

Abstract
Reversible transcriptional silencing of genes located near telomeres, termed the telomere position effect (TPE), is well characterized in Saccharomyces cerevisiae. TPE has also been observed in human tumor cell lines, but its function remains unknown. To investigate TPE in normal mammalian cells, we developed clones of mouse embryonic stem (ES) cells that contain single-copy marker genes integrated adjacent to different telomeres. Analysis of these telomeric transgenes demonstrated that they were expressed at very low levels compared to the same transgenes integrated at interstitial sites. Similar to the situation in yeast, but in contrast to studies with human tumor cell lines, TPE in mouse ES cells was not reversed with trichostatin A. Prolonged culturing without selection resulted in extensive DNA methylation and complete silencing of telomeric transgenes, which could be reversed by treatment with 5-azacytidine. Thus, complete silencing of the telomeric transgenes appears to involve a two-step process in which the initial repression is reinforced by DNA methylation. Extensive methylation of the telomeric transgenes was also observed in various tissues and embryonic fibroblasts isolated from transgenic mice. In contrast, telomeric transgenes were not silenced in ES cell lines isolated from 3-day-old preimplantation embryos, consistent with the hypothesis that TPE plays a role in the development of the embryo.
Telomeres are composed of short repeat sequences added onto the ends of chromosomes by telomerase (9, 37). Along with their associated proteins, these telomeric repeat sequences form a cap at the end of the chromosome that serves multiple essential functions, including disguising the ends from appearing as double-strand breaks and preventing chromosome fusion. While telomeres are maintained in germ line cells, they shorten with age in most somatic human cells due to a lack of sufficient telomerase activity (15). Extensive telomere shortening in somatic cells is a signal for replicative cell senescence (15), which results from failure to form a protective cap (29).
In addition to their role in protecting eukaryotic chromosomes, telomeres can also influence the expression of nearby genes. The reversible silencing of genes near telomeres, termed the telomere position effect (TPE), has been extensively studied in Saccharomyces cerevisiae (52). Yeast TPE was found to involve changes in chromatin conformation, to be dependent on both the distance from the telomere and the telomere length, and to be mediated through Sir2p, a type III histone deacetylase (21, 24, 52). Many of these studies were conducted using transgenes integrated near telomeres on truncated chromosome ends (i.e., missing much of the subtelomeric sequences) (2, 3, 20, 44). Subsequent studies of either transgenes integrated at yeast native telomeres or endogenous yeast genes, however, revealed a somewhat different picture, with some genes escaping TPE (18, 43, 55). This variability in the response to TPE has been demonstrated to result from the presence of certain DNA elements which function as insulators within subtelomeric regions of some yeast native telomeres (18, 43).
For mammalian cells, the loss of TPE as a result of telomere shortening was originally proposed to be a mechanism for mediating senescence (23). More recently, however, it has been proposed to be responsible for progressive changes in the gene expression profile as a function of replicative age (57). Furthermore, TPE may play a role in human genetic disease as a result of repositioning of active genes near telomeres or subtelomeric sequences following chromosome rearrangements, such as those seen in individuals with ring chromosomes (26, 54). Programmed silencing of genes in mammalian cells is known to play a critical role in development, genomic imprinting, and inactivation of the X chromosome (25). This type of silencing involves alterations in histone N-terminal tails, including deacetylation and methylation, which result in increased chromatin condensation. In addition to these changes in chromatin conformation, silencing in mammalian cells also involves methylation of DNA. Such methylation occurs primarily at CpG sites and subsequent to changes in chromatin structure to institute a stable shutdown of gene expression (8).
Initial studies with mammalian cells failed to find evidence for TPE or an altered chromatin conformation near a newly formed telomere in human cells (42) or for the repression of telomeric transgenes in a human/hamster hybrid cell line (6). Another study found no apparent influence of telomere length on the expression of an adjacent transgene in a human cell line (50). Later studies, however, demonstrated TPE in human cancer cell lines by using transgenes located adjacent to telomeres, similar to the approach used with yeast (5, 31). As in the case for yeast, Baur et al. found an increase in TPE in cells with longer telomeres, whereas Koering et al. found no such correlation. Both groups, however, found that TPE was reduced by trichostatin A (TSA), an inhibitor of type I and II histone deacetylases (36), indicating that alterations in chromatin conformation were involved. Koering and colleagues also reported that 5-azacytidine (5-AzaC), an inhibitor of DNA methyltransferases (28), had no effect. Therefore, DNA methylation, which is commonly associated with silencing of genes in mammalian cells (8, 25), does not appear to be involved in TPE in human cancer cell lines.
More recently, an investigation of the expression of endogenous genes located near telomeres in human fibroblasts found significant alterations in expression of a large number of genes in senescent cells (41). It was demonstrated, however, that telomere length alone was not sufficient to determine the expression status of these endogenous subtelomeric genes. More specifically, an examination of eight tandem telomeric genes on a single chromosome revealed a discontinuous pattern of altered gene expression during telomere shortening. In some respects, the results of this study are similar to the findings with yeast, in which endogenous subtelomeric genes also show variable expression levels, depending on the presence of DNA elements that could insulate them from TPE (18, 43).
For this study, we have investigated the influence of adjacent telomeres on the expression of two different transgenes, transcribed in opposite directions and from two different promoters, in both mouse embryonic stem (ES) cells and transgenic mice. This approach provides an opportunity to study TPE in normal mammalian cells both in culture and in vivo as well as to address how various genetic factors might influence this process.
MATERIALS AND METHODS
Cell lines and culture conditions.
The mouse ES cell line JM-1 was obtained from Roger Pedersen (University of California, San Francisco). JM-1 cells were initially grown and expanded on feeder layers of irradiated STO cells as previously described (46). These ES cells were subsequently adapted to growth without feeder layers by the addition of leukemia inhibitory factor (ESGRO; Chemicon, Temecula, CA) to the culture medium at 1,000 U/ml. The medium was also supplemented with ES-cell-qualified 8% fetal bovine serum (HyClone, Logan, UT), 8% newborn bovine serum (Invitrogen, Carlsbad, CA), and a nucleoside mixture (Specialty Media). Cultures were trypsinized and passaged every 3 to 4 days, as colonies reached ∼80% confluence. ES cells in culture were periodically (every five passages) monitored by karyotyping and cytogenetic analysis. At least every five passages, cells were frozen down in ES medium containing 12% dimethyl sulfoxide (Sigma, St. Louis, MO) and supplemented with 15% fetal bovine serum, and pellets were prepared for the isolation of genomic DNA and total RNA.
Plasmids.
We have previously described the construction of two plasmids, pNPT-tel and pNPT2-tel, which were used here for the generation of ES cell clones with telomeric or interstitial integration sites (35). Briefly, both plasmids were constructed from the pSXneo-1.6T2AG3 plasmid, which had been previously shown to be capable of seeding new telomeres upon integration (22). The pNPT-tel and pNPT2-tel plasmids contain an ampicillin resistance gene, a neomycin resistance (neo) gene, the herpes simplex virus thymidine kinase (HSV tk) gene, and 1.6 kb of telomeric repeat sequences. The neo gene has a promoter from the HSV tk gene with a polyomavirus enhancer (53), while the HSV tk gene has a mouse phosphoglycerate kinase gene (pgk) promoter for efficient expression in ES cells (48). Expression of the HSV tk gene makes cells sensitive to ganciclovir (10, 38). In pNPT-tel, an 18-bp I-SceI endonuclease recognition site was introduced between the HSV tk and neo genes, whereas in pNPT2-tel, the I-SceI site was introduced at a BstEII site in the 3′-untranslated region of the HSV tk gene near the telomeric repeat sequences. Transfections were performed with 20 μg of plasmid DNA linearized with the NotI restriction enzyme, which positions the telomeric repeat sequences in the proper orientation at one end.
Characterization of transfected ES cell clones.
All ES cell clones used for this study were generated by transfection via electroporation using linearized plasmid DNAs, as described previously (35, 49). Stably transfected clones, containing both intact neo and HSV tk genes, were selected in medium containing 300 μg/ml G418 (Invitrogen, Carlsbad, CA), followed by testing for sensitivity to 2 μM ganciclovir (Sigma, St. Louis, MO). The isolation and characterization of the telomeric A211 and A405 ES cell clones were previously described in detail (35). A third telomeric clone, A353, and a panel of clones with interstitial integration sites were isolated and characterized in a similar fashion. Genomic DNA from each clone was purified as described previously (39) and digested with appropriate restriction enzymes. Samples were then fractionated by agarose gel electrophoresis using standard protocols or pulsed-field gel electrophoresis with 1% agarose gels in 0.5 × TBE (0.045 M Tris-borate, 0.001 M EDTA) at 200 V at 10°C, with pulsing at 2- to 3.6-second intervals. Southern blot analysis was performed as described previously (17, 35, 49). Clones demonstrating restriction fragments consistent with a single copy of the integrated plasmid, either at a telomere or at an interstitial site, were picked for further analysis. The exact genomic locations of the plasmid sequences for telomeric clones A211 and A405 were determined by fluorescence in situ hybridization (FISH) and cloning of cellular DNA at the integration site as described previously (35). For late-passage telomeric clones, in addition to the routine cytogenetic and Southern blot examinations mentioned earlier, genomic DNAs were analyzed using BAL-31 nuclease (New England Biolabs, Ipswich, MA) to confirm that the transgenes had maintained their telomeric locations. Briefly, genomic DNAs were digested with BAL-31 (0.1 U/μg DNA) according to the manufacturer's instructions, and aliquots were removed at 0-, 15-, 30-, 60-, and 120-min intervals. The DNA samples were then extracted with phenol-chloroform and digested with either HpaI (A211) or BamHI (A405). Samples were then fractionated by either standard agarose gel electrophoresis or pulsed-field gel electrophoresis and subjected to Southern blot analysis as described above.
Derivation of FS-1, FS-2, and FS-3 subclones from clones A211 and A405.
Subclones FS-1 and FS-2, derived from clone A211, and subclone FS-3, derived from clone A405, were isolated as G418-resistant, ganciclovir-resistant colonies following the introduction of double-strand breaks at the I-SceI site within the plasmid sequences, as described previously (35). Briefly, transient expression of the I-SceI gene was achieved by transfection of the pCBASce plasmid containing the I-SceI gene with a chicken β-actin promoter (kindly provided by Maria Jasin and Sloan Kettering), which has been found to provide a high efficiency of cutting at I-SceI sites in mammalian cells (45).
Cytogenetic and FISH analyses.
Cytogenetic analysis and FISH of various ES cell clones were carried out periodically as described previously (35). FISH signals were detected using antidigoxigenin-fluorescein isothiocyanate (Roche) or antiavidin-rhodamine (Roche) according to the manufacturer's protocols. Slides were mounted on Vectashield (Vector Laboratories) with 0.1 μg/ml of DAPI (4′,6′-diamidino-2-phenylindole; Sigma) as a counterstain. For clone A353, the identity of the chromosome into which the plasmid sequences were integrated was determined using a chromosome 12-specific probe (Cambio, Cambridge, United Kingdom). FISH for analysis of the integrated plasmid sequences was performed as described previously (16, 33).
Treatment of cells with inhibitors of DNA methylation and histone deacetylases.
Treatment of the ES cells with 5-AzaC (Sigma, St. Louis, MO) was performed with 2 × 106 log-phase cells plated out in gelatin-coated T-75 flasks in duplicate. Cultures were then incubated for 96 h, with daily changes of fresh medium either with or without 3 μg/ml 5-AzaC. Cells were then rinsed with fresh medium and allowed to recover in the absence of the drug for 2 days prior to the preparation of genomic DNA and total RNA. Treatment with TSA (Sigma, St. Louis, MO) was performed on duplicate cultures in T-75 flasks that were plated the day before. Cultures were incubated either with or without TSA (300 nM) for 24 h, with the treatment resulting in the death of approximately 70 to 80% of the cells. The medium containing TSA was then removed, and cells were allowed to recover for 24 h in medium without TSA prior to genomic DNA and total RNA preparations.
Colony survival assays.
ES cell clones were passaged in parallel cultures either with or without selection in G418. To periodically monitor the expression of the neo and HSV tk transgenes, 2,000 log-phase cells from each culture were plated out in triplicate in gelatin-coated T-25 tissue culture flasks containing medium alone (to determine the plating efficiency) or medium containing either G418 (300 μg/ml) or ganciclovir (2 μM). After 7 days, colonies were fixed, stained (34), and scored. The numbers of G418- and ganciclovir-resistant colonies were then normalized to the plating efficiencies, and percentages of survival were calculated. The inverse of the percentage for G418-resistant colonies was plotted to monitor the loss of neo gene expression (G418-sensitive cells). ES cell clones in culture were monitored every five passages by cytogenetic analysis to monitor the chromosome number. In addition, ganciclovir-resistant colonies were periodically isolated and examined by PCR and DNA sequencing to screen for base mutations and/or deletions of the transgenes.
DNA methylation assays.
The methylation status of the DNA at the plasmid integration sites was determined by first digesting 20 μg of genomic DNA in duplicate with SacI at 37°C for 5 h. SacI-treated DNAs were then incubated with either MspI or HpaII at 37°C overnight. Both MspI and HpaII cleave DNA at 5′-CCGG-3′ sites; however, HpaII is sensitive to DNA methylation. Incubation was continued for an additional 2 h after the addition of another 5 units of the enzymes to ensure complete digestion. Southern blot analysis was then performed on the genomic DNA as described previously (35), using a 32P-labeled probe consisting of the pNTP plasmid, which lacks telomeric repeat sequences.
Analysis of transgene expression by q-RT-PCR.
Total RNA from each ES cell clone was prepared using a Midi purification kit (Invitrogen, Carlsbad, CA). Samples were treated with a DNA-free kit (Ambion, Austin, TX) to remove traces of genomic DNA contamination. Preparation of cDNAs from these RNA samples was done as described previously (14), with the following modifications: reactions were incubated for 40 min at 48°C, using 7.5 mM MgCl2, a 1 mM concentration of each deoxynucleoside triphosphate, 5 μM hexamers (random primers; Invitrogen, Carlsbad, CA), and 2.5 U/μl of Moloney murine leukemia virus reverse transcriptase (RT; Invitrogen, Carlsbad, CA). To control for genomic DNA contamination, a mock cDNA preparation of each sample was prepared in a parallel reaction without the presence of RT. Real-time quantitative RT-PCR (q-RT-PCR) was performed by the UCSF Cancer Center Genome Analysis core facility, using TaqMan dual-label probes (Perkin-Elmer, Norwalk, CN) and specific primers on an ABI 7700 Prism real-time thermocycler (PE Biosystems, Foster City, CA). Quantitative PCRs (in triplicate) and data analysis were performed as previously described (14). The linearity of the RT reactions was initially tested by using four different amounts of RNA (500, 250, 125, and 62.5 ng), and the PCR efficiency was optimized to nearly 100% by using sample test runs. Expression of the mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control for normalization of the target genes, using a GAPDH-specific forward primer (5′-TGCACCACCAACTGCTTAG-3′), reverse primer (5′-GGATGCAGGGATGATGTTC-3′), and TaqMan probe (5′-FAM-CAGAAGACTGTGGATGGCCCCTC-BHQ1-3′). Expression of the neo gene was determined using a neo-specific forward primer (5′-CTTGCTCCTGCCGAGAAAGT-3′), reverse primer (5′-TTCGCTTGGTGGTCGAATG-3′), and TaqMan probe (5′-6FAM-CAGCCGCCGCATTGCATCA-TAMRA-3′). To determine the expression of the HSV tk gene, the TaqMan amplicon was designed to span the junction of the phosphoglycerate kinase promoter and the HSV tk coding sequence, using a forward primer (5′-TCTTGGTGGCGTGAAACTCC-3′), reverse primer (5′-AGACGCGTGTTGATGGCAG-3′), and TaqMan probe (5′-FAM-TGTAGAAGCGCGTATGGCTTCGTACCC-TAMRA-3′).
Transgenic mice.
The A405 transgenic mouse strain was established from ES cell clone A405 in conjunction with the UCSF Cancer Center Transgenic Core Facility. A405 cells were injected into 50 blastocysts from C57BL/6J mice, resulting in the generation of four chimeric mice. Breeding these mice with C57BL/6J mice generated agouti mice derived from the ES clone A405. Southern blot analysis of tail DNAs demonstrated that approximately half of these mice contained the telomeric plasmid sequences. A405 mice containing a single copy of the telomeric plasmid were used for methylation analysis of plasmid sequences in tissues, isolation of primary cells, and establishment of ES cell lines (see below).
Isolation of ES cell lines.
The ES cell lines derived from the A405 transgenic mouse strain were established as previously described (40). Briefly, blastocysts were collected by flushing out the uterine horns of mice at 3.5 day postcoitum with M2 medium (Sigma). These blastocysts were then transferred individually to 10-mm tissue culture wells containing a layer of PMEF feeder cells (Specialty Media) in ES medium and were cultured at 37°C in a 5% CO2 humidified incubator for approximately 10 days. The blastocysts were then removed using a finely drawn micropipette, and the inner-cell-mass-derived outgrowths were trypsinized and transferred to new tissue culture wells containing PMEF feeder cells. After 4 days in culture, the ES cell-like clumps were selected, expanded, and frozen for future use.
RESULTS
Selection of mouse ES cell clones containing telomeric integration sites.
Mouse ES cell clones containing selectable marker genes located adjacent to a telomere were used to monitor TPE. These clones were obtained by transfection of a mouse ES cell line with linearized plasmids containing telomeric repeat sequences on one end (Fig. 1A), as previously described (35, 49). The plasmids used contain (i) a neo gene with an HSV tk promoter and a polyomavirus enhancer and (ii) an HSV tk gene with a mouse pgk promoter. The promoters are immediately adjacent to one another, with transcription outward in opposite directions. The neo gene is used for positive selection in medium containing G418, and the HSV tk gene is used for negative selection in ganciclovir. In addition to the selectable marker genes, these plasmids contain an 18-bp recognition site for the introduction of double-strand breaks with the I-SceI endonuclease. The two plasmids used in these studies, pNPT-tel and pNPT2-tel, are identical except for the location of the I-SceI site, which in pNPT-tel is located between the promoters for the neo and HSV tk genes, whereas in pNPT2-tel it is located at the 3′ end of the HSV tk gene. Most of the plasmids are integrated at apparently random interstitial sites. At some integration sites, however, the telomeric repeat sequences in the plasmid act as a seed for the formation of a new telomere and are elongated to a length similar to other telomeres within the cell (35, 49). Of a total of 1,005 clones that were resistant to G418 and sensitive to ganciclovir, only 5 were found to contain a plasmid located at a telomere when analyzed by Southern blot and cytogenetic analyses (35, 49).
FIG.1.

Generation and characterization of ES cell clones for analysis of TPE. (A) Strategy for obtaining a single chromosome marked with unique plasmid sequences at a telomere. Plasmid constructs used in the experiments included the following general characteristics: (i) two selectable marker genes, one for positive (neo and one for negative (HSV tk) selection; (ii) a stretch of telomeric repeats to seed a telomere; and (iii) a recognition site for the rare-cutting I-SceI endonuclease to cut off the telomere. NotI-linearized plasmid fragments were then used for transfection of mouse embryonic stem cells. Plasmid integration is random and occurs by nonhomologous end joining (NHEJ). At some integration sites, telomeric repeat sequences at the 3′ end of the plasmid are used by telomerase as a template for seeding a new telomere. (B) Physical maps of integrated plasmid sequences in ES cell clones. The genomic organizations of the telomeric plasmids in the parental clones A211 and A405 are compared with the inverted repeats in the FS-1, FS-2, and FS-3 subclones. Also presented are physical maps of the telomeric clone A353 and an example of an interstitial clone (Interstitial) retaining a portion of the telomeric plasmid at the integration site. Locations of EcoRI (E), HindIII (H), and SacI (S) recognition sites are indicated for clones A211, A405, and A353 and subclones FS-1, FS-2, and FS-3. Similarly, BglII (B) restriction sites for the interstitial clone are shown, along with the restriction fragments expected to hybridize with the pgk-poly(A) probe (dark horizontal line). For each clone, the adjacent cellular DNA (chromosome 18 for A211, chromosome 15 for A405, and chromosome 12 for A353), vector sequences (amp/ori), neo and HSV tk genes, and telomeric repeat sequences (arrowheads) are shown. The I-SceI recognition sites and locations and directions of the promoters for the neo and HSV tk genes are also indicated. (C) FISH analysis of telomeric plasmid integration site in ES cell clone A353. (Top) The location of the integration site in clone A353 (solid arrow) was determined by hybridization with the pNPT-Δ plasmid (yellow). Chromosomes were counterstained with propidium iodide. (Bottom) The identity of the chromosome containing the integration site was determined by hybridization with a chromosome 12-specific painting probe (arrows), while chromosomes were counterstained with DAPI.
Three ES cell clones, A211, A405, and A353, which were found to contain a single copy of the plasmid located at a telomere (Fig. 1B), were used for analysis of TPE. Two of these clones, A211 and A405, were previously described in studies on the consequences of telomere loss (35, 49). Clone A211 contains a single copy of pNPT-tel located at the telomere on the long arm of chromosome 18, while clone A405 contains a single copy of pNPT2-tel located at the telomere on the long arm of chromosome 15. Rescue of the plasmid sequences from clone A405 demonstrated that it is integrated in the middle of the promoter of the hnRNP A1 gene (GenBank accession no. DQ077812). This region is normally transcriptionally active in dividing cells (7). The establishment of a transgenic mouse with clone A405 allowed us to study TPE in vivo. Clone A353 contains a single copy of the pNPT2-tel plasmid located at the telomere on the long arm of chromosome 12 (Fig. 1B). The location of the integrated plasmid in clone A353 was revealed with FISH, using a probe consisting of the pNPT plasmid lacking telomeric repeat sequences (Fig. 1C). Southern blot analysis demonstrated that the integrated plasmid in A353 contains both of the marker genes but is missing much of the plasmid sequences containing the ampicillin resistance gene and the bacterial origin of replication (data not shown).
As controls, we utilized a number of other ES cell clones, named I-1 through I-7 (Fig. 1B), each of which has a single copy of the pNPT-tel plasmid integrated at an interstitial site, as determined by Southern blot analysis (data not shown). Careful examination of these interstitial clones revealed that four of them (I-1, I-2, I-3, and I-7) had retained a portion of the plasmid telomeric repeats at the integration site (Fig. 2). In contrast, interstitial clones I-4, I-5, and I-6 had lost most or all of the telomeric repeat sequences prior to integration.
FIG. 2.

Analysis of the presence of telomeric repeat sequences at interstitial integration sites. Genomic DNAs from clones I-1 through I-7 were digested with BglII and examined by Southern blot analysis. The probes used consisted of either the pgk-poly(A) sequence contained on the end of the HSV tk gene (left panel) or telomeric repeat sequences (right panel). Because BglII cuts in the center of the pgk-poly(A) fragment (Fig. 1B), the pgk-poly(A) probe hybridizes to a 2.2-kb fragment containing the HSV tk gene as well as an additional fragment containing the remaining 3′ portion of the linearized plasmid and the adjacent cellular DNA that is unique to each clone (fragments a to f). In addition, the pgk-poly(A) probe also hybridizes to a 1.9-kb fragment originating from the endogenous pgk gene (arrow). Hybridization with the telomeric repeat sequence probe demonstrated that only the integration sites in clones I-1, I-2, I-3, and I-7 contained sufficient telomeric repeat sequences to be detectable (a, b, c, and f).
In addition to these interstitial clones, we also utilized as controls subclones derived from A211 and A405 in which the plasmid sequences are no longer located adjacent to a telomere. These clones, FS-1, FS-2 (both derived from A211), and FS-3 (derived from A405), were isolated following the introduction of double-strand breaks into the plasmid sequences with the I-SceI endonuclease (35). Rescue of the plasmid sequences and FISH demonstrated that these clones had lost the HSV tk gene and that the remaining plasmid sequences were no longer adjacent to the telomere. Instead, the remaining neo reporter gene was now flanked by large inverted repeats extending from the breakpoints and was located far away from the chromosome end (Fig. 1B). These subclones often showed an increase in copy number of the plasmid sequences due to DNA amplification. A comparison of hybridization levels of plasmid sequences relative to endogenous cellular sequences by Southern blot analysis demonstrated that on average, subclone FS-1 contains one copy of the neo gene, subclone FS-3 contains two copies, and subclone FS-2 contains three copies (Fig. 3). The absence of a telomere adjacent to the neo gene in subclones FS-1, FS-2, and FS-3 provides a control for the effects of the cellular sequences proximal to the telomeric integration site.
FIG. 3.
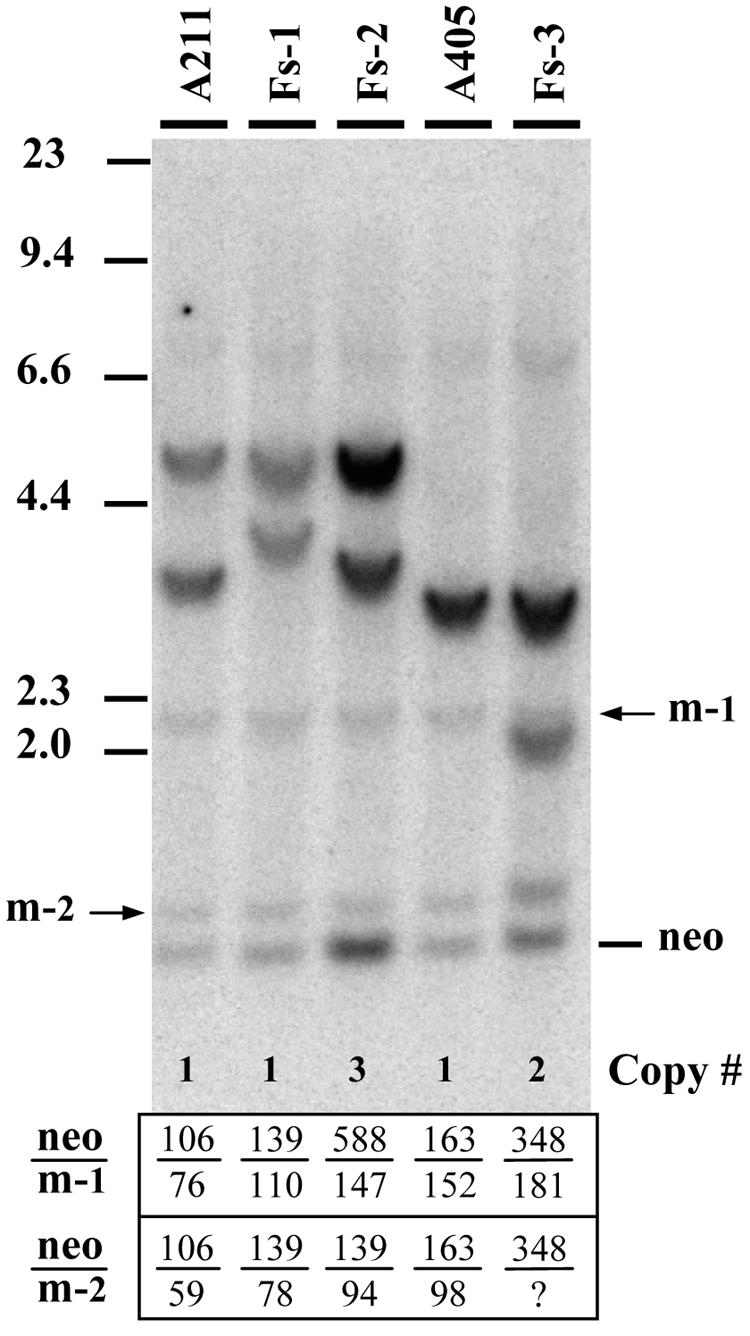
Analysis of the structures and copy numbers of the integrated plasmid sequences in clones A211 and A405 and their subclones that lost the telomeres following the introduction of double-strand breaks with the I-SceI endonuclease. Genomic DNAs from clone A211 and its subclones, FS-1 and FS-2, and from clone A405 and its subclone, FS-3, were digested with both EcoRI and HindIII and subjected to Southern blot analysis. The probe consisted of the pNPT plasmid, which lacks telomeric repeat sequences. The sizes of the bands and analysis of cloned fragments (35) are consistent with the structures shown in Fig. 1B. The average number of copies of the neo gene for each cell line (Copy #) was estimated by first determining the ratio of the intensity (in counts per minute) of hybridization of the neo gene to the intensity of hybridization to either of the two endogenous mouse pgk bands (arrows m-1 and m-2). This value was then compared to the value obtained for either clone A211 (for clones FS-1 and -2) or A405 (for clone FS-3) to determine the average number of copies for the neo gene in that cell line. The neo-to-pgk intensity ratios are shown in the table at the bottom of the figure. The question mark indicates that the intensity of the lower pgk band for clone FS-3 was uncertain because of superimposition of one of the plasmid bands.
Telomeric transgenes are expressed at low levels.
We initially examined the neo gene expression in our three telomeric clones, A211, A405, and A353, by Northern blot analysis. However, despite a strong signal for an internal actin control, the strength of the signal for the neo gene proved too weak to be detected in all three clones (data not shown). We therefore measured the expression of both the neo and HSV tk genes by using a sensitive real-time quantitative RT-PCR method employing TaqMan technology. The expression levels of the transgenes varied widely in the interstitial clones (Fig. 4; see Table S1 in the supplemental material). The three interstitial clones that contained few, if any, telomeric repeats at the integration site, I-4, I-5, and I-6, showed high expression levels, while three of the four interstitial clones with telomeric repeats at the integration sites (I-1, I-3, and I-7) showed very low expression levels. In contrast, interstitial clone I-2 showed high expression levels. However, this clone contained the least amount of telomeric repeat sequences at the integration site, as shown by the lower level of hybridization and the small size of the BglII fragment containing the telomeric repeat sequences (Fig. 2, fragment b). Thus, the presence of telomeric repeats at interstitial sites appears to either negatively influence the expression level of the transgenes or perhaps favor selection of already repressed loci (i.e., heterochromatin regions).
FIG. 4.

Comparative analysis of transgene expression levels in ES cell clones by q-RT-PCR. Expression levels of the neo and HSV tk genes, plotted relative to that of the mouse GAPDH gene, are shown for clones with interstitial integration sites (Int.) with (⧫) and without (⋄) detectable telomeric repeat sequences and for clones with telomeric integration sites (Tel.) selected with (•) and without (○) prescreening for sensitivity to ganciclovir. The expression levels of clone I-2, which contains only a small number of telomeric repeat sequences at the integration site, are indicated.
In contrast to the interstitial clones, all of the clones with telomeric integration sites showed low expression levels of the transgenes. On average, the expression levels of the neo and HSV tk genes in clones A211, A405, and A353 were 4.5-fold lower than their levels in the seven interstitial clones. However, this difference in expression levels becomes even more dramatic when the comparison is limited to clones I-4, I-5, and I-6, which do not contain any detectable adjacent telomeric repeat sequences, with average expression levels of the neo and HSV tk genes in the three telomeric clones being 7- and 13-fold lower, respectively. These results are consistent with the observation for HeLa cells made by Baur et al., who reported that the average expression level of a reporter gene at telomeric integration sites was approximately 10-fold lower than that at interstitial sites, which did not contain telomeric repeat sequences (5, 31).
Further evidence for TPE in mouse ES cells was found in our efforts to modify the conditions for selection of ES cell clones containing telomeric integration sites. Based on the premise that TPE in ES cells reduces expression of the transgenes located near telomeres, we eliminated the step involving prescreening for ganciclovir sensitivity and obtained a 10-fold higher frequency of clones containing telomeric integration sites (5 of 100 versus 5 of 1,005). As we suspected, these new clones demonstrated levels of expression of the neo and HSV tk genes that were even lower than those observed in clones A211, A405, and A353, which were preselected for their sensitivity to ganciclovir (Fig. 4; see Tables S1 and S2 in the supplemental material).
We also determined the expression levels of the neo gene in the three I-SceI-derived subclones of the telomeric clones, i.e., FS-1, FS-2 (both derived from A211), and FS-3 (derived from A405). As mentioned earlier, as a result of I-SceI-generated double-strand breaks, these subclones no longer contain the HSV tk gene, and the remaining neo gene is no longer adjacent to a telomere. After normalization for copy number (Fig. 3), the neo gene expression levels in all three subclones were initially similar to those in their parental clones (Fig. 5). However, while the expression levels of the telomeric neo gene decreased in clones A211 and A405 upon passaging, the expression levels in subclones FS-1, FS-2, and FS-3 increased with passaging in the absence of G418 selection. In fact, by passage 20, subclones FS-1 and FS-2 expressed the neo gene at approximately seven- and eightfold higher levels, respectively, than their parental A211 clone. Similarly, subclone FS-3 expressed the neo gene at fourfold higher levels than its parental A405 clone. This increased expression was not due to changes in copy number or chromosome rearrangement, as demonstrated by Southern blot and cytogenetic analyses (data not shown). Thus, passage in culture without selection results in a further drop in expression of the transgenes located adjacent to a telomere, while an increase in expression is seen after the loss of the telomere. This is consistent with a repressive role for the telomere, and it further indicates that while TPE in ES cells is inheritable, it is also reversible upon the loss of the telomere.
FIG. 5.

Telomeric transgenes are gradually derepressed after the loss of telomeres. q-RT-PCR analysis was performed to compare the initial expression levels (gray bars) of the neo gene in clones A211 and A405 and their subclones, which had lost the telomeres following expression of the I-SceI endonuclease, with expression levels after 20 passages (black bars).
Repression of telomeric transgenes is not alleviated by TSA.
DNA sequencing of reporter genes in the telomeric clones showed that low levels of expression were not due to any base changes or DNA damage (data not shown). Silencing of genes through heterochromatin formation in mammalian cells is often mediated by enzymatic deacetylation of the histone tails (8). To determine whether histone deacetylation is involved in the repression of telomeric transgenes, ES cell clones were treated with TSA, a highly specific inhibitor of class I and II histone deacetylases (36). In contrast to the results reported for TPE in human tumor cell lines (5, 31), the expression levels of the telomeric transgenes in mouse ES cell clones showed little or no increase following TSA treatment. Among the three telomeric clones, the expression of the neo and HSV tk genes in clones A211 and A353 was not affected by TSA treatment (Fig. 6). In clone A405, the expression levels of the marker genes increased approximately twofold; however, the levels of expression were still far below those of clones with interstitial transgenes lacking detectable telomeric repeats (Fig. 4). Thus, unlike the case in human cancer cell lines, deacetylation of histones by type I or II histone deacetylases does not appear to play a major role in TPE in mouse ES cells.
FIG. 6.

Effect of TSA on expression levels of transgenes in telomeric clones versus clones with interstitial integration sites containing telomeric repeat sequences. q-RT-PCR using total RNAs isolated from telomeric clones A211, A405, and A353 and interstitial clones I-1, I-2, I-3, and I-7 was used to determine the expression levels of the neo and HSV tk genes relative to that of the mouse GAPDH gene, with (black bars) or without (gray bars) TSA treatment. Note that clone I-2 contains the smallest number of adjacent internal telomeric repeats among the four interstitial clones shown here.
In contrast, ES cell clones containing interstitial integration sites showed a heterogeneous response to TSA treatment (Fig. 6). Similar to the results of an earlier study (5), TSA treatment had little effect on the expression levels of interstitial transgenes without adjacent telomeric repeat sequences (data not shown). This was not surprising since these interstitial transgenes all showed high levels of expression even without TSA treatment (Fig. 4). Similarly, TSA had little effect on the expression of interstitial transgenes in clone I-2, which contains minimal telomeric repeat sequences and exhibits high levels of expression (Fig. 6). In contrast, clones I-1, 1-3, and I-7, which also contain some portion of the repeats at the integration sites but express the transgenes at low levels, showed variable responses. While TSA treatment did not enhance expression of the transgenes in clone I-1, it significantly enhanced their expression levels in clones I-3 and I-7. In fact, the expression levels of both the neo and HSV tk genes in clones I-3 and I-7 increased 1.5- and 3-fold, respectively, reaching expression levels that were nearly half those observed in the clones with interstitial transgenes lacking detectable adjacent repeats. Thus, the repression of some interstitial integration sites containing telomeric repeat sequences appears to be mediated, at least partially, through type I and/or II histone deacetylases. This variation in the mechanism of repression for transgenes at interstitial sites containing telomeric repeat sequences may result from differences in the cellular DNA surrounding the integration sites, which has previously been shown to influence the silencing of transgenes in mammalian cells (56).
Telomeric transgenes are expressed at minimal levels required for selection and are fully silenced upon growth without selection.
Our initial studies indicated that clones containing telomeric transgenes gradually lost resistance to G418 and sensitivity to ganciclovir when grown in the absence of selection. Thus, in addition to their already low levels of expression, telomeric transgenes appeared to undergo complete silencing in the absence of selection. To further investigate this apparent silencing of the telomeric transgenes, clones A211 and A405 were tested for the ability to form colonies in G418 and ganciclovir at various times after growth without selection (Fig. 7A). The results demonstrated that, when grown without selection, clones A211 and A405 both show decreased expression of the neo and HSV tk genes, as shown by a decrease in the number of G418-resistant colonies (plotted as the inverse, G418-sensitive colonies) and an increase in the number of ganciclovir-resistant colonies. For both clones, the increase in the percentage of G418-sensitive colonies began immediately after the removal of selection, whereas there was a slight delay in the increase in ganciclovir-resistant colonies. This difference in timing could reflect a lower rate of silencing for the HSV tk gene or the fact that changes in the neo gene expression level are more critical for colony formation than changes in HSV tk gene expression. Regardless of the mechanism, the percentages of both G418-sensitive and ganciclovir-resistant colonies eventually leveled off. This leveling off was due to cells in the population that did not silence the telomeric transgenes, as shown by the fact that G418-resistant subclones selected from these plateau populations failed to silence the transgenes upon further growth without selection (unpublished observation). The percentages of cells in the population that failed to silence the transgenes gradually increased when clones A211 and A405 were kept under selection, suggesting that cells that do not silence have a selective advantage when grown in medium containing G418. It should be noted that throughout the culturing procedure, A211 and A405 ES cells maintained the plasmid sequences at their respective telomeric locations, as confirmed by cytogenetic examination, digestion with BAL-31 nuclease, and Southern blot analysis (see Materials and Methods). However, we also found that colony formation at later passages becomes highly erratic after the appearance of extensive aneuploidy in these ES cell lines (Fig. 7A), suggesting that the fluctuation in colony number was a consequence of increased chromosome instability in the late-passage ES cells (data not shown).
FIG. 7.

Decreasing expression of telomeric neo and HSV tk genes with increasing passages in culture. (A) Clones A211 (left) and A405 (right), grown for various numbers of passages in culture, were plated in medium containing either G418 or ganciclovir, and the numbers of resistant colonies were determined. Changes in expression levels are shown by increases in ganciclovir-resistant colonies (⋄) or decreases in G418-resisant colonies (•). Note that for the sake of comparison, the data on the loss of resistance to G418 has been plotted as the inverse. The timing of the appearance of aneuploidy and chromosome instability is designated by dashed lines, after which the numbers of G418- and ganciclovir-resistant colonies become unreliable. (B) q-RT-PCR was used to determine the expression levels of the neo (gray bars) and HSV tk (black bars) genes relative to that of the mouse GAPDH gene in clones A211 and A405 after various numbers of passages in culture.
To determine how increased sensitivity to G418 and resistance to ganciclovir corresponds to changes in gene expression, we next used q-RT-PCR to determine the expression levels of the neo and HSV tk genes after various numbers of passages in medium without G418 selection (Fig. 7B). Consistent with the studies on colony formation, the expression levels of both marker genes dropped off significantly with passaging for both A211 and A405. Moreover, in view of the fact that some cells in the population that did not silence the transgenes had a selective advantage, it appears likely that most cells eventually lose expression of both transgenes. Consistent with this conclusion, individual subclones selected for resistance to ganciclovir showed undetectable expression levels of both the neo and HSV tk genes (data not shown). A reduction in expression levels of the transgenes was also seen in clones with interstitial integration sites without telomeric repeat sequences after growth without selection (see Table S1 in the supplemental material). However, this did not result in a significant loss of resistance to G418 or sensitivity to ganciclovir after a similar number of passages (data not shown). This is likely to reflect the fact that expression at interstitial integration sites was initially high, so a more substantial drop in expression of the neo and HSV tk genes was required for a loss of resistance to G418 or sensitivity to ganciclovir. Taken together, these results suggest that expression levels of the telomeric transgenes are close to the minimal levels required for selection.
DNA methylation of transgenes.
We previously noted that for clone A211, the loss of resistance to G418 upon growth without selection correlated with partial digestion of the plasmid sequences with EcoRI (49). Genomic DNA from clone A211 grown under selection was completely digested with EcoRI, as shown by the presence of two bands on Southern blots, with one at 3 kb and one at 11 kb (Fig. 8, left panel). However, with an A211 culture grown without selection, a larger band appeared due to a failure to cut at an EcoRI site within the neo gene promoter. Sequence analysis of the DNA surrounding this EcoRI site (underlined) revealed that it was flanked on both ends by CpG residues (…CGAATTCG…), which are preferred sites for methylation. An identical EcoRI site has been previously reported to become resistant to EcoRI digestion due to methylation of these CpG sites (30). To further investigate the involvement of DNA methylation, an A211 culture in which about 50% of the cells in the population had lost their resistance to G418 was treated with 5-AzaC, an inhibitor of DNA methyltransferases that promotes demethylation of DNA (28). Following treatment with 5-AzaC, the cells regained their resistance to G418, and the 14-kb partial-digestion EcoRI fragment disappeared (Fig. 8, right panel). In addition, subclones isolated from A211 cultivated without selection revealed various extents of partial EcoRI digestion that showed a direct correlation with their sensitivities to G418 (Fig. 8, middle panel). Thus, methylation of the plasmid sequences appeared to be responsible for the further loss of neo gene expression when cells were grown without selection. It should be noted that although 5-AzaC treatment reversed the DNA methylation and complete silencing of the transgenes in cells cultured without selection, it did not alleviate the initial repression observed at the time of selection. In other words, treatment with 5-AzaC returned the expression of the telomeric transgenes to close to their original levels prior to culturing of the cells without selection (90% and 75% of the original levels for the neo and HSV tk genes, respectively).
FIG. 8.
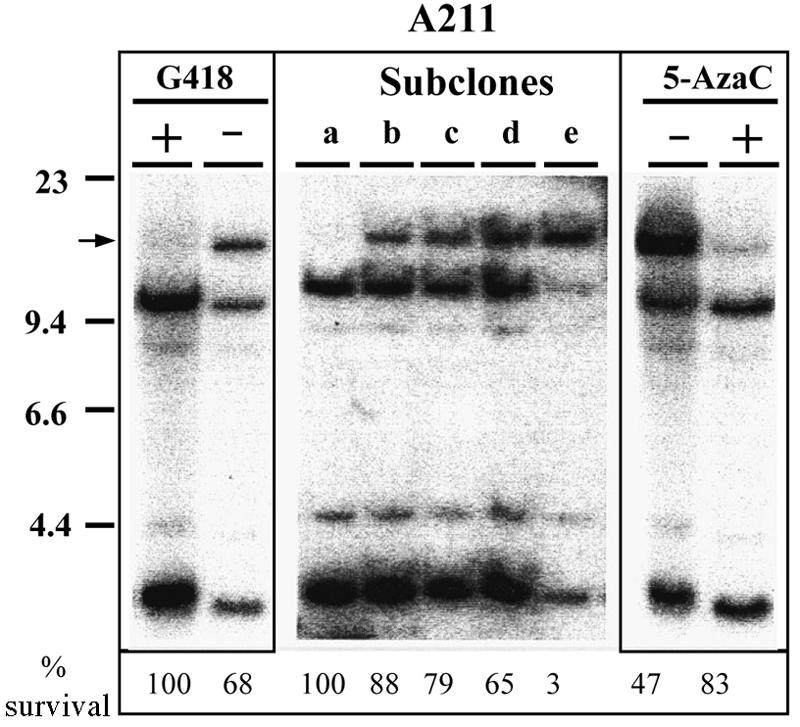
Partial digestion of an EcoRI site in clone A211 correlates with sensitivity to G418 and is a result of DNA methylation. Southern blot analysis was performed with EcoRI-digested genomic DNAs isolated from clone A211 grown with (+) or without (−) selection (left panel), individual A211 subclones selected after prolonged growth of clone A211 without G418 selection (middle panel), and clone A211 grown without selection and treated with (+) or without (−) 5-AzaC (right panel). Hybridization was performed using the pNPT plasmid lacking telomeric repeat sequences. The position of the EcoRI-resistant band is shown (arrow). The percent survival of each cell line plated in G418, normalized to its plating efficiency in the absence of selection, is shown at the bottom.
To further investigate the role of DNA methylation in silencing, genomic DNAs isolated from A211 and A405 cultures grown without selection for various numbers of passages were analyzed by Southern blotting using MspI and HpaII. These restriction enzymes, which are isoschizomers, both recognize a 4-bp sequence (CCGG). However, only HpaII is sensitive to methylation of the recognition site. For this analysis, genomic DNAs were first digested with SacI, which recognizes a 6-bp sequence and is not sensitive to DNA methylation (Fig. 9). The results clearly demonstrate a progressive methylation of plasmid sequences upon passage in culture without selection, as evidenced by the gradual appearance of SacI-specific bands. The timing of methylation corresponds to the timing of silencing for the neo and HSV tk genes (Fig. 7A), with full methylation of the transgenes reached by 20 passages (∼70 days) for clone A211 and 30 passages (∼100 days) for clone A405, suggesting that DNA methylation was responsible for the complete silencing of telomeric transgenes. Further evidence for this conclusion was found in the observation that treatment with 5-AzaC resulted in an increase in G418 resistance (Fig. 8), demonstrating that complete silencing was reversible and dependent on DNA methylation. Therefore, growth of clones A211 and A405 in G418 selects against methylation of the telomeric transgenes, although their methylation in some cells is evident even by the first passage after the removal of G418, as shown by the partial digestion of DNA with HpaII compared to digestion with MspI (Fig. 9).
FIG. 9.

Southern blot analysis demonstrating increasing methylation of telomeric plasmid sequences in clones A211 and A405 upon passaging in culture. Genomic DNAs isolated from A211 and A405 cultures, grown for various numbers of passages after removal of G418 selection, were digested with SacI, followed by digestion with either HpaII, which does not cut DNA methylated at the restriction site, or MspI, which is insensitive to DNA methylation. Hybridization was performed with the pNPT plasmid lacking telomeric repeat sequences. Digestion with SacI generated plasmid-containing fragments of 3 and 4 kb with clone A211 and of 1.9 and 8 kb with clone A405 (arrows). These large fragments disappeared if the DNA was cut with either MspI or HpaII. The presence of these bands in DNA digested with HpaII denotes its failure to cut due to extensive methylation of its recognition sites. In contrast, the presence of small plasmid-containing fragments (bracket) demonstrates extensive digestion of the same DNA preparations with MspI.
We also analyzed individual G418-resistant subclones that were isolated from A211 and A405 cultures grown for prolonged periods without selection. As mentioned earlier, these subclones represent a subpopulation that fails to silence the transgenes even when not under selection. Even after another 10 passages in culture, these subclones maintained their resistance to G418 and sensitivity to ganciclovir, and no methylation of the plasmid sequences was detected (data not shown). Thus, they appear to be defective in de novo methylation of the telomeric plasmid sequences. Whether this defect in de novo methylation is specific for telomeric transgenes or pertains to all transgenes or endogenous genes in these clones is not known. Regardless, these results provide additional evidence that DNA methylation is responsible for the extensive silencing of telomeric transgenes that is observed in cells grown without selection. In addition, the low expression levels of the telomeric transgenes in these methylation-defective subclones (1.37% and 0.78% of GAPDH levels for the neo and HSV tk genes, respectively) support the conclusion that the low expression levels of the telomeric transgenes in cultures grown under selection are not due to methylation but most likely reflect changes in chromatin conformation (8).
Silencing and DNA methylation in transgenic mice.
Making a transgenic mouse strain from ES clone A405 (see Materials and Methods) provided us with an opportunity to investigate the extent of repression of telomeric transgenes in vivo. Genomic DNAs isolated from lungs, livers, and kidneys were subjected to Southern blot analysis using either SacI/MspI or SacI/HpaII digestion to determine the extent of methylation. The results demonstrated extensive methylation of DNA in all three of the tissues studied (Fig. 10). To further investigate the extent of methylation in vivo, we also isolated mouse embryonic fibroblasts (MEFs) from the A405 mice. Similar to genomic DNAs from tissues of the A405 mice, genomic DNAs isolated from these fibroblast cultures demonstrated extensive methylation of the telomeric transgenes (data not shown). Furthermore, these fibroblast cultures failed to form any colonies in the presence of G418 but grew well in the presence of ganciclovir, demonstrating an extensive silencing of both telomeric transgenes. As with the ES cells, treatment of these fibroblast cultures with 5-AzaC resulted in a large number of cells (approximately 1 in 103) with resistance to G418 which were also sensitive to ganciclovir, indicating that both transgenes were intact. Thus, similar to ES cells grown without selection, cells in vivo also showed extensive silencing and methylation of the telomeric transgenes. In stark contrast, telomeric transgenes in the ES cell lines isolated from 3.5-day-old blastocysts obtained from A405 mice were not silenced, as demonstrated by their growth in G418 and their sensitivity to ganciclovir. However, similar to the case for clones A211 and A405, telomeric transgenes were expressed at very low levels in the ES cell lines derived from A405 mice. In two independently isolated lines, expression of the neo gene was 0.29 and 0.51% that of GAPDH, while expression of the HSV tk gene was 0.53 and 1.15% that of GAPDH, respectively (see Table S2 in the supplemental material). In addition, like clones A211 and A405, ES cells isolated from A405 mice showed a gradual loss of resistance to G418 and increased resistance to ganciclovir upon growth in culture without selection. The expression of these transgenes in ES cells from early embryos, but not in somatic tissues from older embryos or adult mice, is consistent with the well-established global demethylation of DNA in preimplantation embryos (25). It also indicates that silencing of the telomeric transgenes through DNA methylation is reversible under physiological conditions in the developing embryo.
FIG. 10.

Southern blot analysis demonstrating methylation of telomeric plasmid sequences in tissues of mice established from the ES cell clone A405. Genomic DNAs from kidneys, livers, and lungs were isolated from A405 mice containing telomeric plasmid sequences. Digestion with restriction enzymes and Southern blot analysis were then performed as described in the legend to Fig. 9.
DISCUSSION
The low expression levels of transgenes adjacent to telomeres compared to the levels of the same transgenes integrated at interstitial sites in our study demonstrates TPE in mouse ES cells, the first such report for normal mammalian cells. Following initial selection, expression levels of telomeric transgenes were, on average, 4.5-fold lower than their levels of expression at interstitial sites. Furthermore, expression levels of the neo and HSV tk genes at telomeric sites were 7- and 13-fold lower, respectively, than their expression levels from interstitial sites that did not contain telomeric repeat sequences. Further evidence for TPE in mouse ES cells was provided by the increased expression of the neo gene in the FS-1, FS-2, and FS-3 subclones, in which the transient expression of the I-SceI endonuclease had resulted in the loss of adjacent telomeres (Fig. 5). This rules out a role for proximal mouse sequences in repression of the telomeric transgenes, since the mouse DNA sequences originally proximal to the telomeric plasmid now flank the integration site on both sides in these subclones.
Our results also demonstrate that complete silencing of the telomeric transgenes occurs in the absence of selection and that this silencing involves extensive methylation of DNA (Fig. 8, 9, and 10). However, DNA methylation appears to be a secondary mechanism for suppression of the telomeric transgenes which is distinct from the mechanism responsible for their low levels of expression in cells kept under selection. This conclusion is supported by several lines of evidence. First, the very low expression levels of the telomeric transgenes in clones A211 and A405 occur even though there is no detectable or minimal methylation of the plasmid sequences. This also holds true for ES cell lines isolated from 3-day-old preimplantation embryos. Second, subclones of A211 and A405 that do not perform de novo methylation of the plasmid DNA and fail to silence the transgenes after prolonged growth without selection still express the transgenes at very low levels. Third, while treatment of cells with 5-AzaC reverses the DNA methylation and complete silencing of the telomeric transgenes, it does not alleviate their initial high level of repression. Finally, the increased expression of the neo gene in the FS-1, FS-2, and FS-3 subclones following the loss of the adjacent telomere (Fig. 5) is not accompanied by a decrease in DNA methylation (data not shown). Based on these observations, we believe that the complete silencing of genes located near telomeres in mouse ES cells occurs through a two-step mechanism, with an initial repression step mediated through a telomeric-specific chromatin conformation, which is then followed by complete silencing mediated by DNA methylation, similar to silencing in genomic imprinting and X chromosome inactivation (8). Like these other forms of epigenetic silencing, the complete silencing of telomeric transgenes is also reversible, as demonstrated by the fact that demethylation of DNA following treatment with 5-AzaC results in both an increase in survival in G418 and an increase in expression levels.
We also demonstrated extensive silencing and DNA methylation of the telomeric transgenes in tissues and MEFs isolated from A405 transgenic mice. The telomeric transgenes are silenced in essentially all of the cells in these tissues since the DNA containing the transgenes is completely resistant to digestion with methylation-sensitive restriction enzymes, and MEFs were unable to form any colonies in G418 but were able to form large numbers of colonies in ganciclovir. As in the case with ES cell clones A211 and A405, this silencing was reversible, as MEFs were capable of forming colonies in G418 after treatment with 5-AzaC. In contrast, ES cell lines established from early embryos of the A405 transgenic mice grew well in G418 and were sensitive to ganciclovir. This indicates that the DNA in these ES cells was not extensively methylated in vivo and therefore appears to have been demethylated, similar to the rest of the genomic DNA in the preimplantation embryo (8). However, the expression levels of the telomeric transgenes in these ES cell lines were still very low, similar to the levels expressed in clones A211 and A405. Combined, these results demonstrate the presence of a strong TPE in the mouse, which in combination with DNA methylation can lead to complete silencing of genes located near telomeres in both ES cells in culture and a variety of other cell types in vivo.
In some respects, our findings are consistent with studies of human tumor cell lines, which also utilized telomeric transgenes to demonstrate the presence of a substantial TPE (4, 31). However, we also found several significant differences between TPE in mice and that reported for human tumor cells. The TPE in human tumor cells was reversible by treatment with TSA, indicating that deacetylation of histones by type I and/or II histone deacetylases was involved. In contrast, we found that TSA did not produce a significant reversal of TPE in mouse ES cells. TSA did produce a slight increase in expression of the telomeric transgenes in clone A405. However, it is important that the transgenes in clone A405 are integrated into the promoter of the hnRNP A1 gene (unpublished observation), which is actively expressed in proliferating cells (7) and therefore might influence the effect of TSA on TPE. Regardless, the increase in expression level of the transgenes after TSA treatment (2.2- and 1.5-fold for the neo and HSV tk genes, respectively) was still much less than the 10- to 50-fold increase in expression reported for the telomeric transgenes in a human tumor cell line (4). This difference in response between mouse ES cells and human tumor cells to TSA may be an indication of different mechanisms for generating TPE in these cells, although it may also reflect differences in metabolism or uptake of TSA. One possible explanation is that the mouse homolog of yeast Sir2, Sir2α, is involved in TPE in mouse ES cells but not in human tumor cells. Like its counterpart in yeast, Sir2α is a class III histone deacetylase and is therefore not sensitive to TSA, and it yields the same array of products in vitro (1, 21). An inhibitor of Sir2, sirtinol, was found to have no effect on TPE in a human tumor cell line (31). However, because the ability of sirtinol to inhibit mammalian Sir2 is not well characterized, the potential role of histone deacetylation by Sir2 in mammalian TPE remains to be investigated. Also, a likely scenario is the involvement of histone methyltransferases in mouse TPE. A recent report from María Blasco's laboratory demonstrates an epigenetic regulation of telomere length in the mouse through changes in the methylation status of histone H3 Lys9 and concomitant changes in the binding of certain chromobox proteins (19).
Another striking difference between our results with mouse ES cells and studies of human tumor cell lines is in the role of DNA methylation in silencing of the telomeric transgenes. Although the expression levels of the telomeric transgenes in the mouse ES cells were very low even without DNA methylation, the complete silencing that occurred after growth without selection was accompanied by extensive DNA methylation. Similarly, complete silencing and extensive DNA methylation of the transgenes were also observed in vivo in various mouse tissues. The relationship between this complete silencing and DNA methylation was further demonstrated by the reversal of silencing upon treatment with 5-AzaC. In contrast, DNA methylation did not appear to play a role in TPE in the human tumor cell line studied by Koering and colleagues (31). This lack of response may have been due to the relatively low level of 5-AzaC that was used in that study (1 μM versus the 3 μM concentration used in our study). Moreover, this difference in response to 5-AzaC might also reflect basic differences in the ability of mouse ES cells and human tumor cell lines to perform de novo methylation of DNA.
There are several possible explanations for the observed differences in TPE/silencing between mouse ES cells and human tumor cells. First, it is possible that there is a fundamental difference in the mechanisms of TPE/silencing in humans and mice. For example, the difference in the lengths of telomeres in inbred laboratory mice and humans (47) could influence the mechanism of TPE/silencing. A more likely explanation, however, would be that the variations in the mechanism of TPE/silencing reflect fundamental differences between ES cells and somatic cells or between normal and tumor cells. It is clear from our data on MEFs and tissues obtained from A405 transgenic mice that mouse somatic cells are capable of maintaining the methylation patterns of the telomeric transgenes set at the embryonic stage. However, this is not an indication of de novo methylation capability in these cells. De novo methylation activity is present at high levels in mouse ES cells, as shown by the extensive DNA methylation of transgenes (32, 51). In contrast, somatic cells show limited de novo DNA methylation capabilities, which corresponds to the reduced expression of methyltransferase enzymes involved in de novo DNA methylation (12, 13, 32). Moreover, a decrease in global DNA methylation is also observed in cancer cells (27, 58), which also have reduced levels of DNA methyltransferases involved in de novo methylation (13). Thus, in view of the reduced de novo DNA methylation capability in somatic and cancer cells, it is not surprising that repression of the telomeric transgenes in human tumor cells was not followed by extensive DNA methylation (31). Similarly, the difference in response between mouse ES cells and human tumor cells to TSA might reflect the fact that a homolog of yeast Sir2, Sir2α, is involved in TPE in ES cells but not in somatic and/or tumor cells.
Finally, an intriguing observation of our study is that telomeric transgenes are completely silenced and extensively methylated in vivo in somatic mouse tissue. Although DNA methylation and silencing of transgenes are common in transgenic mice, DNA methylation-associated inactivation of interstitial transgenes generally exhibits mosaicism, the frequency of which depends on the integration site and the nature of the integrated sequences (56). The absence of mosaicism in the methylation and expression of the telomeric transgenes therefore shows that silencing at telomeres is exceptionally efficient. Consistent with this conclusion, the isolation of methylated DNA from human cells showed enrichment for subtelomeric DNA, demonstrating that the regions around telomeres are heavily methylated (11). In contrast to the tissues in adult mice or MEFs derived from 13-day-old embryos, ES cells isolated from preimplantation embryos initially showed little or no silencing of the telomeric transgenes. In fact, prior to implantation, DNA in the early embryo is known to undergo extensive demethylation. Then, between implantation and gastrulation, the embryo undergoes global de novo methylation to reestablish the DNA methylation pattern, which will be maintained throughout the life of the somatic cells in the animal (25). This expression of genes in the early embryo as a result of demethylation and their progressive silencing afterwards have been proposed to play an important role in development (25). The observed expression of telomeric transgenes in ES cells isolated from preimplantation embryos is therefore consistent with the hypothesis that the regulation of expression of genes through TPE has a role in early development. The lack of methylation and complete silencing of the telomeric transgenes in human tumor cell lines may also mean that telomeric genes normally expressed early in development are also expressed in cancer cells, although, as mentioned above, this may also just reflect the lack of de novo methylation in human tumor cells. Understanding the molecular mechanisms responsible for TPE and silencing of telomeric genes may therefore provide new insights into both human development and cancer.
Supplementary Material
Acknowledgments
We thank Michael Siu for helping with the initial screening of the second set of ES cell clones with telomeric plasmid integrations and Laure Sabatier for help with cytogenetic analysis of the telomeric clone A353. We also thank Bijan Fouladi for helpful discussions and John Donelson for proofreading the manuscript.
This work was supported by grant number RO1 ES008427 from the National Institute of Environmental Health Sciences, NIH.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Alcendor, R. R., L. A. Kirshenbaum, S. Imai, S. F. Vatner, and J. Sadoshima. 2004. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 95:971-980. [DOI] [PubMed] [Google Scholar]
- 2.Aparicio, O. M., B. L. Billington, and D. E. Gottschling. 1991. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell 66:1279-1287. [DOI] [PubMed] [Google Scholar]
- 3.Aparicio, O. M., and D. E. Gottschling. 1994. Overcoming telomeric silencing: a trans-activator competes to establish gene expression in a cell cycle-dependent way. Genes Dev. 8:1133-1146. [DOI] [PubMed] [Google Scholar]
- 4.Baer, R., A. Heppell, A. M. Taylor, P. H. Rabbitts, B. Boullier, and T. H. Rabbitts. 1987. The breakpoint of an inversion of chromosome 14 in a T cell leukemia: sequences downstream of the immunoglobulin heavy chain locus are implicated in tumorigenesis. Proc. Natl. Acad. Sci. USA 84:9069-9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baur, J. A., Y. Zou, J. W. Shay, and W. E. Wright. 2001. Telomere position effect in human cells. Science 292:2075-2077. [DOI] [PubMed] [Google Scholar]
- 6.Bayne, R. A. L., D. Broccoli, M. H. Taggart, E. J. Thomson, C. J. Farr, and H. J. Cooke. 1994. Sandwiching of a gene within 12 kb of a functional telomere and alpha satellite does not result in silencing. Hum. Mol. Genet. 3:539-546. [DOI] [PubMed] [Google Scholar]
- 7.Biamonti, G., M. T. Bassi, L. Cartegni, F. Mechta, M. Buvoli, F. Cobianchi, and S. Riva. 1993. Human hnRNP protein A1 gene expression. Structural and functional characterization of the promoter. J. Mol. Biol. 230:77-89. [DOI] [PubMed] [Google Scholar]
- 8.Bird, A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16:6-21. [DOI] [PubMed] [Google Scholar]
- 9.Blackburn, E. H. 2001. Switching and signaling at the telomere. Cell 106:661-673. [DOI] [PubMed] [Google Scholar]
- 10.Brisebois, J. J., and M. S. DuBow. 1993. Selection for spontaneous null mutations in a chromosomally-integrated HSV-1 thymidine kinase gene yields deletions and a mutation caused by intragenic illegitimate recombination. Mutat. Res. 287:191-205. [DOI] [PubMed] [Google Scholar]
- 11.Brock, G. J., J. Charlton, and A. Bird. 1999. Densely methylated sequences that are preferentially localized at telomere-proximal regions of human chromosomes. Gene 240:269-277. [DOI] [PubMed] [Google Scholar]
- 12.Chen, Z., K. S. Kowneman, and D. R. Corey. 2003. Consequences of telomerase inhibition and combination treatments for the proliferation of cancer cells. Cancer Res. 63:5917-5925. [PubMed] [Google Scholar]
- 13.Chen, Z., B. P. Monia, and D. R. Corey. 2002. Telomerase inhibition, telomere shortening, and decreased cell proliferation by cell permeable 2′-O-methoxyethyl oligonucleotides. J. Med. Chem. 45:5423-5425. [DOI] [PubMed] [Google Scholar]
- 14.Collins, C., J. M. Rommens, D. Kowbel, T. Godfrey, M. Tanner, S.-I. Hwang, D. Polikoff, G. Nonet, J. Cochran, K. Myambo, K. E. Jay, J. Froula, T. Cloutier, W.-L. Kou, P. Yaswen, S. Dairkee, J. Giovanola, G. B. Hutchinson, J. Isola, O.-P. Kallioniemi, M. Palazzolo, C. Martin, C. Ericsson, D. Pinkel, D. Albertson, W.-B. Li, and J. W. Gray. 1998. Positional cloning of ZNF217 and NABC1: genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc. Natl. Acad. Sci. USA 95:8703-8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Lange, T. 1995. Telomere dynamics and genome instability in human cancer, p. 265-293. In E. H. Blackburn and C. W. Greider (ed.), Telomeres. Cold Spring Harbor Press, Plainview, N.Y.
- 16.Dutrillaux, B., and J. Couturier. 1981. La pratique de l'analysee chromosomique. Masson, Paris, France.
- 17.Fouladi, B., D. Miller, L. Sabatier, and J. P. Murnane. 2000. The relationship between spontaneous telomere loss and chromosome instability in a human tumor cell line. Neoplasia 2:540-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fourel, G., E. Revardel, C. E. Koering, and E. Gilson. 1999. Cohabitation of insulators and silencing elements in yeast subtelomeric regions. EMBO J. 18:2522-2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Cao, M., R. O'Sullivan, A. H. Peters, T. Jenuwein, and M. A. Blasco. 2004. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 36:94-99. [DOI] [PubMed] [Google Scholar]
- 20.Gottschling, D. E., O. M. Aparicio, B. L. Billington, and V. A. Zakian. 1990. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63:751-762. [DOI] [PubMed] [Google Scholar]
- 21.Guarente, L. 2000. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 14:1021-1026. [PubMed] [Google Scholar]
- 22.Hanish, J. P., J. L. Yanowitz, and T. De Lange. 1994. Stringent sequence requirements for the formation of human telomeres. Proc. Natl. Acad. Sci. USA 91:8861-8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harley, C. B., A. B. Futcher, and C. W. Greider. 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345:458-460. [DOI] [PubMed] [Google Scholar]
- 24.Imai, S.-I., C. M. Armstrong, M. Kaeberlein, and L. Guarente. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795-799. [DOI] [PubMed] [Google Scholar]
- 25.Jaenisch, R., and A. Bird. 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33:245-254. [DOI] [PubMed] [Google Scholar]
- 26.Jalal, S. M., A. R. Harwood, G. S. Sekhon, C. Pham Lorentz, R. P. Ketterling, D. Babovic-Vuksanovic, R. G. Meyer, R. Ensenauer, M. H. Anderson, Jr., and V. V. Michels. 2003. Utility of subtelomeric fluorescent DNA probes for detection of chromosome anomalies in 425 patients. Genet. Med. 5:28-34. [DOI] [PubMed] [Google Scholar]
- 27.Jones, P. A., and P. W. Laird. 1999. Cancer epigenetics comes of age. Nat. Genet. 21:163-167. [DOI] [PubMed] [Google Scholar]
- 28.Juttermann, R., E. Li, and R. Jaenisch. 1994. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA methylation. Proc. Natl. Acad. Sci. USA 91:11797-11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karlseder, J., A. Smogorzewska, and T. de Lange. 2002. Senescence induced by altered telomere state, not telomere loss. Science 295:2446-2449. [DOI] [PubMed] [Google Scholar]
- 30.Kleymenova, E. V., X. Yuan, M. E. LaBate, and C. L. Walker. 1998. Identification of a tumor-specific methylation site in the Wilms tumor suppressor gene. Oncogene 16:713-720. [DOI] [PubMed] [Google Scholar]
- 31.Koering, C. E., A. Pollice, M. P. Zibella, S. Bauwens, A. Puisieux, M. Brunori, C. Brun, L. Martins, L. Sabatier, J. F. Pulitzer, and E. Gilson. 2002. Human telomeric position effect is determined by chromosomal context and telomeric chromatin integrity. EMBO Rep. 3:1055-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lei, H., S. P. Oh, M. Okano, R. Juttermann, K. A. Goss, and R. Jaenisch. 1996. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 122:3195-3205. [DOI] [PubMed] [Google Scholar]
- 33.Lemieux, N., B. Dutrillaux, and E. Viegas-Pequignot. 1992. A simple method for simultaneous R- or G-banding and fluorescence in situ hybridization of small single-copy genes. Cytogenet. Cell Genet. 59:311-312. [DOI] [PubMed] [Google Scholar]
- 34.Leonhardt, E. A., M. Trinh, K. Chu, and W. C. Dewey. 2000. Mutations induced in the HPRT gene by X-irradiation during G1 and S: analysis of base pair alterations, small deletions and splice errors. Mutat. Res. 471:7-19. [DOI] [PubMed] [Google Scholar]
- 35.Lo, A. W. I., C. N. Sprung, B. Fouladi, M. Pedram, L. Sabatier, M. Ricoul, G. E. Reynolds, and J. P. Murnane. 2002. Chromosome instability as a result of double-strand breaks near telomeres in mouse embryonic stem cells. Mol. Cell. Biol. 22:4836-4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marks, P. A., V. M. Richon, T. Miller, and W. K. Kelly. 2004. Histone deacetylase inhibitors. Adv. Cancer Res. 91:137-168. [DOI] [PubMed] [Google Scholar]
- 37.McEachern, M. J., A. Krauskopf, and E. H. Blackburn. 2000. Telomeres and their control. Annu. Rev. Genet. 34:331-358. [DOI] [PubMed] [Google Scholar]
- 38.Murata, S., T. Matsuzaki, S. Takai, H. Yaoita, and M. Noda. 1995. A new retroviral vector for detecting mutations and chromosomal instability in mammalian cells. Mutat. Res. 334:375-383. [DOI] [PubMed] [Google Scholar]
- 39.Murnane, J. P., L. F. Fuller, and R. B. Painter. 1985. Establishment and characterization of a permanent pSVori-transformed ataxia-telangiectasia cell line. Exp. Cell Res. 158:119-126. [DOI] [PubMed] [Google Scholar]
- 40.Nagy, A., M. Gertsenstein, K. Vinersten, and R. Behringer. 2003. Manipulating the mouse embryo: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 41.Ning, Y., J. F. Xu, Y. Li, L. Chavez, H. C. Riethman, P. M. Lansdorp, and N. P. Weng. 2003. Telomere length and the expression of natural telomeric genes in human fibroblasts. Hum. Mol. Genet. 12:1329-1336. [DOI] [PubMed] [Google Scholar]
- 42.Ofir, R., A. C. Wong, H. E. McDermid, K. L. Skorecki, and S. Selig. 1999. Position effect of human telomeric repeats on replication timing. Proc. Natl. Acad. Sci. USA 96:11434-11439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pryde, F. E., and E. J. Louis. 1999. Limitations of silencing at native yeast telomeres. EMBO J. 18:2538-2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Renauld, H., O. M. Aparicio, P. D. Zierath, B. L. Billington, S. K. Chhablani, and D. E. Gottschling. 1993. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev. 7:1133-1145. [DOI] [PubMed] [Google Scholar]
- 45.Richardson, C., M. E. Moynahan, and M. Jasin. 1998. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 12:3831-3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robertson, E. J. 1987. Embryo-derived stem cells, p. 71-112. In E. J. Robertson (ed.), Teratocarcinomas and embryonic stem cells: a practical approach. Oxford University Press, Oxford, United Kingdom.
- 47.Shay, J. W., and W. D. Wright. 2001. Telomeres and telomerase: implications for cancer and aging. Radiat. Res. 155:188-193. [DOI] [PubMed] [Google Scholar]
- 48.Soriano, P., C. Montgomery, R. Geske, and A. Bradley. 1991. Targeted disruption of the c-src proto-oncogene leads to osteoporosis in mice. Cell 64:693-702. [DOI] [PubMed] [Google Scholar]
- 49.Sprung, C. N., G. E. Reynolds, M. Jasin, and J. P. Murnane. 1999. Chromosome healing in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 96:6781-6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sprung, C. N., L. Sabatier, and J. P. Murnane. 1996. Effect of telomere length on telomeric gene expression. Nucleic Acids Res. 24:4336-4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stewart, C., H. Stuhlmann, D. Jahner, and R. Jaenisch. 1982. De novo methylation, expression, and infectivity of retroviral genomes introduced into embryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 79:4098-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tham, W. H., and V. A. Zakian. 2002. Transcriptional silencing at Saccharomyces telomeres: implications for other organisms. Oncogene 21:512-521. [DOI] [PubMed] [Google Scholar]
- 53.Thomas, K. R., and M. R. Capecchi. 1987. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51:503-512. [DOI] [PubMed] [Google Scholar]
- 54.van Karnebeek, C. D., S. Quik, S. Sluijter, M. M. Hulsbeek, J. M. Hoovers, and R. C. Hennekam. 2002. Further delineation of the chromosome 14q terminal deletion syndrome. Am. J. Med. Genet. 110:65-72. [DOI] [PubMed] [Google Scholar]
- 55.Vega-Palas, M. A., E. Martin-Figueroa, and F. J. Florencio. 2000. Telomeric silencing of a natural subtelomeric gene. Mol. Gen. Genet. 263:287-291. [DOI] [PubMed] [Google Scholar]
- 56.Whitelaw, E., H. Sutherland, M. Kearns, H. Morgan, L. Weaving, and D. Garrick. 2001. Epigenetic effects on transgene expression. Methods Mol. Biol. 158:351-368. [DOI] [PubMed] [Google Scholar]
- 57.Wood, J. G., and D. A. Sinclair. 2002. TPE or not TPE? It's no longer a question. Trends Pharmacol. Sci. 23:1-4. [DOI] [PubMed] [Google Scholar]
- 58.Yuasa, Y. 2002. DNA methylation in cancer and ageing. Mech. Ageing Dev. 123:1649-1654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.