. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell 83:1079-1089. [DOI] [PubMed] [Google Scholar]
- 26.Liang, M., F. Melchior, X. H. Feng, and X. Lin. 2004. Regulation of Smad4 sumoylation and transforming growth factor-beta signaling by protein inhibitor of activated STAT1. J. Biol. Chem. 279:22857-22865. [DOI] [PubMed] [Google Scholar]
- 27.Lieber, M. R., Y. Ma, U. Pannicke, and K. Schwarz. 2003. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell. Biol. 4:712-720. [DOI] [PubMed] [Google Scholar]
- 28.Lieber, M. R., Y. Ma, U. Pannicke, and K. Schwarz. 2004. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair 3:817-826. [DOI] [PubMed] [Google Scholar]
- 29.Mao, Y., S. D. Desai, and L. F. Liu. 2000. SUMO-1 conjugation to human DNA topoisomerase II isozymes. J. Biol. Chem. 275:26066-26073. [DOI] [PubMed] [Google Scholar]
- 30.Melchior, F., M. Schergaut, and A. Pichler. 2003. SUMO: ligases, isopeptidases, and nuclear pores. Trends Biochem. Sci. 28:612-618. [DOI] [PubMed] [Google Scholar]
- 31.Miyauchi, Y., S. Yogosawa, R. Honda, T. Nishida, and H. Yasuda. 2002. Sumoylation of Mdm2 by protein inhibitor of activated STAT (PIAS) and RanBP2 enzymes. J. Biol. Chem. 277:50131-50136. [DOI] [PubMed] [Google Scholar]
- 32.Mizuta, R., H. L. Cheng, Y. Gao, and F. W. Alt. 1997. Molecular genetic characterization of XRCC4 function. Int. Immunol. 9:1607-1613. [DOI] [PubMed] [Google Scholar]
- 33.Mizuta, R., M. Mizuta, S. Araki, and D. Kitamura. 2002. RAG2 is down-regulated by cytoplasmic sequestration and ubiquitin-dependent degradation. J. Biol. Chem. 277:41423-41427. [DOI] [PubMed] [Google Scholar]
- 34.Modesti, M., J. E. Hesse, and M. Gellert. 1999. DNA binding of Xrcc4 protein is associated with V(D)J. recombination but not with stimulation of DNA ligase IV activity. EMBO J. 18:2008-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modesti, M., M. S. Junop, R. Ghirlando, M. van de Rakt, M. Gellert, W. Yang, and R. Kanaar. 2003. Tetramerization and DNA ligase IV interaction of the DNA double-strand break repair protein XRCC4 are mutually exclusive. J. Mol. Biol. 334:215-228. [DOI] [PubMed] [Google Scholar]
- 36.Muller, S., A. Ledl, and D. Schmidt. 2004. SUMO: a regulator of gene expression and genome integrity. Oncogene 23:1998-2008. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa, K., and H. Yokosawa. 2002. PIAS3 induces SUMO-1 modification and transcriptional repression of IRF-1. FEBS Lett. 530:204-208. [DOI] [PubMed] [Google Scholar]
- 38.Nishida, T., and H. Yasuda. 2002. PIAS1 and PIASxalpha function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem. 277:41311-41317. [DOI] [PubMed] [Google Scholar]
- 39.Pickart, C. M. 2004. Back to the future with ubiquitin. Cell 116:181-190. [DOI] [PubMed] [Google Scholar]
- 40.Rallabhandi, P., K. Hashimoto, Y. Y. Mo, W. T. Beck, P. K. Moitra, and P. D'Arpa. 2002. Sumoylation of topoisomerase I is involved in its partitioning between nucleoli and nucleoplasm and its clearing from nucleoli in response to camptothecin. J. Biol. Chem. 277:40020-40026. [DOI] [PubMed] [Google Scholar]
- 41.Renzing, J., and D. P. Lane. 1995. p53-dependent growth arrest following calcium phosphate-mediated transfection of murine fibroblasts. Oncogene 10:1865-1868. [PubMed] [Google Scholar]
- 42.Robins, P., and T. Lindahl. 1996. DNA ligase IV from HeLa cell nuclei. J. Biol. Chem. 271:24257-24261. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez, M. S., C. Dargemont, and R. T. Hay. 2001. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276:12654-12659. [DOI] [PubMed] [Google Scholar]
- 44.Rooney, S., J. Chaudhuri, and F. W. Alt. 2004. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol. Rev. 200:115-131. [DOI] [PubMed] [Google Scholar]
- 45.Sadofsky, M. J. 2004. Recombination-activating gene proteins: more regulation, please. Immunol. Rev. 200:83-89. [DOI] [PubMed] [Google Scholar]
- 46.Sadofsky, M. J., J. E. Hesse, J. F. McBlane, and M. Gellert. 1993. Expression and V(D)J. recombination activity of mutated RAG-1 proteins. Nucleic Acids Res. 21:5644-5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sampson, D. A., M. Wang, and M. J. Matunis. 2001. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 276:21664-21669. [DOI] [PubMed] [Google Scholar]
- 48.Sancar, A., L. A. Lindsey-Boltz, K. Unsal-Kaccmaz, and S. Linn. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73:39-85. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz, D. C., and M. Hochstrasser. 2003. A superfamily of protein tags: ubiquitin, SUMO and related modifiers. Trends Biochem. Sci. 28:321-328. [DOI] [PubMed] [Google Scholar]
- 50.Sibanda, B. L., S. E. Critchlow, J. Begun, X. Y. Pei, S. P. Jackson, T. L. Blundell, and L. Pellegrini. 2001. Crystal structure of an Xrcc4-DNA ligase IV complex. Nat. Struct. Biol. 8:1015-1019. [DOI] [PubMed] [Google Scholar]
- 51.Stamato, T. D., R. Weinstein, A. Giaccia, and L. Mackenzie. 1983. Isolation of cell cycle-dependent gamma ray-sensitive Chinese hamster ovary cell. Somat. Cell Genet. 9:165-173. [DOI] [PubMed] [Google Scholar]
- 52.Steinacher, R., and P. Schar. 2005. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr. Biol. 15:616-623. [DOI] [PubMed] [Google Scholar]
- 53.Sugasawa, K., Y. Okuda, M. Saijo, R. Nishi, N. Matsuda, G. Chu, T. Mori, S. Iwai, K. Tanaka, K. Tanaka, and F. Hanaoka. 2005. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121:387-400. [DOI] [PubMed] [Google Scholar]
- 54.Tatham, M. H., E. Jaffray, O. A. Vaughan, J. M. Desterro, C. H. Botting, J. H. Naismith, and R. T. Hay. 2001. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276:35368-35374. [DOI] [PubMed] [Google Scholar]
- 55.Tatham, M. H., S. Kim, E. Jaffray, J. Song, Y. Chen, and R. T. Hay. 2005. Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mechanism for SUMO paralog selection. Nat. Struct. Mol. Biol. 12:67-74. [DOI] [PubMed] [Google Scholar]
- 56.Tatham, M. H., S. Kim, B. Yu, E. Jaffray, J. Song, J. Zheng, M. S. Rodriguez, R. T. Hay, and Y. Chen. 2003. Role of an N-terminal site of Ubc9 in SUMO-1, -2, and -3 binding and conjugation. Biochemistry 42:9959-9969. [DOI] [PubMed] [Google Scholar]
- 57.Terui, Y., N. Saad, S. Jia, F. McKeon, and J. Yuan. 2004. Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J. Biol. Chem. 279:28257-28265. [DOI] [PubMed] [Google Scholar]
- 58.Tsai, R. Y., and R. R. Reed. 1997. Using a eukaryotic GST fusion vector for proteins difficult to express in Escherichia coli. BioTechniques 23:794-800. [DOI] [PubMed] [Google Scholar]
- 59.Wanzel, M., D. Kleine-Kohlbrecher, S. Herold, A. Hock, K. Berns, J. Park, B. Hemmings, and M. Eilers. 2005. Akt and 14-3-3eta regulate Miz1 to control cell-cycle arrest after DNA damage. Nat. Cell Biol. 7:30-41. [DOI] [PubMed] [Google Scholar]
- 60.Weissman, A. M. 2001. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell. Biol. 2:169-178. [DOI] [PubMed] [Google Scholar]
- 61.Yu, Y., W. Wang, Q. Ding, R. Ye, D. Chen, D. Merkle, D. Schriemer, K. Meek, and S. P. Lees-Miller. 2003. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair 2:1239-1252. [DOI] [PubMed] [Google Scholar]
- 62.Yurchenko, V., Z. Xue, and M. Sadofsky. 2003. The RAG1 N-terminal domain is an E3 ubiquitin ligase. Genes Dev. 17:581-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang, H., H. Saitoh, and M. J. Matunis. 2002. Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol. Cell. Biol. 22:6498-6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao, X., and G. Blobel. 2005. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. USA 102:4777-4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.