

Abstract
Signaling through the insulin-like growth factor I receptor (IGF-IR) axis is essential for transformation by many dominantly acting oncoproteins. However, the mechanism by which IGF-IR contributes to oncogenesis remains unknown. To examine this, we compared transformation properties of the oncogenic ETV6-NTRK3 (EN) chimeric tyrosine kinase in IGF-IR-null R− mouse embryo fibroblasts with R− cells engineered to reexpress IGF-IR (R+ cells). We previously showed that R− cells expressing EN (R− EN cells) are resistant to transformation but that transformation is restored in R+ cells. We now show that while R− EN cells have intact Ras-extracellular signal-regulated kinase signaling and cell cycle progression, they are defective in phosphatidylinositol-3-kinase (PI3K)-Akt activation and undergo detachment-induced apoptosis (anoikis) under anchorage-independent conditions. In contrast, R+ cells expressing EN (R+ EN cells) suppress anoikis and are fully transformed. The requirement for IGF-IR in R− EN cells is overcome by ectopic expression of either activated Akt or a membrane-targeted form of EN. Moreover, compared to R− EN cells, R+ EN cells show a dramatic increase in membrane localization of insulin receptor substrate 1 (IRS-1) in association with EN. Since EN is known to bind IRS-1 as an adaptor protein, our findings suggest that IGF-IR may function to localize EN/IRS-1 complexes to cell membranes, in turn facilitating PI3K-Akt activation and suppression of anoikis.
The insulin-like growth factor I (IGF-I) receptor (IGF-IR) is a membrane protein tyrosine kinase (PTK) that binds the insulin-like growth factors IGF-I and IGF-II (reviewed in reference 38). Ligand binding autoactivates the PTK domain and links the receptor to downstream signaling pathways including the Ras-mitogen-activated protein kinase (Ras-extracellular signal-regulated kinase [ERK]) cascade (20) and the phosphatidylinositol-3-kinase (PI3K)-Akt pathway (23, 24). IGF-IR has a multitude of physiological functions in normal cells, including mitogenesis, suppression of apoptosis, cell adhesion, and regulation of cell size and life span (43, 46). In addition, abundant literature points to a critical role for the IGF-IR axis in cellular transformation (6, 7). Antibodies to IGF-IR (27) or IGFs (26), antisense receptor or IGF (14, 37), or dominant-negative IGF-IR mutants (26) all reverse the transformed phenotype or inhibit tumorigenesis. IGF-IR overexpression transforms mouse fibroblasts (28), and IGF-IR gene amplification has been reported in human malignancies including breast cancer (1, 9, 45). IGF-II overexpression is well documented in both pediatric and adult tumors (reviewed in reference 64). Recently, selective inhibition of the IGF-IR PTK was shown to block tumor growth in vivo in animal studies (25).
Perhaps most compelling evidence for a role of IGF-IR in oncogenesis is the observation that many dominantly acting oncoproteins such as activated Ras, c-Src, simian virus 40 large T antigen, and overexpressed receptor PTKs fail to transform cells lacking IGF-IR (reviewed in reference 5). These studies analyzed transforming properties of the respective oncoproteins in R− cells, which are IGF-IR−/− mouse embryo fibroblasts derived from mice with a targeted disruption of the IGF-IR locus (3, 40). Transformation can be restored by reintroduction of IGF-IR (44, 57) but not by overexpression of the major IGF-IR adaptor, insulin receptor substrate 1 (IRS-1) (5). These observations have led to speculation that IGF-IR activation provides a second, or complementary, signal that is essential for oncogenesis by dominantly acting transforming proteins (6, 7).
Many hypotheses have been put forth to explain how IGF-IR might contribute to oncoprotein function, including enhancement of mitogenesis or suppression of apoptosis (reviewed in reference 4). However, other studies indicated that the mitogenic and antiapoptotic functions of IGF-IR are distinct from its transformation activity (48, 52). Indeed, no theory fully explains the phenomenon, and the mechanism by which the loss of IGF-IR signaling blocks transformation remains contentious (reviewed in reference 7). Prevailing evidence suggests that resistance to apoptosis is at least partially involved, as inhibition of this pathway leads to massive apoptosis of tumor cells but much less so of normal cells (6). The IGF-IR axis may be particularly important for survival of tumor cells under anchorage-independent conditions, i.e., for suppression of anoikis (reviewed in reference 7). IGF-IR signaling protects suspended cells from anoikis (50, 66), and inhibition of the IGF-IR axis blocks anchorage-independent soft agar colony formation of tumor cells (56). Moreover, while antisense inhibition of IGF-IR had little effect on the survival of melanoma anchorage-dependent monolayer cultures, it profoundly blocked soft agar colony formation and tumorigenicity of the same cells (51). This is consistent with previous reports indicating that the apoptotic effect of the IGF-IR blockade is more pronounced in metastatic cells (21, 41), as it is hypothesized that the ability to survive under anchorage-independent conditions is a requirement of metastatic cells.
To specifically address how IGF-IR contributes to transformation by dominantly acting oncoproteins, we assessed its ability to complement the transforming properties of the ETV6-NTRK3 (EN) chimeric tyrosine kinase. This protein is encoded by the t(12;15)(p13;q25)-associated ETV6-NTRK3 gene fusion of congenital fibrosarcoma (31), mesoblastic nephroma (30, 53), acute myeloid leukemia (22), and human secretory breast carcinoma (62). EN is therefore unique among fusion oncoproteins in having potent transforming activities across multiple cell lineages (reviewed in reference 35). EN contains the sterile alpha motif (SAM) oligomerization domain of the ETV6 (or TEL) transcription factor linked to the PTK domain of the neurotrophin 3 receptor NTRK3 (TRKC) (31). It is a predominantly cytosolic resident protein that undergoes ligand-independent PTK activation through SAM domain-mediated polymerization (63), leading to the constitutive induction of cyclin D1 and aberrant cell cycle progression (61). EN constitutively activates two of the major effector pathways of wild-type NTRK3, namely, the Ras-ERK and PI3K-Akt cascades, and both are required for EN transformation (61). EN utilizes the IRS-1 protein as an adaptor to link to these pathways; the C terminus of EN forms a direct interaction with the phosphotyrosine binding domain of IRS-1, and IRS-1 binding is essential for EN transformation (34). We previously showed that EN fails to transform R− cells and that transformation is restored in R− cells engineered to reexpress IGF-IR (R+ cells) (44). Therefore, we have now utilized EN-expressing R− and R+ cells as a model system in which to study the mechanism by which IGF-IR complements EN in transformation. We find that while the absence of IGF-IR does not affect Ras-ERK induction of cyclin D1 and cell cycle progression, this receptor appears to be essential for anchorage-independent growth and Akt activation in EN-transformed cells. Moreover, targeting of the EN molecule to the plasma membrane through N-myristoylation increases membrane-associated IRS-1, activates Akt signaling, and induces transformation in the absence of IGF-IR, suggesting a role for this receptor in EN membrane targeting.
MATERIALS AND METHODS
Cell culture and transfection.
R− IGF-IR knockout mouse embryonic fibroblasts were engineered to overexpress IGF-IR and designated R+ cells, as described previously (44), and were grown in 9% fetal bovine serum (FBS) (Invitrogen) and Dulbecco's modified Eagle's medium (DMEM). The BOSC23 packaging cell line was obtained from Rob Kay (Terry Fox Laboratory, Vancouver, British Columbia, Canada) and grown in 9% FBS and DMEM. Confluent monolayers were trypsinized, resuspended as single cells, and replated at a concentration of 1.0 × 105 cells/ml on tissue culture dishes that had been coated with sterile 1.4% agar for anchorage-independent suspension cultures. Cells were isolated by centrifugation (1,000 rpm) every 48 h and replated in new medium on fresh agar-coated plates. Starvation of both monolayer and spheroid cells was accomplished by replacing the high-serum (9% FBS) medium with serum-free (0.25% FBS) medium for 20 to 24 h prior to harvesting of cells. Harvested cells were subsequently analyzed for protein expression as described below.
Infections of R− and R+ fibroblasts were carried out using the murine stem cell virus (MSCV) retroviral expression system (Clontech). Briefly, the BOSC23 packaging cell line was transfected with target plasmids using the calcium phosphate method as described previously (61). Cells coexpressing two different constructs were made by transfecting and selecting for the control (MSCV) or ETV6-NTRK3 constructs first and then coexpressing and selecting for the second construct (i.e., wild-type and mutant IGF-IR). Cells that survived selection were grown up to establish polyclonal, stably expressing cell lines. Expression of all proteins was confirmed by Western blotting.
DNA constructs.
The cDNA encoding N-myristoylated EN was produced via PCR amplification of EN residues 43 to 633 using a 5′ primer containing an HpaI restriction site and the N-myristoylation sequence from the Lck tyrosine kinase with a 3′ primer containing an EcoRI restriction site. 5′ hemagglutinin (HA)-tagged versions of wild-type and N-myristoylated EN cDNA were produced by PCR amplification of EN residues 43 to 633 using 5′ primers containing an HpaI restriction site and two tandem HA DNA sequences {for wild-type HA-tagged EN [(HA)EN]} or an HpaI restriction site, the Lck N-myristoylation sequence, and two tandem HA DNA sequences [for N-myristoylated (HA)EN] with a 3′ primer containing an EcoRI restriction site. PCR was performed using high-fidelity Pfu DNA polymerase (Stratagene). All PCR products were digested with HpaI/EcoRI and cloned into MSCVpuro. IGF-IR(Y950F) in the MSCV neomycin plasmid was a kind gift of Renato Baserga. IGF-IR(K1003A) was excised from pBPV(KA) (Renato Baserga) using SacI and NotI, treated with mung bean nuclease to give blunt DNA ends, and cloned into the HpaI site of MSCV neomycin. Myc-tagged Akt cDNA was excised from pUSE(Akt-myr) using XbaI, treated with mung bean nuclease, and cloned into the HpaI site of MSCV neomycin. All constructs were verified by restriction digest mapping and DNA sequencing.
Antibodies and Western blotting.
Monolayer cells grown to ∼90% confluence were rinsed once with phosphate-buffered saline (PBS) and lysed with 1,000 μl of phosphorylation solubilization buffer (50 mM HEPES, 100 mM NaF, 10 mM Na4P2O7, 2 mM Na3VO4, 2 mM EDTA, 2 mM NaMoO4, and 0.5% NP-40) containing protease and phosphatase inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, and 250 μM phenylmethylsulfonyl fluoride). Anchorage-independent cultures were spun down at 1,000 rpm, washed once with PBS, and lysed in 250 μl of the same buffer with 15 passages through a 22-gauge syringe to break apart spheroid structures. All cells were solubilized for 30 min at 4°C on a shaking platform. Lysates were cleared by centrifugation at 12,000 × g for 10 min at 4°C. Protein quantification of the lysates was performed using a detergent-compatible protein assay kit from Bio-Rad. Total cell lysate (30 μg) was mixed with Laemmli buffer and electrophoresed on 7.5 to 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels according to standard methods. Electrophoresed proteins were transferred onto nitrocellulose membranes (Bio-Rad) before immunoblot analysis with the indicated antibodies. The antibodies used were as follows: phospho-Akt Ser-473 rabbit monoclonal antibody (immunobolt [IB] 1:1,000; Cell Signaling), total Akt rabbit polyclonal antibody (IB 1:1,000; Cell Signaling); phospho-MEK1/2 Ser-217/221 rabbit polyclonal antibody (IB 1:1,000; Cell Signaling), cyclin D1 mouse monoclonal antibody (IB 1:2,000; Cell Signaling), actin goat polyclonal antibody (IB 1:2,000; Santa Cruz), phospho-glycogen synthase kinase 3β (GSK-3β) rabbit polyclonal antibody (IB 1:1,000; Cell Signaling), HA mouse monoclonal antibody (IB 1:1,000; BABCO), IRS-1 rabbit polyclonal antibody (IB 1:1,000, immunoprecipitation 2 μg/ml; Upstate Biotechnology), 4G10 antiphosphotyrosine antibody (IB 1:5,000; Upstate Biotechnology), Grb2 mouse monoclonal antibody (IB 1:5,000; BD Transduction Laboratories), caveolin 1 (IB 1:1,000; BD Transduction Laboratories), IGF-IR β-subunit rabbit polyclonal antibody (IB 1:1,000; Santa Cruz), NTRK3 rabbit polyclonal antibody (IB 1:1,000, immunoprecipitation 1 μg/ml; Santa Cruz), and poly(ADP-ribose) polymerase (PARP) (IB 1:1,000; Cell Signaling).
Soft agar colony assays.
Soft agar assays were performed as described previously (44). Briefly, cells were seeded in triplicate at a concentration of 8 × 103 cells/35-mm dish. Bottom layers were made up of 0.4% agar in 9% FBS and DMEM. Cells were resuspended in a top layer of 0.2% agar in 9% FBS and DMEM. Cells were fed every other day by placing 2 drops of medium on the top layer. After 2 weeks at 37°C, the number of single cells and colonies/high-power field were counted. Results were formulated as a percentage of macroscopic (>0.1-mm) colonies formed/total number of cells plated. Cell lines were examined at five sites per well for a total of 15 fields in a minimum of five separate soft agar experiments. The statistical significance of differences in the respective groups was evaluated using the Student's t test; P values of <0.05 were considered to be of statistical significance.
In vivo tumor growth in nude mice.
Pathogen-free male athymic nude mice, 6 to 8 weeks old, were obtained from Harlan. One million R− and derivative cells were injected subcutaneously at three sites/animal (five animals/group), for a total of 15 monitored sites for each cell line. Animals were housed in laminar flow racks and microisolator cages under specific pathogen-free conditions and received autoclaved food and water. Nude mice were evaluated for tumor growth periodically until 20 days after injection, a time point at which tumor growth in the group of R+ cells expressing EN (R+ EN cells) necessitated the termination of the experiment. Tumor volume was estimated using the following formula: tumor length × (tumor width)2 × 0.5236. The statistical significance of differences in tumor size in the respective groups was evaluated using the Mann-Whitney rank test; P values of <0.05 were considered to be of statistical significance.
Immunofluorescence microscopy.
Fibroblast cells exponentially growing on coverslips were rinsed with PBS and fixed with cold methanol at −20°C for 10 min. To detect HA-tagged EN [(HA)EN] or the N-myristoylated HA-tagged EN construct [(HA)ENmyr], coverslips were incubated overnight with mouse anti-HA antibody (1:1,000 dilution) followed by the secondary antibody Oregon green 514-conjugated goat anti-mouse antibody (Molecular Probes). Slides were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) and analyzed using a Zeiss Axioplan epifluorescent microscope equipped with a COHU charge-coupled-device camera. For confocal images, fibroblast cells grown on coverslips were starved overnight (16 h) in 0.25% serum medium, rinsed with PBS, and fixed with 4% paraformaldehyde for 10 min followed by permeabilization with 0.01% Triton X-100 for 10 min. Coverslips were incubated for 1.5 h with mouse anti-HA antibody (1:200 dilution) followed by incubation with Oregon green 514-conjugated goat anti-mouse antibody. Slides were counterstained with DAPI, and images were obtained using a Leica DM IRE2 inverted confocal microscope. For each immunofluorescence experiment, control staining in the absence of primary antibodies or using preimmune antibodies showed no signals (data not shown).
Subcellular fractionation.
R− (HA)EN, R+ (HA)EN, R− (HA)ENmyr, and R+ (HA)ENmyr monolayers at ∼90% confluence in 15-cm dishes were starved overnight (16 h) in 0.25% serum medium, washed with PBS, and then resuspended in 1.5 ml of homogenization buffer (20 mM HEPES, 50 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol, 0.25% NP-40, and protease and phosphatase inhibitors). Cells were incubated in homogenization buffer on ice for 10 min and then lysed with 10 passages through a 22-gauge needle. Samples were centrifuged at 800 × g for 10 min at 4°C to remove nuclei and cell debris. Supernatants were collected and centrifuged at 15,000 × g for 15 min at 4°C. The resulting supernatant was removed and used as the cytoplasmic fraction. The heavy membrane pellet was resuspended in homogenization buffer and designated the membrane fraction. Thirty micrograms of total protein from each fraction was loaded onto SDS-polyacrylamide gel electrophoresis gels and immunoblotted with anti-HA antibodies. The purity of the cytoplasmic and membrane fractions was validated with anti-Grb2 and anticaveolin antibodies, respectively.
BrdU proliferation assays.
R− EN and R+ EN spheroids were grown in medium containing 9% FBS for the times indicated. Fresh medium containing 9% FBS and 100 μM bromodeoxyuridine (BrdU) (Sigma) was then added for a further 24 h prior to harvesting. Cells were pelleted, formalin fixed, and embedded in paraffin according to standard protocols. Immunostaining with an anti-BrdU antibody (Sigma) was performed according to the manufacturer's instructions. All conditions were repeated in triplicate, and the percentage of BrdU-positive cells was calculated by counting cells in five representative high-power fields for each condition (approximately 200 to 400 cells/high-power field).
Fluorescence-activated cell sorter (FACS) analysis.
Monolayer and spheroid cells were resuspended in a 1:1 solution of PBS and Accumax cell dissociation reagent (Innovative Cell Technologies) and incubated for 15 min at 37°C with periodic vortexing to break apart cell clumps. Cells were then passed through a 70-μm nylon strainer, fixed in 70% ethanol, treated with 100 g/ml RNase, and stained with 50 g/ml propidium iodide, and live cells were analyzed for DNA content using the FACSCalibur flow cytometry system with CellQuest and Modfit LT analytic software (Becton Dickinson).
Caspase 3 activity assays.
The activity of caspase 3 was determined by using the fluorogenic caspase 3 substrate (Z-Asp-Glu-Val-Asp)2-rhodamine 110-bisamide (Calbiochem). Fibroblast cells were plated as a monolayer or spheroids in 10-cm dishes and grown for the times and conditions indicated. Cells were collected by centrifugation at 1,000 rpm, washed once in PBS, and lysed in caspase 3 lysis buffer {10 mM HEPES, pH 7.4, 50 mM NaCl, 2 mM MgCl2, 5 mM EGTA, 0.2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), and protease inhibitors as described above}. One hundred micrograms of protein lysate was combined with dithiothreitol-containing 2× reaction buffer and 1 mM caspase 3 substrate and incubated at 37°C for 1 h. The fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm with a Wallac 1420 multilabel counter (Perkin-Elmer). The relative amount of caspase 3 activity was expressed as the fluorescence ratio between the various treatments and the untreated R+ EN fibroblast monolayer grown to 80% confluence.
RESULTS
Anchorage-independent growth of ETV6-NTRK3-expressing cells requires IGF-IR.
Several studies have postulated a role for IGF-IR in supporting anchorage-independent growth of transformed cells (reviewed in reference 7). We previously reported that when cultured under anchorage-dependent monolayer conditions, EN-expressing R− fibroblasts (R− EN cells) have similar rates of growth and cell viability as EN-expressing R− cells engineered to reexpress IGF-IR (R+ EN cells) (44). However, R− EN cells are defective in the ability to form colonies in soft agar compared to R+ EN cells (44). Since soft agar colony formation, widely used as a criterion for cell transformation, is a measure of a cell's capacity to grow under anchorage-independent conditions (49), we hypothesized that IGF-IR may complement EN by conferring nonadherent growth ability to EN-transformed cells. To model anchorage-independent growth, we previously described the culturing of Ewing tumor cell lines as multicellular spheroids (36). This approach utilizes tissue culture plates overlaid with a nonadherent 1.4% agar coating to prevent attachment of cells to the plastic plate surfaces (54). Whereas most tumor cell lines form matrix-deficient multicellular spheroids through cell-cell adhesion under these conditions, nonmalignant cells generally fail to do so and undergo massive cell death in culture (8). We therefore cultured cell lines on agar-coated plates for 24 to 96 h in low (0.25%)- and high (9%)-serum medium as described previously (36). As shown in Fig. 1A and B, R− fibroblasts undergo rapid cell death as single cells, while R+ fibroblasts form only tiny clusters that undergo morphological changes suggestive of cell death after several days in culture. Although R− EN fibroblasts initially formed loosely formed larger spheroid structures over the first 24 h, we observed that by 96 h, R− EN cultures consisted of poorly formed aggregates of dead or dying cells (Fig. 1B). In contrast, R+ EN cells readily form multicellular spheroids within 6 to 24 h that are stable indefinitely in culture (data not shown). Therefore, the ability of fibroblasts to grow as anchorage-independent multicellular spheroids requires the presence of both EN and IGF-IR.
FIG. 1.

ETV6-NTRK3-induced growth of multicellular spheroids requires IGF-IR expression. Adherent monolayer cultures of mouse embryo fibroblasts negative (R−) or positive (R+) for IGF-IR and transduced with ETV6-NTRK3 (R−EN and R+EN, respectively) or constructs with vector alone (R−MSCV and R+MSCV, respectively) were grown to confluence, trypsinized, resuspended, and then replated on agar-coated dishes at a density of 1.0 × 105 cells/ml. Photographs of anchorage-independent cultures (magnification, ×100) were taken at 24 h (A) or 96 h (B) after plating on agar-coated plates.
ETV6-NTRK3-expressing cells lacking IGF-IR are defective in Akt activation.
We previously showed that EN strongly activates the Ras-ERK pathway, leading to cyclin D1 up-regulation and aberrant cell cycle progression as well as the PI3K-Akt cell survival cascade (61). Since both cascades are essential for EN transformation (61), we wondered whether IGF-IR might contribute to one or both pathways in EN-transformed cells. Previous studies of anchorage-dependent monolayer cultures failed to detect differences in the activation of these pathways in R− EN cells compared to R+ EN cells under serum-free conditions (44). However, we did observe a deficiency in the ability of R− EN cells to fully activate Akt after serum stimulation, while MEK activation was less obviously affected (44). Given the requirements for IGF-IR in anchorage-independent growth as shown above, we next wished to determine whether these pathways were differentially activated in cells grown as spheroid cultures. R+, R− EN, and R+ EN cell lines (R− cells alone are not viable under these conditions) were cultured on agar-coated plates for 48 h in low (0.25%)- and high (9%)-serum medium as described above. Cell lysates were then immunoblotted for phosphorylated MEK1/2, cyclin D1, and phosphorylated Akt (Ser-473). As shown in Fig. 2A, R+ spheroid cultures grown in low-serum medium show minimal activation of MEK1/2 compared to R− EN and R+ EN cell lines. All cell lines responded to serum stimulation by enhanced MEK activation, indicating that the Ras-ERK pathway is intact in these cells (Fig. 2B). R− EN and R+ EN spheroids also both showed serum-independent cyclin D1 up-regulation, while in R+ spheroid cultures, cyclin D1 expression remained low. Therefore, MEK activation and cyclin D1 induction both appear to be serum and IGF-IR independent in EN-expressing spheroids. This is consistent with our findings in monolayer cultures of EN-transformed fibroblasts that show serum-independent MEK and cyclin D1 induction (44, 65). However, while R+ and R+ EN spheroid cultures both showed Akt activation (as detected by Ser-473 phosphorylation) that could be enhanced by serum stimulation, Akt phosphorylation was severely impaired in R− EN spheroids in both low and high concentrations in serum. Moreover, Akt activation in these cells could not be rescued by higher serum concentrations or over a longer time course of serum stimulation (data not shown). To confirm that defects in phosphorylation of Akt at serine 473 represented impaired Akt kinase activity, cell lysates from R+, R− EN, and R+ EN cells grown as spheroids in low-serum medium were examined for their level of Ser-9 phosphorylation of GSK-3β, a known Akt substrate (58). GSK-3β showed significantly lower levels of phosphorylation in R− EN spheroids compared to either R+ or R− EN in the absence of serum, indicating a functional loss of Akt activity in this cell line. From these observations, we conclude that the transformation-deficient R− EN cell line is impaired in the activation of the PI3K-Akt pathway under anchorage-independent conditions. However, Ras-ERK pathway activation and cyclin D1 up-regulation appear to be intact in these cells, indicating that the induction of this cascade is dependent on EN expression but is independent of IGF-IR in nonadherent cultures.
FIG. 2.
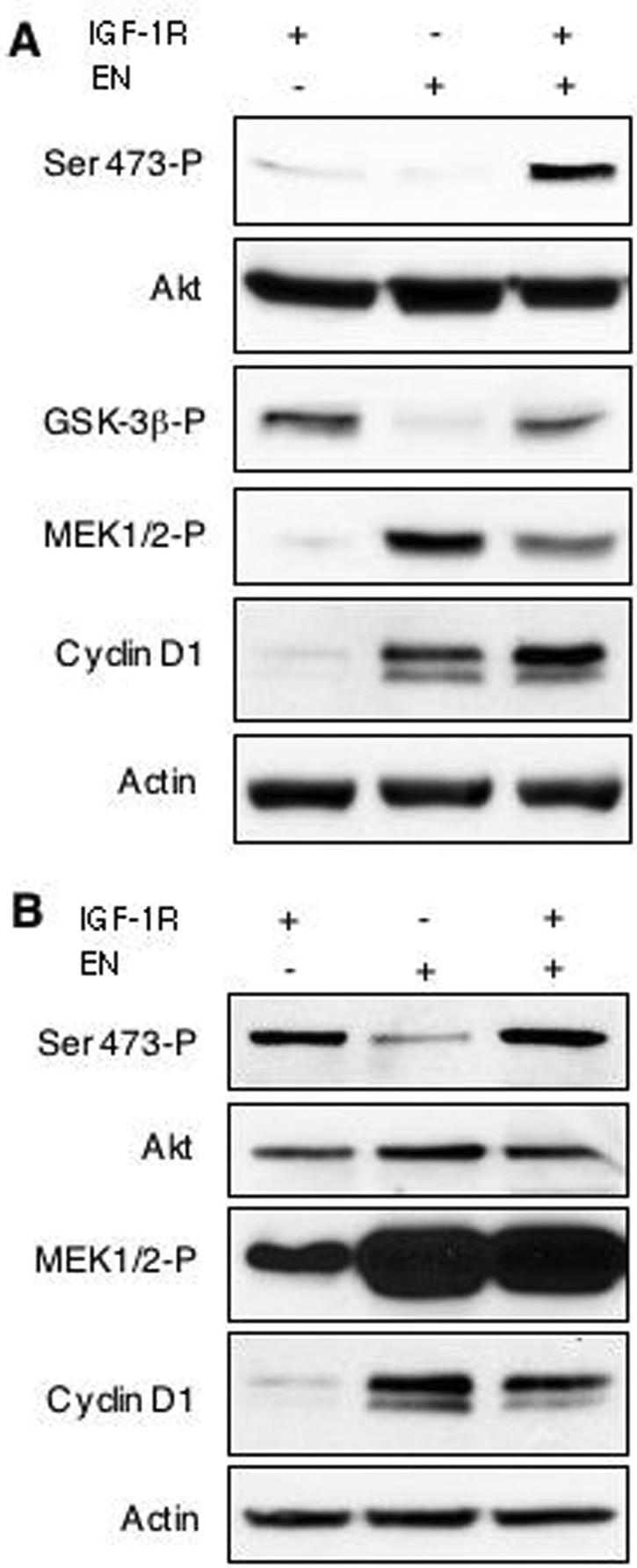
Akt activation in EN-expressing cells requires IGF-IR. R+, R− EN, and R+ EN mouse embryo fibroblasts (IGF-IR + or −, R+ or R− cells, respectively; EN + or −, with or without EN expression, respectively) were cultured under anchorage-independent conditions using agar-coated plates in either 0.25% (A) or 9% (B) serum for 48 h. Cells were then lysed and subjected to immunoblotting using antibodies to the indicated proteins. Activated Akt (Ser-473-P), GSK-3β (GSK-3β-P), and MEK1/2 (MEK1/2-P) were assessed using antibodies to phosphorylated proteins. Detection of actin was used as a loading control.
Expression of activated Akt restores anchorage-independent growth and transformation activity to ETV6-NTRK3-expressing IGF-IR-null fibroblasts.
To determine whether the critical deficiency preventing R− EN cells from anchorage-independent growth and cellular transformation is their inability to activate the PI3K-Akt pathway, we expressed a constitutively active myristoylated Akt construct (Akt-myr) (2) in these cells. Stable expression of Akt-myr in R− and R− EN cell lines was documented in Western blots as a slight band shift upwards in mobility due to the presence of a Myc epitope tag on the exogenous protein. Despite the fact that total Akt immunoblotting indicated only a small increase in overall Akt expression due to the presence of Akt-myr, we saw a significant increase in Akt phosphorylation at Ser-473 (almost exclusively attributed to the exogenous band), indicating the restoration of PI3K-Akt signaling in R− EN/Akt-myr cells. Increased phosphorylation of the downstream Akt substrate GSK-3β correlated with Akt-myr expression, indicating an increase in Akt kinase activity. In contrast, expression of Akt-myr had no effect on the Ras-ERK pathway, as MEK activation remained low in the R−/Akt-myr cells and comparable for R− EN/Akt-myr, R− EN, and R+ EN cells (Fig. 3A, compare lanes 2, 3, and 5).
FIG. 3.

Restoration of Akt activation in R− EN cells rescues anchorage-independent growth and in vivo transformation. (A) Myristoylated (constitutively active) Akt was expressed (Akt-myr +) in R− (IGF-IR −) or R+ (IGF-IR +) cells transduced with EN (EN +) or vector alone (EN −). Spheroid cultures were grown in 0.25% serum, lysed, and immunoblotted. Expression of the Akt-myr protein was detected as an upward band shift in both phosphorylated Akt (Ser 473-P) and total Akt immunoblots due to the presence of a Myc tag. Phosphorylated Akt substrate GSK-3β (GSK-3β-P) was detected using a phospho-specific antibody, as was activated MEK1/2 (MEK1/2-P). Detection of actin was used as a loading control. (B) Anchorage-independent spheroid growth of Akt-myr-expressing R− and R− EN cells at 24 and 96 h as indicated. (C) Soft agar colony-forming assays of R+, R− EN, R+ EN, and R− cells expressing myristoylated Akt (R−/Akt-myr), and R− EN cells expressing myristoylated Akt (R−EN/Akt-myr). Monolayer cells were trypsinized, washed, and plated in medium containing 0.2% agar to assess anchorage-independent growth. Results are presented as the number of macroscopic colonies formed (>0.1 mm in diameter) at 14 days after plating divided by the number of cells originally plated. R− EN/Akt-myr cells formed a significantly greater percentage of colonies than both R− EN and R−/Akt-myr cells. Statistical analysis was performed using the Student's t test (* represents P < 0.0001). (D) Tumorigenesis in nude mice injected with R− and R+ fibroblast cell lines. R+, R− EN, R+ EN, R− ENmyr, R−/Akt-myr, and R− EN/Akt-myr cells were injected subcutaneously into five mice per cell line and three sites per mouse. The y axis represents the average size of tumors from all sites for all mice in each given group, as measured every 2 to 3 days. Statistical analysis of tumor volume at day 20 was performed using the Mann-Whitney rank test, with the P value comparing R− EN and R+ EN cells (at P < 0.0001).
R− EN/Akt-myr cells readily formed stable spheroids under anchorage-independent conditions (Fig. 3B), and soft agar colony formation was restored to levels comparable to those of R+ EN cells (Fig. 3C). However, expression of Akt-myr in the absence of EN was insufficient to support spheroid (Fig. 3B) or soft agar colony formation (Fig. 3C) of R− fibroblasts. Similarly, R+ fibroblasts were resistant to transformation by Akt-myr (data not shown). We did notice a small decrease in average soft agar colony size in R− EN/Akt-myr cells compared to that in R+ EN cells (data not shown), possibly indicating that the myristoylated Akt construct is expressed at suboptimal levels or that additional functions of the EN complex are important for this process. However, these data provide strong evidence that the activation of PI3K-Akt signaling is critical for EN-induced anchorage-independent growth.
To determine if the observed effects of Akt activation on anchorage-independent growth correlate with transformation activity in EN-expressing cells, we next assessed the ability of the various cell lines to form tumors in nude mice. R+ EN cells readily formed large palpable tumors after subcutaneous injection at rates comparable to those previously reported for EN-expressing NIH 3T3 cells (62) (Fig. 3D). In contrast, R− EN cells along with R− and R+ control cells failed to form tumors even 4 weeks after injection. However, expression of Akt-myr in R− EN cells restored tumorigenic activity; R− EN/Akt-myr tumors grew to sizes similar to those of R+ EN tumors (average volume, >2,000 mm3), although there was a lag of 2 to 4 days in attaining comparable tumor volumes in the R− EN/Akt-myr mice. Morphologically, the tumors observed with each cell line were identical and showed a fibrosarcoma phenotype (data not shown). These data confirm that the ability of activated Akt to restore anchorage-independent growth to R− EN cells correlates with tumorigenesis and therefore with transformation activity.
IGF-IR mutants lacking IRS-1 binding ability or kinase activity fail to support ETV6-NTRK3 transformation.
We previously demonstrated that IRS-1 acts as an adaptor to link EN to both the Ras-ERK and PI3K-Akt pathways through Grb2 and PI3K p85 subunit binding, respectively, but that IRS-1 association with EN and its tyrosine phosphorylation are IGF-IR independent (34, 44). In fact, EN/IRS-1 complexes retain Grb2 and PI3K p85 subunit binding activity in R− cells even though these cells are nontransformed (44). We therefore reasoned that a possible function of IGF-IR in EN oncogenesis may be to localize putative EN/IRS-1/Grb2/p85 complexes at sites of activation of downstream pathways through the well-established interaction between IGF-IR and IRS-1. Such a process would likely require the kinase activity of IGF-IR, which autophosphorylates tyrosine residue 950 that is required for IRS-1 (and Shc) binding (59). We therefore tested whether stable expression of a tyrosine 950-to-phenylalanine IGF-IR mutant [IGF-IR(Y950F)] (59) or a kinase-dead IGF-IR mutant [IGF-IR(K1003A)] lacking the ATP-binding site (60) could restore transformation activity of EN to R− cells. Cells were plated in spheroid cultures for 48 h in 0.25% (Fig. 4A), or 9% (data not shown) serum and then lysed to determine the relative activation of the Ras-ERK and PI3K-Akt pathways. As seen in Fig. 4A, MEK activation in cells coexpressing EN plus either IGF-IR(Y950F) or IGF-IR(K1003A) was similar to that in R− EN and R+ EN cells and enhanced compared to lines lacking EN. As before, Akt phosphorylation was impaired in R− EN cells compared to R+ EN cells, and this was not rescued by expression of IGF-IR(Y950F) or IGF-IR(K1003A). Immunoblotting using antibodies directed against the IGF-IR β-subunit showed equivalent expression of wild-type IGF-IR and the two mutant constructs. The ability of the IGF-IR mutants to restore transformation activity to R− EN fibroblasts was next assessed by soft agar colony assays. Figure 4B demonstrates that unlike wild-type IGF-IR, the Y950F and K1003A mutants were unable to support macroscopic colony formation of R− EN cells. These findings indicate that a kinase-active form of IGF-IR, and one that retains IRS-1 binding, is required for EN-associated Akt activation and transformation.
FIG. 4.

IGF-IR(Y950F) and IGF-IR(K1003A) mutants fail to restore Akt activation and anchorage-independent growth to R− EN fibroblasts. (A) R− fibroblasts were engineered to express EN (EN +) and wild-type IGF-IR (IGF-IR +), IGF-IR with a tyrosine-to-phenylalanine mutation at residue 950 (Y950F +), or IGF-IR with a lysine-to-alanine mutation at residue 1003 (K1003A +) as indicated. These cells along with R+ and R+ EN cells were grown in serum-free spheroid cultures for 24 h, lysed, and subjected to immunoblotting using antibodies to the indicated proteins as described in the legend of Fig. 2. Detection of actin was used as a loading control. (B) Soft agar colony-forming assays were performed for each of these cell lines as described in the legend of Fig. 3C. * represents a P value of <0.0001 using the Student's t test to compare R+ EN to R− Y950F EN and R− K1003A EN.
ETV6-NTRK3 and IRS-1 membrane localization is increased by IGF-IR expression.
Since IGF-IR is a plasma membrane receptor, one possibility suggested by the above-described findings is that IGF-IR functions to localize EN/IRS-1 complexes to the plasma membrane at sites of activation of the PI3K-Akt cascade. We therefore tested the effects of expressing a membrane-targeted EN construct in IGF-IR-null cells. An N-myristoylated, HA-tagged EN construct [(HA)ENmyr] was generated (Fig. 5A), and expressed in both R− and R+ fibroblasts, and its cellular localization was assessed and compared to that of HA-tagged EN [(HA)EN] alone. (HA)EN proteins showed a diffuse cytosolic distribution in R+ cells (Fig. 5C) as previously shown for EN-transformed NIH 3T3 cells (63), while a punctate perinuclear and cytosolic pattern was observed in R− cells (Fig. 5B). (HA)ENmyr showed a prominent plasma membrane distribution in both R− (Fig. 5D) and R+ (data not shown) cells. Using confocal microscopy to more precisely examine cell membranes, localization of (HA)EN to the plasma membrane was readily observed in cells expressing IGF-IR (Fig. 5F), while (HA)EN localized exclusively within the cytoplasm in R− EN cells (Fig. 5E). Of particular note, (HA)EN localized to small membrane extensions in R+ cells, which was not evident in R− cells expressing this construct. Extensive plasma membrane localization of (HA)ENmyr was confirmed by confocal microscopy in both the absence (Fig. 5G) and presence (data not shown) of IGF-IR expression. Interestingly, (HA)EN association with the membrane was observed in both the presence and absence of serum (data not shown), suggesting that its localization may be serum independent.
FIG. 5.

Membrane localization of ETV6-NTRK3 in the presence of IGF-IR or following N-terminal myristoylation of ETV6-NTRK3. (A) Schematic of HA-tagged N-terminal myristoylated (N-myr) ETV6-NTRK3 (ENmyr) possessing the Lck myristoylation sequence and two tandem HA epitope tags at the N terminus of the EN molecule. (B to D) Immunofluorescence detection of EN molecules. Stable expression of HA-tagged EN or HA-tagged ENmyr in R− or R+ mouse embryo fibroblasts is depicted. (B) R− (HA)EN, (C) R+ (HA)EN, and (D) R− (HA)ENmyr cells were grown on coverslips in 9% serum, fixed, and subjected to immunofluorescence using anti-HA antibodies (green labeling). Cell nuclei were stained with DAPI (blue staining). (E to G) Confocal microscopic images of similarly stained R− (HA)EN (E), R+ (HA)EN (F), and R− (HA)ENmyr (G) fibroblasts grown in low (0.25%)-serum conditions. White arrow represents 100 μm.
These findings were confirmed by subcellular fractionation studies of the (HA)EN and (HA)ENmyr molecules. R− and R+ cells expressing these constructs were starved overnight in 0.25% serum, lysed, and separated into cytoplasmic and membrane-associated fractions using differential centrifugation (see Materials and Methods). Total protein aliquots from each fraction were resolved by SDS-polyacrylamide gel electrophoresis and immunoblotted with anti-HA antibodies to determine the relative abundance of EN molecules in each fraction (Fig. 6A). R− cells showed virtually no membrane-associated (HA)EN, while a small but significant proportion of (HA)EN could be detected in R+ cell plasma membrane fractions, which was reproducible over multiple experimental replicates (Fig. 6A, lane 6, and data not shown). (HA)ENmyr demonstrated strong membrane localization regardless of IGF-IR expression, with a two- to threefold increase compared to cytoplasmic fractions in both R− and R+ cells. Interestingly, a slight band shift occurs in membrane-associated (HA)EN or (HA)ENmyr compared to cytosolic proteins, potentially indicating multiple tyrosine phosphorylation events for this pool of EN molecules. Anti-Grb2 and anti-caveolin immunoblotting confirmed the identity of the cytoplasmic and membrane fractions, respectively.
FIG. 6.

IGF-IR expression or myristoylation of EN leads to membrane localization of EN and tyrosine-phosphorylated IRS-1. (A) EN and IRS-1 subcellular localization. Monolayer R− or R+ cells expressing (HA)EN or (HA)ENmyr were starved for 16 h in medium containing 0.25% serum, washed, and harvested. Cell lysates were separated into cytoplasmic (C) and membrane (M) fractions by differential centrifugation, followed by immunoblotting with antibodies directed against HA, IRS-1, Grb2 (as a cytosolic marker), and caveolin 1 (as a membrane marker). (B) Immunoprecipitation of IRS-1 from membrane fractions. Aliquots of 500 μg protein isolated from membrane fractions of cell lines treated as in C were subjected to anti-IRS-1 immunoprecipitation, followed by immunoblotting with antiphosphotyrosine (P-Tyr), anti-IRS-1, or anti-NTRK3 antibody as indicated. Approximate sizes of protein bands (in kilodaltons) are shown to the right of each panel.
We next determined whether IRS-1 was also differentially localized in R− EN cells compared to R+ EN cells. Using anti-IRS-1 antibodies, we detected abundant cytosolic IRS-1 in all cell lines tested (Fig. 6A), as expected based on previous studies (33). However, compared to R− EN cells, the proportion of IRS-1 associated with membrane fractions was dramatically elevated in R+ EN cells and in either R− or R+ cells expressing ENmyr (Fig. 6A, compare lane 4 to lanes 6, 8, and 10). To confirm this, we performed immunoprecipitation from membrane fractions from cells starved overnight using antibodies to IRS-1 followed by immunoblotting using anti-phosphotyrosine antibodies. This showed prominent tyrosine phosphorylation of an ∼185-kDa band only in membranes from R+ EN, R− ENmyr, and R+ ENmyr cells, which was confirmed to be IRS-1 by reprobing with anti-IRS-1 antibodies (Fig. 6B). Moreover, tyrosine-phosphorylated EN could readily be pulled down in association with IRS-1 from these fractions but not from those of R− EN or R+ cells (Fig. 6B). This indicates that at least a proportion of membrane-localized EN and IRS-1 molecules are associated with each other in R+ EN, R− ENmyr, and R+ ENmyr cells. From these studies, we conclude that (i) EN localization to membranes is dependent on IGF-IR expression or N-myristoylation, with the latter possibly being a more efficient mechanism, and (ii) IRS-1 membrane localization is enhanced in R+ cells by the presence of EN or in R− and R+ cells by the expression of ENmyr.
Membrane-targeted ETV6-NTRK3 confers anchorage-independent growth and transforms IGF-IR-null fibroblasts.
If localization of EN and IRS-1 to the plasma membrane requires IGF-IR and is important for transformation, the expression of membrane-targeted EN should circumvent the requirement for IGF-IR in transformation-related functions of EN. We therefore first assessed the effects of myristoylated EN on the ability of R− and R+ cells to grow under anchorage-independent conditions. ENmyr was expressed at levels similar to those of EN and showed significant tyrosine phosphorylation when expressed in each cell type (Fig. 7A). As shown in Fig. 7B, either R− or R+ cells engineered to express ENmyr readily formed multicellular spheroids identical to those of R+ EN cells, and R− ENmyr spheroids are stable indefinitely in anchorage-independent cultures (data not shown). To confirm this, we next performed soft agar colony assays as shown in Fig. 7C. Similar to previous observations (44), R+ and R− EN cell lines showed minimal rates of macroscopic colony formation (averages of 6.9% and 3.7%, respectively), while R+ EN fibroblasts demonstrated potent colony-forming ability, with an average of 33.0% of cells forming macroscopic colonies. Moreover, myristoylated EN was able to induce anchorage-independent growth regardless of IGF-IR expression, and reexpression of IGF-IR did not significantly affect the rate of colony formation (43.1% and 41.5% macroscopic colony formation for R− ENmyr and R+ ENmyr cells, respectively). Finally, expression of ENmyr was highly tumorigenic even in cells lacking IGF-IR, as subcutaneously injected R− ENmyr cells rapidly formed large tumors at rates comparable to those of R+ EN cells (Fig. 3D).
FIG. 7.

ENmyr restores Akt activation and anchorage-independent growth to IGF-IR-null cells. (A) Anti-NTRK3 immunoprecipitation on whole-cell lysates of EN- and ENmyr-expressing cells shows the level of oncogene expression. Blots were immunoblotted for antiphosphotyrosine, stripped, and reprobed with anti-NTRK3. (B) R− ENmyr and R+ ENmyr readily formed viable multicellular spheroids after 96 h in anchorage-independent culture (magnification, ×100). (C) Soft agar colony-forming assays of EN- and ENmyr-expressing cells. Cells were plated in soft agar as described in the legend of Fig. 3C. R− EN cells form a significantly lower percentage of colonies than R+ EN, R− ENmyr, and R+ ENmyr cells. * represents a P value of <0.0001, as assessed by the Student's t test. Spheroid cultures of R− (IGF-IR −) or R+ (IGF-IR +) cells expressing EN (EN +), ENmyr (ENmyr +), or vector alone (EN −, ENmyr −) were grown in 0.25% serum (D) or 9% serum (E) for 48 h, lysed, and subjected to immunoblotting as described in the legend of Fig. 2. Total levels of cyclin D1 protein were detected using a specific antibody, while activated Akt (Ser-473-P), GSK-3β (GSK-3β-P), and MEK1/2 (MEK1/2-P) were assessed using antibodies to phosphorylated proteins. Detection of actin was used as a loading control.
We next tested the signaling properties of ENmyr-expressing cells. As shown in Fig. 7D and E, lanes 4 and 5, R− ENmyr and R+ ENmyr spheroids showed similar constitutive activation of MEK and cyclin D1 induction in low- and high-serum media compared to R+ EN cells. Of particular note, ENmyr was able to restore Akt activation in R− cells to levels comparable to those observed in R+ EN cells under either low- or high-serum conditions. The ability of ENmyr to activate these pathways was also observed in R− ENmyr cells grown in monolayer cultures (data not shown). These studies indicate that membrane targeting of EN induces cellular Akt activation, anchorage-independent growth, soft agar colony formation, and tumorigenesis of cells lacking IGF-IR. This supports our hypothesis that an important function of IGF-IR in EN oncogenesis is to enhance EN membrane localization and that this likely involves the IRS-1 protein.
IGF-IR suppresses anoikis in anchorage-independent ETV6-NTRK3-transformed fibroblasts.
The above-described data indicate that IGF-IR contributes to maximal Akt activation in EN-transformed cells. We next wished to determine whether this predominantly influences cell proliferation or survival, as Akt has been linked to both functions (67). R− EN and R+ EN cells have similar growth and survival rates in monolayer cultures (44). To characterize these processes under anchorage-independent conditions, spheroid cultures of the various cell lines were assessed for indices of cell proliferation and apoptosis. FACS analysis failed to detect major differences in cell cycle progression between R− EN and R+ EN cells at 6 h (data not shown), 48 h, or 96 h in culture (Fig. 8A). This is in keeping with the comparable levels of MEK activation and cyclin D1 observed in R− EN and R+ EN spheroid cultures (Fig. 2), as Ras-ERK induction of cyclin D1 is thought to underlie aberrant cell cycle progression in EN-transformed cells (61). After 96 h in spheroid culture, we observed a significant proportion of sub-G1 DNA content in R− EN cells, a phenomenon often associated with late-stage apoptosis (39). To confirm these data, cells were assessed for uptake of BrdU as a measure of proliferation. As shown in Fig. 8B and C, the proliferative index of R− EN spheroids was identical to that of R+ EN spheroids after 24 h in culture, with each cell line showing ∼41% BrdU-positive nuclei. Interestingly, proliferating cells were distributed evenly throughout the spheroid structures for both cell lines at this time point. At the 48- and 96-h time points, R+ EN spheroids had a slightly increased proliferative index (at 48 h, 27.6% for R− EN versus 34.1% for R+ EN [P = 0.055]; at 96 h, 24.0% for R− EN versus 29.4% for R+ EN [P = 0.03]) (Fig. 8C). However, R− EN spheroids at both 48 h (data not shown) and 96 h (Fig. 8B) showed obvious structural disintegration, likely indicating that cell death was contributing to the observed differences in proliferation.
FIG. 8.

R− EN fibroblasts proliferate in anchorage-independent cultures. (A) R− EN and R+ EN fibroblasts were cultured as spheroids for 48 and 96 h as indicated. Multicellular spheroid and aggregates were separated into single cells, immediately fixed, stained with propidium iodide, and subjected to FACS analysis. Numbers represent the approximate percentages of cells in G1/S/G2, respectively. FL2-H, intensity of fluorescence channel 2. (B) R− EN and R+ EN spheroids were grown for the times indicated in the presence of BrdU, harvested, fixed, and embedded in paraffin. Sections from the spheroids were cut, and the samples were immunostained with anti-BrdU antibodies. Pictures were taken from a typical field at ×100 magnification. Dark staining indicates BrdU positivity. (C) BrdU-positive nuclei were counted and are represented as a percentage of total cells (n = 15).
We next compared apoptotic rates in spheroids, since we observed prominent morphological features of cell death in R− EN cells after extended days in anchorage-independent cultures (Fig. 1B and 8B). Cell lysates were collected from R− EN, R+ EN, R− ENmyr, and R− EN/Akt-myr spheroid cultures in a time course from 24 to 96 h after plating, and caspase 3 activation was measured by fluorometry. As shown in Fig. 8A, R− EN spheroid cells showed significant increases in caspase 3 activation by 24 h relative to R+ EN monolayers, which were maintained until 72 h. By 96 h after plating, caspase 3 activity levels returned to baseline, presumably because most cells were nonviable or in late stages of apoptosis. In contrast, R+ EN and R− ENmyr spheroids showed no detectable increases in caspase 3 activation (Fig. 9A), even after 7 days in culture (data not shown). Similarly, no enhanced caspase 3 activation was observed in R− EN/Akt-myr cells (Fig. 9A), and these cells remained viable in long-term spheroid cultures (data not shown). This difference was not observed in monolayer cultures, where R+, R− EN, and R+ EN cells all demonstrated only low-level caspase 3 activation (data not shown). To further document increased apoptosis in R− EN spheroids compared to R+ EN spheroids, we performed anti-PARP immunoblots on cell lysates from R− EN, R+ EN, R− ENmyr, or R− EN/Akt-myr fibroblasts grown for 24 or 48 h in anchorage-independent cultures (Fig. 9B). Lysates from R+ EN monolayers treated with 10 μg/ml doxorubicin were included as a positive control for PARP cleavage. R− EN cells showed significant levels of cleaved PARP at both 24 and 48 h of anchorage-independent culture, whereby ∼50% of the total cellular amount was of the cleaved form, indicating that significant induction of caspase 3-mediated apoptosis was occurring. In contrast, cleaved PARP was undetectable or present at very low levels relative to the uncleaved form in R+ EN, R− ENmyr, and R− EN/Akt-myr cells at both time points tested. These findings strongly indicate that the IGF-IR axis is not essential for cell cycle progression in EN-transformed cells but instead supports cell survival through enhanced Akt activation. Since this is only observed in cells placed under anchorage-independent conditions, we conclude that IGF-IR signaling functions to suppress anoikis in EN-transformed cells.
FIG. 9.

R− EN fibroblasts undergo apoptotic death in anchorage-independent cultures. (A) Caspase 3 activation in spheroid cultures of R− and R+ fibroblasts. R− EN, R+ EN, R− ENmyr, and R− EN/Akt-myr cells were plated in 9% serum under anchorage-independent conditions for 0 to 96 h as indicated, lysed, and assayed for caspase 3 activity by fluorometry as described in Materials and Methods. The relative level of caspase 3 activation was normalized to a value of 1.0 for R+ EN fibroblasts growing in subconfluent monolayer cultures. Error bars represent ±1 standard deviation. (B) R− EN, R+ EN, R− ENmyr, and R− EN/Akt-myr fibroblasts were grown in spheroid cultures for 24 or 48 h as indicated, lysed, and subjected to anti-PARP and antiactin immunoblotting. The 116-kDa and 89-kDa bands represent uncleaved and cleaved PARP, respectively, as indicated. R+ EN monolayers treated with 10 μg/ml doxorubicin (R+EN ML + dox) are included as a positive control for PARP cleavage. Detection of actin was used as a loading control.
PI3K inhibitors block ETV6-NTRK3-induced anchorage-independent growth.
If PI3K-Akt signaling is crucial for suppression of anoikis in EN-transformed cells, then blocking this pathway should inhibit EN-induced anchorage-independent growth. Previous studies have shown that the PI3K inhibitor LY294002 blocks soft agar colony formation of EN-transformed NIH 3T3 fibroblasts (61). We first confirmed that LY294002 also impairs soft agar colony formation of R+ EN, R− ENmyr, and R− EN/Akt-myr cells, which it does in a dose-dependent manner to as low as 2.5 μM (Fig. 10B). As expected, EN myristoylation does not confer resistance to LY294002. Soft agar colony formation of the R− EN/Akt-myr cell line was also inhibited by LY294002, since activation of the Akt-myr construct is dependent on PI3K to activate PDK1 and PDK2 functions (2). Inhibition of colony formation by LY294002 correlates with a dose-dependent loss of Akt phosphorylation, as shown for R+ EN cells in Fig. 10A. Moreover, LY294002 inhibits spheroid growth of EN-transformed fibroblasts (see Fig. 10C for R+ EN cells), while the U0126 MEK1/2 inhibitor, which effectively blocked Erk1/2 phosphorylation (Fig. 10A), had no demonstrable effect on spheroid formation or morphology (Fig. 10C). We next plated R+ EN fibroblasts in either monolayer or nonadherent cultures and treated cells with various doses of LY294002 for 24 h and assessed caspase 3 activation as a measure of apoptotic cell death. Interestingly, we found that while LY294002 treatment specifically induces apoptosis in anchorage-independent cultures, monolayer cells remain unaffected (Fig. 10D). For example, treatment of R+ EN spheroids with 10 μM LY294002, which effectively inhibits Akt Ser-473 phosphorylation (Fig. 10A), induces similar levels of caspase 3 activation as observed in R− EN spheroid cells grown for 24 h in spheroid culture with no treatment (Fig. 9A). Although U0126 induced low-level caspase 3 activation in R+ EN spheroids, similar effects were observed with R− ENmyr spheroids (data not shown), arguing that this effect is IGF-IR independent. Therefore, while we cannot rule out a contribution of the Ras-ERK pathway to suppression of anoikis in EN-transformed fibroblasts, we conclude that the inhibition of Akt signaling through a PI3K blockade can significantly impair anchorage-independent growth of these cells.
FIG. 10.

Inhibition of PI3K-Akt signaling blocks anchorage-independent growth induced by EN. (A) R+ EN fibroblasts were grown under anchorage-independent culture conditions in medium containing 9% serum and were treated with various doses of LY294002, vehicle control (dimethyl sulfoxide [DMSO]), or 25 μM U0126 for 4 h. Similarly cultured R− EN cells were treated with vehicle control only. Cells were then lysed and immunoblotted using antibodies to the indicated proteins. Detection of actin was used as a loading control. (B) R+, R− EN, R+ EN, R− ENmyr, and R− EN/Akt-myr fibroblasts were grown in soft agar as described in the legend of Fig. 3B except that various doses of LY294002 or vehicle control were included. Cultures were fed every 2 days with 2 drops of medium containing the appropriate concentration of LY294002 (LY) or vehicle control. R+ EN, R− ENmyr, and R− EN/Akt-myr all showed significant differences when DMSO treatment was compared to 10 μM LY294002 treatment (* represents P < 0.0001 by Student's t test) (C) Photomicrographs (magnification, ×100) of the effects of LY294002 on R+ EN spheroid formation after 24 and 72 h. R+ EN cells were plated under anchorage-independent conditions and treated with 25 μM LY294002 or 25 μM U0126 at the time of plating. Cells were spun down and replated with drug-containing medium every 24 h. (D) LY294002 induces apoptosis in R+ EN spheroid cultures but not in monolayers. R+ EN cells were plated in either anchorage-independent cultures or subconfluent monolayers and treated with vehicle control (DMSO), 10 μg/ml doxorubicin (dox), or various doses of LY294002 for 24 h. Cells were then collected, lysed, and assayed for caspase 3 activity as described in the legend of Fig. 7. Caspase 3 activation is plotted relative to untreated R+ EN monolayer cells. Error bars represent ±1 standard deviation, with data representing three separate experiments.
DISCUSSION
A large amount of literature points to an essential role for the IGF-I receptor in transformation by dominantly acting oncogenes. Besides ETV6-NTRK3, other childhood tumor fusion oncoproteins such as the Ewing tumor EWS-FLI1 molecule and PAX3-FKHR of alveolar rhabdomyosarcoma also require an intact IGF-IR axis for oncogenesis (44, 65, 68). In this study, we show that IGF-IR is necessary for suppression of anoikis in anchorage-independent EN-transformed murine fibroblasts. IGF-IR-null R− EN cells fail to form colonies in soft agar and do not survive in anchorage-independent spheroid cultures. This correlates with the inability of these cells to form tumors when injected into nude mice, compared to R+ EN cells, which are highly tumorigenic. We found that expression of EN in R− cells is specifically associated with a defect in Akt activation, while induction of the Ras-ERK pathway leading to cyclin D1 up-regulation and cell cycle progression are not impaired. Soft agar colony formation, spheroid formation, and tumorigenesis are all restored by ectopic expression of an activated form of Akt in R− EN cells but not in R− cells expressing activated Akt alone. The inability of EN to transform IGF-IR-null cells could be circumvented by membrane targeting of the chimeric oncoprotein through N-terminal myristoylation, suggesting that IGF-IR may function at least in part to localize EN to the plasma membrane for optimal activation of downstream signaling pathways. Membrane localization of EN correlated with a dramatic increase in the amount of IRS-1 at the plasma membrane, and EN and IRS-1 were associated with each other in this compartment. Expression of the ENmyr molecule in IGF-IR-null cells is sufficient for stimulating the full spectrum of EN downstream signals including IRS-1 membrane targeting and Akt activation, suggesting that localization of EN along with IRS-1 to membranes may be particularly important for the induction of the PI3K-Akt cascade.
In order to survive under anchorage-independent conditions and ultimately to metastasize, cancer cells must be capable of suppressing detachment-induced apoptosis, or anoikis. Many studies point to a critical role for the prosurvival activity of Akt in suppressing anoikis (16). Akt substrates such as Bad (17, 18) and caspase 9 (12) are direct apoptotic effectors that are inactivated by phosphorylation, while Akt phosphorylation of Forkhead transcription factors prevents their nuclear localization and subsequent up-regulation of proapoptotic genes (10, 32). Akt activation through the expression of oncogenic Ras (29), or ectopic expression of constitutively active Akt (55), is sufficient to prevent anoikis. Others have reported that inhibitors of PI3K-Akt signaling, but not of the Ras-ERK pathway, inhibit anchorage-independent growth of fibroblasts induced by the v-Ros and epidermal growth factor receptor-Ros oncogenes (47). Interestingly, in a recent report, a genome-wide screen for suppressors of anoikis identified the TrkB (NTRK2) neurotrophin receptor as a key molecule in this process (19). Ectopic expression of TrkB in rat intestinal epithelial cells led to the up-regulation of phosphorylated Akt and the prevention of caspase 3-mediated apoptosis, and these cells could be maintained under anchorage-independent conditions. Whether this involves IGF-IR was not evaluated. The NTRK3 kinase domain shares 79% identity with that of NTRK2, and therefore, EN may promote anchorage-independent survival of fibroblasts in a related fashion. In the current study, growth of R− EN cells in nonadherent cultures resulted in marked caspase 3 activation and cell death that was reversed by ectopic expression of myristoylated Akt. We also found that treatment of R+ EN cell spheroids with LY294002 resulted in a significant induction of caspase 3 activity that was not observed in monolayer cells treated in parallel (Fig. 10B). The latter findings require further validation, such as with the use of dominant-negative PI3K p110, since interpretations of LY294002 experiments can be unreliable. Taken together, our observations establish an essential role for Akt activation in maintaining the survival of EN-expressing fibroblasts under anchorage-independent conditions and implicate IGF-IR in this process.
Impaired Akt activation in R− EN spheroids persisted even in the presence of serum. This is surprising since these cells presumably possess other growth factor receptors that would be expected to activate the PI3K-Akt pathway in the absence of IGF-IR. One possible explanation is suggested by a recent study indicating that monomeric p85 forms cytosolic sequestration complexes with IRS-1 that act to dampen IRS-1/PI3K p110 signaling (42). We previously observed comparable or even increased binding of the PI3K p85 regulatory subunit to phospho-IRS-1 in serum-stimulated R− EN cells compared to that in R+ EN cells (44). In the absence of IGF-IR to localize EN and IRS-1 to the membrane, EN/IRS-1/p85 complexes might remain predominantly cytosolic to function in a dominant-negative fashion to sequester IRS-1 and the PI3K p110 catalytic subunit away from the plasma membrane at sites of phosphoinositol-3,4,5-trisphosphate (PIP3) and phosphoinositol-3,4-bisphosphate (PIP2) generation (11). This would impact Akt activation, which requires recruitment to the plasma membrane through an association of the Akt pleckstrin homology domain with PIP3 and PIP2. It is tempting to speculate that an active IGF-IR axis (or membrane-targeted EN) may modulate IRS-1 in such a way as to shift its equilibrium to the plasma membrane at sites of p110 function and Akt activation. Previous reports of a membrane-targeted IRS-1 molecule showed increased PI3K-Akt signaling, emphasizing the importance of proper IRS-1 localization for the activation of this pathway (33).
Our finding that Ras-ERK pathway signaling, cyclin D1 induction, and cell cycle progression are unaffected in R− EN cells implies that activation of this cascade may be independent of EN/IRS-1 complex localization to the plasma membrane. Whereas PI3K-Akt signaling occurs almost exclusively at the plasma membrane, Ras-ERK signaling can take place on Golgi and endoplasmic reticulum membranes (13). HA-tagged EN showed a perinuclear expression pattern in R− cells (Fig. 5A), which is indicative of localization to internal membranes. It is therefore possible that Ras-ERK signaling is activated predominantly on endomembranes in EN-expressing cells.
In summary, we demonstrate here that a kinase-active IGF-IR molecule that retains IRS-1 binding complements EN oncogenesis by contributing optimal activation of the PI3K-Akt cascade, a pathway that is essential for transformation by the EN oncoprotein. In contrast, Ras-ERK activation, cyclin D1 induction, and cell cycle progression appear to be IGF-IR independent in EN-transformed cells. Our data suggest that localization of EN and IRS-1 to the plasma membrane is key for Akt activation during EN transformation and that such localization is enhanced by IGF-IR. Whether IGF-IR-mediated IRS-1 membrane localization is also essential for transformation by other fusion oncoproteins remains to be determined. The presence of IGF-IR confers an anchorage-independent growth ability to EN-expressing cells through the suppression of anoikis, likely by facilitating maximal PI3K-Akt activation. Whether the Ras-ERK cascade also plays a role in protecting EN cells from anoikis, potentially by inducing cell cycle arrest through ERK-mediated Bim suppression, as recently described (15), remains unknown. Based on our results, inhibition of the IGF-IR pathway may represent a clinically relevant strategy for tumors that express EN.
Acknowledgments
We thank Renato Baserga for the R− cells and DNA constructs, Jill Kucab and Joan Mathers for technical advisement, and Cristina Tognon, Markus Warmuth, Michael Cox, and Sandra Dunn for helpful discussions.
This work was supported by funds from the Canadian Institutes of Health Research (to P.H.B.S.) and the Johal Program in Pediatric Oncology Basic and Translational Research. M.J.M. was funded by a Canadian Institutes of Health Research doctoral award.
REFERENCES
- 1.Almeida, A., M. Muleris, B. Dutrillaux, and B. Malfoy. 1994. The insulin-like growth factor I receptor gene is the target for the 15q26 amplicon in breast cancer. Genes Chromosomes Cancer 11:63-65. [DOI] [PubMed] [Google Scholar]
- 2.Andjelkovic, M., D. R. Alessi, R. Meier, A. Fernandez, N. J. Lamb, M. Frech, P. Cron, P. Cohen, J. M. Lucocq, and B. A. Hemmings. 1997. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272:31515-31524. [DOI] [PubMed] [Google Scholar]
- 3.Baker, J., J. P. Liu, E. J. Robertson, and A. Efstratiadis. 1993. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75:73-82. [PubMed] [Google Scholar]
- 4.Baserga, R. 2000. The contradictions of the insulin-like growth factor 1 receptor. Oncogene 19:5574-5581. [DOI] [PubMed] [Google Scholar]
- 5.Baserga, R. 1999. The IGF-I receptor in cancer research. Exp. Cell Res. 253:1-6. [DOI] [PubMed] [Google Scholar]
- 6.Baserga, R. 1995. The insulin-like growth factor I receptor: a key to tumor growth? Cancer Res. 55:249-252. [PubMed] [Google Scholar]
- 7.Baserga, R., F. Peruzzi, and K. Reiss. 2003. The IGF-1 receptor in cancer biology. Int. J. Cancer 107:873-877. [DOI] [PubMed] [Google Scholar]
- 8.Bates, R. C., N. S. Edwards, and J. D. Yates. 2000. Spheroids and cell survival. Crit. Rev. Oncol. Hematol. 36:61-74. [DOI] [PubMed] [Google Scholar]
- 9.Berns, E. M., J. G. Klijn, I. L. van Staveren, H. Portengen, and J. A. Foekens. 1992. Sporadic amplification of the insulin-like growth factor 1 receptor gene in human breast tumors. Cancer Res. 52:1036-1039. [PubMed] [Google Scholar]
- 10.Brunet, A., A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson, K. C. Arden, J. Blenis, and M. E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857-868. [DOI] [PubMed] [Google Scholar]
- 11.Cantley, L. C. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655-1657. [DOI] [PubMed] [Google Scholar]
- 12.Cardone, M. H., N. Roy, H. R. Stennicke, G. S. Salvesen, T. F. Franke, E. Stanbridge, S. Frisch, and J. C. Reed. 1998. Regulation of cell death protease caspase-9 by phosphorylation. Science 282:1318-1321. [DOI] [PubMed] [Google Scholar]
- 13.Chiu, V. K., T. Bivona, A. Hach, J. B. Sajous, J. Silletti, H. Wiener, R. L. Johnson II, A. D. Cox, and M. R. Philips. 2002. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 4:343-350. [DOI] [PubMed] [Google Scholar]
- 14.Christofori, G., P. Naik, and D. Hanahan. 1994. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 369:414-418. [DOI] [PubMed] [Google Scholar]
- 15.Collins, N. L., M. J. Reginato, J. K. Paulus, D. C. Sgroi, J. Labaer, and J. S. Brugge. 2005. G1/S cell cycle arrest provides anoikis resistance through Erk-mediated Bim suppression. Mol. Cell. Biol. 25:5282-5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905-2927. [DOI] [PubMed] [Google Scholar]
- 17.Datta, S. R., H. Dudek, X. Tao, S. Masters, H. Fu, Y. Gotoh, and M. E. Greenberg. 1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231-241. [DOI] [PubMed] [Google Scholar]
- 18.del Peso, L., M. Gonzalez-Garcia, C. Page, R. Herrera, and G. Nunez. 1997. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278:687-689. [DOI] [PubMed] [Google Scholar]
- 19.Douma, S., T. Van Laar, J. Zevenhoven, R. Meuwissen, E. Van Garderen, and D. S. Peeper. 2004. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 430:1034-1039. [DOI] [PubMed] [Google Scholar]
- 20.Downward, J. 1996. Control of ras activation. Cancer Surv. 27:87-100. [PubMed] [Google Scholar]
- 21.Dunn, S. E., M. Ehrlich, N. J. Sharp, K. Reiss, G. Solomon, R. Hawkins, R. Baserga, and J. C. Barrett. 1998. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 58:3353-3361. [PubMed] [Google Scholar]
- 22.Eguchi, M., M. Eguchi-Ishimae, A. Tojo, K. Morishita, K. Suzuki, Y. Sato, S. Kudoh, K. Tanaka, M. Setoyama, F. Nagamura, S. Asano, and N. Kamada. 1999. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood 93:1355-1363. [PubMed] [Google Scholar]
- 23.Franke, T. F., D. R. Kaplan, and L. C. Cantley. 1997. PI3K: downstream AKTion blocks apoptosis. Cell 88:435-437. [DOI] [PubMed] [Google Scholar]
- 24.Franke, T. F., D. R. Kaplan, L. C. Cantley, and A. Toker. 1997. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275:665-668. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Echeverria, C., M. A. Pearson, A. Marti, T. Meyer, J. Mestan, J. Zimmermann, J. Gao, J. Brueggen, H. G. Capraro, R. Cozens, D. B. Evans, D. Fabbro, P. Furet, D. G. Porta, J. Liebetanz, G. Martiny-Baron, S. Ruetz, and F. Hofmann. 2004. In vivo antitumor activity of NVP-AEW541—a novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 5:231-239. [DOI] [PubMed] [Google Scholar]
- 26.Kalebic, T., V. Blakesley, C. Slade, S. Plasschaert, D. Leroith, and L. J. Helman. 1998. Expression of a kinase-deficient IGF-I-R suppresses tumorigenicity of rhabdomyosarcoma cells constitutively expressing a wild type IGF-I-R. Int. J. Cancer 76:223-227. [DOI] [PubMed] [Google Scholar]
- 27.Kalebic, T., M. Tsokos, and L. J. Helman. 1994. In vivo treatment with antibody against IGF-1 receptor suppresses growth of human rhabdomyosarcoma and down-regulates p34cdc2. Cancer Res. 54:5531-5534. [PubMed] [Google Scholar]
- 28.Kaleko, M., W. J. Rutter, and A. D. Miller. 1990. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol. Cell. Biol. 10:464-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khwaja, A., P. Rodriguez-Viciana, S. Wennstrom, P. H. Warne, and J. Downward. 1997. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16:2783-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knezevich, S. R., M. J. Garnett, T. J. Pysher, J. B. Beckwith, P. E. Grundy, and P. H. Sorensen. 1998. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 58:5046-5048. [PubMed] [Google Scholar]
- 31.Knezevich, S. R., D. E. McFadden, W. Tao, J. F. Lim, and P. H. Sorensen. 1998. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat. Genet. 18:184-187. [DOI] [PubMed] [Google Scholar]
- 32.Kops, G. J., N. D. de Ruiter, A. M. De Vries-Smits, D. R. Powell, J. L. Bos, and B. M. Burgering. 1999. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398:630-634. [DOI] [PubMed] [Google Scholar]
- 33.Kriauciunas, K. M., M. G. Myers, Jr., and C. R. Kahn. 2000. Cellular compartmentalization in insulin action: altered signaling by a lipid-modified IRS-1. Mol. Cell. Biol. 20:6849-6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lannon, C. L., M. J. Martin, C. E. Tognon, W. Jin, S. J. Kim, and P. H. Sorensen. 2004. A highly conserved NTRK3 C-terminal sequence in the ETV6-NTRK3 oncoprotein binds the phosphotyrosine binding domain of insulin receptor substrate-1: an essential interaction for transformation. J. Biol. Chem. 279:6225-6234. [DOI] [PubMed] [Google Scholar]
- 35.Lannon, C. L., and P. H. Sorensen. 2005. ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 15:215-223. [DOI] [PubMed] [Google Scholar]
- 36.Lawlor, E. R., C. Scheel, J. Irving, and P. H. B. Sorensen. 2002. Anchorage-independent multi-cellular spheroids as an in vitro model of growth signaling in Ewing tumors. Oncogene 21:307-318. [DOI] [PubMed] [Google Scholar]
- 37.Lee, C. T., S. Wu, D. Gabrilovich, H. Chen, S. Nadaf-Rahrov, I. F. Ciernik, and D. P. Carbone. 1996. Antitumor effects of an adenovirus expressing antisense insulin-like growth factor I receptor on human lung cancer cell lines. Cancer Res. 56:3038-3041. [PubMed] [Google Scholar]
- 38.LeRoith, D., H. Werner, D. Beitner-Johnson, and C. T. Roberts, Jr. 1995. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 16:143-163. [DOI] [PubMed] [Google Scholar]
- 39.Li, X., J. Gong, E. Feldman, K. Seiter, F. Traganos, and Z. Darzynkiewicz. 1994. Apoptotic cell death during treatment of leukemias. Leuk. Lymphoma 13(Suppl. 1):65-70. [DOI] [PubMed] [Google Scholar]
- 40.Liu, J. P., J. Baker, A. S. Perkins, E. J. Robertson, and A. Efstratiadis. 1993. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75:59-72. [PubMed] [Google Scholar]
- 41.Long, L., R. Rubin, R. Baserga, and P. Brodt. 1995. Loss of the metastatic phenotype in murine carcinoma cells expressing an antisense RNA to the insulin-like growth factor receptor. Cancer Res. 55:1006-1009. [PubMed] [Google Scholar]
- 42.Luo, J., S. J. Field, J. Y. Lee, J. A. Engelman, and L. C. Cantley. 2005. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J. Cell Biol. 170:455-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mauro, L., M. Salerno, C. Morelli, T. Boterberg, M. E. Bracke, and E. Surmacz. 2003. Role of the IGF-I receptor in the regulation of cell-cell adhesion: implications in cancer development and progression. J. Cell Physiol. 194:108-116. [DOI] [PubMed] [Google Scholar]
- 44.Morrison, K. B., C. Tognon, M. J. Garnett, C. Deal, and P. H. B. Sorensen. 2002. ETV6-NTRK3 transformation requires insulin-like growth factor 1 receptor signaling and is associated with constitutive IRS-1 tyrosine phosphorylation. Oncogene 21:5684-5695. [DOI] [PubMed] [Google Scholar]
- 45.Muleris, M., A. Almeida, M. Gerbault-Seureau, B. Malfoy, and B. Dutrillaux. 1994. Detection of DNA amplification in 17 primary breast carcinomas with homogeneously staining regions by a modified comparative genomic hybridization technique. Genes Chromosomes Cancer 10:160-170. [DOI] [PubMed] [Google Scholar]
- 46.Murphy, C. T., S. A. McCarroll, C. I. Bargmann, A. Fraser, R. S. Kamath, J. Ahringer, H. Li, and C. Kenyon. 2003. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424:277-283. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen, K. T., C. S. Zong, S. Uttamsingh, P. Sachdev, M. Bhanot, M. T. Le, J. L. Chan, and L. H. Wang. 2002. The role of phosphatidylinositol 3-kinase, rho family GTPases, and STAT3 in Ros-induced cell transformation. J. Biol. Chem. 277:11107-11115. [DOI] [PubMed] [Google Scholar]
- 48.O'Connor, R., A. Kauffmann-Zeh, Y. Liu, S. Lehar, G. I. Evan, R. Baserga, and W. A. Blattler. 1997. Identification of domains of the insulin-like growth factor I receptor that are required for protection from apoptosis. Mol. Cell. Biol. 17:427-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ponten, J. 1971. Spontaneous and virus induced transformation in cell culture. Virol. Monogr. 8:1-253. [DOI] [PubMed] [Google Scholar]
- 50.Ravid, D., S. Maor, H. Werner, and M. Liscovitch. 2005. Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-I receptors and signaling. Oncogene 24:1338-1347. [DOI] [PubMed] [Google Scholar]
- 51.Resnicoff, M., D. Coppola, C. Sell, R. Rubin, S. Ferrone, and R. Baserga. 1994. Growth inhibition of human melanoma cells in nude mice by antisense strategies to the type 1 insulin-like growth factor receptor. Cancer Res. 54:4848-4850. [PubMed] [Google Scholar]
- 52.Romano, G., M. Prisco, T. Zanocco-Marani, F. Peruzzi, B. Valentinis, and R. Baserga. 1999. Dissociation between resistance to apoptosis and the transformed phenotype in IGF-I receptor signaling. J. Cell. Biochem. 72:294-310. [DOI] [PubMed] [Google Scholar]
- 53.Rubin, B. P., C. J. Chen, T. W. Morgan, S. Xiao, H. E. Grier, H. P. Kozakewich, A. R. Perez-Atayde, and J. A. Fletcher. 1998. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am. J. Pathol. 153:1451-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santini, M. T., and G. Rainaldi. 1999. Three-dimensional spheroid model in tumor biology. Pathobiology 67:148-157. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt, M., S. Hovelmann, and T. L. Beckers. 2002. A novel form of constitutively active farnesylated Akt1 prevents mammary epithelial cells from anoikis and suppresses chemotherapy-induced apoptosis. Br. J. Cancer 87:924-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scotlandi, K., S. Avnet, S. Benini, M. C. Manara, M. Serra, V. Cerisano, S. Perdichizzi, P. L. Lollini, C. De Giovanni, L. Landuzzi, and P. Picci. 2002. Expression of an IGF-I receptor dominant negative mutant induces apoptosis, inhibits tumorigenesis and enhances chemosensitivity in Ewing's sarcoma cells. Int. J. Cancer 101:11-16. [DOI] [PubMed] [Google Scholar]
- 57.Sell, C., G. Dumenil, C. Deveaud, M. Miura, D. Coppola, T. DeAngelis, R. Rubin, A. Efstratiadis, and R. Baserga. 1994. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 14:3604-3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaw, M., P. Cohen, and D. R. Alessi. 1997. Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 416:307-311. [DOI] [PubMed] [Google Scholar]
- 59.Tartare-Deckert, S., D. Sawka-Verhelle, J. Murdaca, and E. Van Obberghen. 1995. Evidence for a differential interaction of SHC and the insulin receptor substrate-1 (IRS-1) with the insulin-like growth factor-I (IGF-I) receptor in the yeast two-hybrid system. J. Biol. Chem. 270:23456-23460. [DOI] [PubMed] [Google Scholar]
- 60.Tennagels, N., C. Hube-Magg, A. Wirth, V. Noelle, and H. W. Klein. 1999. Expression, purification, and characterization of the cytoplasmic domain of the human IGF-1 receptor using a baculovirus expression system. Biochem. Biophys. Res. Commun. 260:724-728. [DOI] [PubMed] [Google Scholar]
- 61.Tognon, C., M. Garnett, E. Kenward, R. Kay, K. Morrison, and P. H. Sorensen. 2001. The chimeric protein tyrosine kinase ETV6-NTRK3 requires both Ras-Erk1/2 and PI3-kinase-Akt signaling for fibroblast transformation. Cancer Res. 61:8909-8916. [PubMed] [Google Scholar]
- 62.Tognon, C., S. R. Knezevich, D. Huntsman, C. D. Roskelley, N. Melnyk, J. A. Mathers, L. Becker, F. Carneiro, N. MacPherson, D. Horsman, C. Poremba, and P. H. Sorensen. 2002. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2:367-376. [DOI] [PubMed] [Google Scholar]
- 63.Tognon, C. E., C. Mackereth, A. M. Somasiri, L. P. McIntosh, and P. H. B. Sorensen. 2004. Mutations in the SAM domain of the ETV6-NTRK3 chimeric tyrosine kinase block polymerization and transformation activity. Mol. Cell. Biol. 24:4636-4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toretsky, J. A., and L. J. Helman. 1996. Involvement of IGF-II in human cancer. J. Endocrinol. 149:367-372. [DOI] [PubMed] [Google Scholar]
- 65.Toretsky, J. A., T. Kalebic, V. Blakesley, D. LeRoith, and L. J. Helman. 1997. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 272:30822-30827. [DOI] [PubMed] [Google Scholar]
- 66.Valentinis, B., A. Morrione, F. Peruzzi, M. Prisco, K. Reiss, and R. Baserga. 1999. Anti-apoptotic signaling of the IGF-I receptor in fibroblasts following loss of matrix adhesion. Oncogene 18:1827-1836. [DOI] [PubMed] [Google Scholar]
- 67.Vivanco, I., and C. L. Sawyers. 2002. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2:489-501. [DOI] [PubMed] [Google Scholar]
- 68.Wang, W., P. Kumar, J. Epstein, L. Helman, J. V. Moore, and S. Kumar. 1998. Insulin-like growth factor II and PAX3-FKHR cooperate in the oncogenesis of rhabdomyosarcoma. Cancer Res. 58:4426-4433. [PubMed] [Google Scholar]