

Abstract
The Atm protein kinase is central to the DNA double-strand break response in mammalian cells. After irradiation, dimeric Atm undergoes autophosphorylation at Ser 1981 and dissociates into active monomers. Atm activation is stimulated by expression of the Mre11/Rad50/nibrin complex. Previously, we showed that a C-terminal fragment of nibrin, containing binding sites for both Mre11 and Atm, was sufficient to provide this stimulatory effect in Nijmegen breakage syndrome (NBS) cells. To discriminate whether nibrin's role in Atm activation is to bind and translocate Mre11/Rad50 to the nucleus or to interact directly with Atm, we expressed an Mre11 transgene with a C-terminal NLS sequence in NBS fibroblasts. The Mre11-NLS protein complexed with Rad50, localized to the nucleus in NBS fibroblasts, and associated with chromatin. However, Atm autophosphorylation was not stimulated in cells expressing Mre11-NLS, nor were downstream Atm targets phosphorylated. To determine whether nibrin-Atm interaction is necessary to stimulate Atm activation, we expressed nibrin transgenes lacking the Atm binding domain in NBS fibroblasts. The nibrin ΔAtm protein interacted with Mre11/Rad50; however, Atm autophosphorylation was dramatically reduced after irradiation in NBS cells expressing the nibrin ΔAtm transgenes relative to wild-type nibrin. These results indicate that nibrin plays an active role in Atm activation beyond translocating Mre11/Rad50 to the nucleus and that this function requires nibrin-Atm interaction.
The response to DNA double-strand breaks (DSBs) in mammalian cells involves an essential signaling cascade that ensures genomic integrity. The response is initiated by detection of DSBs, followed rapidly by transduction of the damage signal throughout the cell to effector proteins involved in apoptosis, cell cycle control, and DNA repair.
Considerable information has been obtained about transduction of the damage signal to downstream targets. For DNA DSBs, the Atm protein kinase, mutated in individuals with the radiosensitivity disorder ataxia-telangiectasia (A-T), is the primary signal transducer (37). Atm exists as inactive dimers in undamaged cells but rapidly undergoes autophosphorylation at serine 1981 after exposure to DSB-inducing agents and dissociates into active monomers (1). The active Atm monomers phosphorylate a collection of critical downstream effector molecules, including nibrin, Mre11, Brca1, MDC1, 53BP1, p53, Chk2, Smc1, and FANC D2 (2, 8, 14, 16, 17, 19, 29, 36, 39). Phosphorylation of some of these downstream effectors by Atm occurs in the nucleoplasm, whereas others are phosphorylated at sites of DNA damage where Atm relocalizes via interaction with the C terminus of nibrin, a member of the Mre11/Rad50/nibrin (MRN) complex (15, 26, 32, 44).
Although the targets of Atm are well established, the mechanism by which DNA DSBs are detected and the Atm signal transduction cascade initiated is less well understood. In their initial report of Atm autophosphorylation, Bakkenist and Kastan (1) observed that chromatin alterations mediated by exposure to chloroquine, hypotonic conditions, or histone deacetylase inhibitors were sufficient to activate Atm in the absence of DNA DSBs. These findings led the investigators to suggest that changes in chromatin structure caused by DNA DSBs were responsible for Atm activation.
Several lines of evidence suggest a specific role for the MRN complex in Atm activation. The MRN complex has well-documented DNA repair and S-phase checkpoint functions in both yeast and mammalian cells (9). Mre11/Rad50 display nuclease activity and can bind free DNA ends, activities that are enhanced in the presence of nibrin (10, 33, 34). Whereas nibrin has no enzymatic activity, the C terminus of nibrin binds Mre11 directly and translocates Mre11/Rad50 to the nucleus (11). After irradiation, nibrin relocalizes to the sites of DNA damage within 5 min and, as mentioned above, binds and relocalizes Atm to these sites (15, 26, 44). Nibrin is phosphorylated by Atm in response to DNA damage, and this phosphorylation event is required for proper S-phase checkpoint activation (16, 25, 43, 46).
Hypomorphic mutations in nibrin and Mre11 result in the radiosensitivity disorders Nijmegen breakage syndrome (NBS) and A-T-like disorder (ATLD), which share many features with A-T (38, 42). Cell lines from patients with NBS or ATLD have delayed kinetics of Atm autophosphorylation at early times after low doses of irradiation or exposure to radiomimetic compounds (5, 18, 20, 41). Similarly, cells in which Mre11 has been degraded by adenovirus infection have deficient Atm activation (4). More recently, Difilippantonio et al. (12) reported that mouse B cells conditionally null for nibrin expression displayed little or no Atm activation.
Previously, we showed that nuclear expression of Mre11/Rad50 complexed with just a C-terminal fragment of nibrin was sufficient to stimulate Atm activation at early times after irradiation (5). In contrast, nuclear expression of a nibrin transgene lacking the C-terminal 100 amino acids was unable to stimulate Atm activation under the same conditions (5, 18). Since the C-terminal 100 amino acids of nibrin contain separate but adjacent binding domains for Mre11 and Atm, nibrin may stimulate Atm autophosphorylation by translocating Mre11/Rad50 to the nucleus or by binding and relocalizing Atm to the sites of DNA damage (15, 44). The former hypothesis is appealing since the MRN complex can process DNA DSBs, an activity that would likely induce chromatin alterations (33, 34). Indeed, ATLD cells complemented with a nuclease-deficient mutant of Mre11 were deficient in Atm activation, and a Rad50 ATP-dependent DNA-unwinding activity mutant was unable to stimulate Atm autophosphorylation in vitro (24, 41). Whether nibrin-Atm interaction enhances Atm activation after irradiation is not clear from the published studies (15, 44). Using cell extracts, You et al. (44) found nibrin-Atm interaction stimulated Atm activation, whereas in an in vivo system, Atm autophosphorylation was found to be independent of nibrin-Atm interaction (15).
To discriminate between these two possible roles of nibrin in Atm activation, we expressed an Mre11 transgene with an artificial C-terminal NLS sequence in NBS fibroblasts in order to deliver Mre11/Rad50 to the nucleus in the absence of nibrin. We compared Atm activation after irradiation in these cells to NBS cells expressing nibrin transgenes lacking the Atm binding domain. Our results indicate that nibrin plays an active role in stimulating Atm autophosphorylation after irradiation beyond translocating Mre11/Rad50 to the nucleus and that this function requires nibrin-Atm interaction.
MATERIALS AND METHODS
Cell lines.
The simian virus 40 transformed fibroblast cell line, NBS-ILB1, was established from an NBS patient homozygous for the 657del5 mutation (21). NBS-ILB1 cells infected with the pLXIN retroviral vector alone (BD Clontech, Palo Alto, CA) or with pLXIN expressing a wild-type nibrin cDNA (NBS1), a nibrin mutant with a C-terminal deletion of 100 amino acids (aa) (Nb652), or a 300-aa C-terminal fragment of nibrin (NbFR5) have been described previously (5, 7, 11). D6809 is a primary fibroblast cell line established from an ATLD patient homozygous for the R633X nonsense mutation (38). GM0637 simian virus 40 transformed normal fibroblasts were used as controls. All cells were grown in Dulbecco modified Eagle medium (Invitrogen, Carlsbad, CA) supplemented with 15% fetal calf serum (HyClone, Logan, UT), 100 U of penicillin/ml, and 100 μg of streptomycin (Invitrogen)/ml. Cells stably expressing pLXIN transgenes were also maintained in 500 μg of G418 (Invitrogen)/ml. For retroviral packaging, Phoenix A cells were grown in Dulbecco modified Eagle medium supplemented with 10% heat-inactivated fetal calf serum with penicillin-streptomycin (www.stanford.edu/group/nolan).
Retroviral gene expression.
Recombinant retroviruses constructed for these experiments included pLXIN Mre11, pLXIN Mre11-NLS, pLXIN NBS1 ΔAtm that lacked the Atm binding domain, and pLXIN NbFR5 ΔAtm. A wild-type Mre11 cDNA previously cloned by PCR (11) was subcloned into pLXIN, upstream of the IRES-neomycin cassette. To construct Mre11-NLS, the stop codon of the Mre11 cDNA was changed to a serine residue (TGA→TCA) by using QuikChange site-directed mutagenesis (Stratagene, Inc., La Jolla, CA), and a hemagglutinin (HA) tag, followed by two artificial NLS sequences, was added to the Mre11 C terminus by PCR. To delete the C-terminal 20 amino acids of nibrin, encompassing the Atm binding region, a lysine residue at position 735 was changed to a stop codon (AAA→TAA) by QuikChange mutagenesis. The K735X mutation was introduced into pLXIN NBS1, as well as pLXIN NbFR5. Bulk cell lines stably expressing these recombinant retroviruses were generated as described previously (7). Briefly, Phoenix A cells were transiently transfected with the retroviral plasmid DNAs. After 48 h, viral supernatants were harvested and used to infect NBS-ILB1 cells or D6809 ATLD cells. Stable cell lines were selected with G418. Cells were plated at a clonal dilution to isolate cloned cell lines.
Immunoblot analysis.
Total cell lysates were prepared from cells with or without irradiation by resuspending cells in EBC buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 1 mM β-mercaptoethanol, 0.5% Nonidet P-40, proteinase inhibitors [Roche Applied Science, Indianapolis, IN]), followed by sonication. Nuclear and cytoplasmic proteins were isolated from cells by using an NE-PER kit (Pierce, Rockford, IL). Protein concentration was determined by the Bradford method (Bio-Rad, Hercules, CA). For immunoprecipitation, 250 to 800 μg of protein was mixed with primary antibodies and protein G-coupled magnetic beads overnight (Dynal Biotech ASA, Oslo, Norway). For direct Western blots, 25 μg of protein or 105 cell equivalents was loaded per lane. Protein samples were denatured in NuPage LDS sample buffer (Invitrogen) and were electrophoresed by using NuPage Tris-acetate gels (Invitrogen) as described previously (6). Proteins were transferred to Immobilon-P nylon membranes (Millipore Corp., Bedford, MA). Immunoblots were probed with a variety of primary antibodies, including rabbit polyclonal antisera specific for nibrin, Mre11, Rad50, or Atm (Novus Biological, Littleton, CO). Mre11 protein was also detected by using a monoclonal anti-Mre11 antibody (a gift from T. Demaggio, Icos Corp., Bothell, WA). Nibrin phosphorylation was detected by using an anti-phosphoserine 343 monoclonal antibody (Upstate Cell Signaling Solutions, Lake Placid, NY), and Atm autophosphorylation was detected with an anti-phosphoserine 1981 monoclonal antibody (Abcam, Cambridge, MA). A monoclonal Chk2 antibody (a gift from D. Delia) was used to detect total Chk2 protein, and Chk2 phosphorylation was detected with a rabbit anti-phosphothreonine 68 antisera (Cell Signaling Technology, Beverly, MA). Total p53 protein was detected by using a monoclonal p53 antibody (Upstate), and a rabbit polyclonal phosphoserine 15 antibody was used to detect p53 phosphorylation (Cell Signaling Technology). The HA tag was detected by using a monoclonal anti-HA antibody (Roche). Primary antibodies were detected with horseradish peroxidase-coupled goat anti-rabbit immunoglobulin G (IgG) and goat anti-mouse IgG (BD Pharmingen, San Diego, CA). Immunoblot staining was detected by chemiluminescence (Perkin-Elmer Life Sciences, Wellesley, MA).
Chromatin association.
Isolation of proteins associated with chromatin was performed according to the method of Mendez and Stillman (30). Briefly, cells were harvested by trypsinization, washed, and resuspended in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol, proteinase inhibitors). After addition of 0.1% Triton X-100, cells were incubated for 5 min on ice, and cytoplasmic proteins were isolated by centrifugation. The nuclear pellet was washed once in buffer A and lysed in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, and proteinase inhibitors) for 30 min on ice. Soluble nuclear proteins were isolated by centrifugation. The insoluble nuclear pellet was then washed sequentially in buffer B supplemented with 125 mM NaCl and then 250 mM NaCl to release proteins associated with chromatin with increasing avidity. The remaining insoluble nuclear pellet was lysed directly in LDS buffer (Invitrogen). Cellular fractions were sonicated, and immunoblot analysis was performed as described above. Rabbit antisera specific for Hsp90 was used as a cytoplasmic protein control (Cell Signaling Technology), and a monoclonal antibody to TRF2 was used as a nuclear protein control (EMD Biosciences, La Jolla, CA).
Immunofluorescence.
Cell lines were grown on glass coverslips overnight (Viromed, Minneapolis, MN), and the following day they were exposed to 0 or 12 Gy of irradiation. Cells were fixed after 6 h with 4% paraformaldehyde-0.1% Triton X-100 and were blocked overnight in 10% fetal calf serum in phosphate-buffered saline. To detect nibrin and Mre11 localization in the cells, unirradiated coverslips were costained with rabbit antinibrin antisera (Novus) and a monoclonal antibody specific for Mre11 (T. Demaggio). To detect irradiation-induced foci, irradiated coverslips were costained with a monoclonal γ-H2AX antibody (Upstate) and a rabbit polyclonal antibody specific for Mre11 (Novus). The primary antibodies were detected with goat anti-rabbit IgG coupled to Alexa 568 and goat anti-mouse IgG coupled to Alexa 488 (Molecular Probes, Eugene, OR). Confocal microscopy was performed with a Nikon fluorescence microscope and a Bio-Rad confocal imaging system using LaserSharp 2000 (Bio-Rad). Individual fields were Z-planed, and images at 488 and 568 nm were stacked and merged.
RESULTS
The Mre11-NLS transgene interacts with Rad50 and localizes to the nucleus.
The endogenous Mre11/Rad50 proteins do not contain any recognizable NLS sequences and, in the absence of nibrin, remain complexed in the cytoplasm (3, 13, 35). To determine whether Mre11/Rad50 alone are capable of stimulating Atm activation, we attached two artificial NLS sequences and an HA tag to the C terminus of an Mre11 cDNA cloned in the pLXIN retroviral vector. These additional sequences added 29 amino acids to the Mre11 protein, changing the predicted molecular weight from approximately 81 to 84 kDa. The Mre11-NLS transgene was expressed in NBS-ILB1 fibroblasts, which do not express full-length nibrin protein, and clonal cell lines expressing Mre11-NLS were isolated. As a control, a wild-type Mre11 cDNA lacking the NLS and HA sequences was expressed in NBS-ILB1 cells.
Western blot analysis showed a higher-molecular-weight species in the Mre11-NLS clones, as well as endogenous Mre11 protein (Fig. 1A). This higher-molecular-weight species corresponded to the band identified by the anti-HA antibody, indicating that the Mre11-NLS protein could be stably expressed. Three clonal lines were chosen for further analysis that expressed low (NLS.11), medium (NLS.8), and high (NLS.12) levels of Mre11-NLS protein relative to control cells.
FIG. 1.
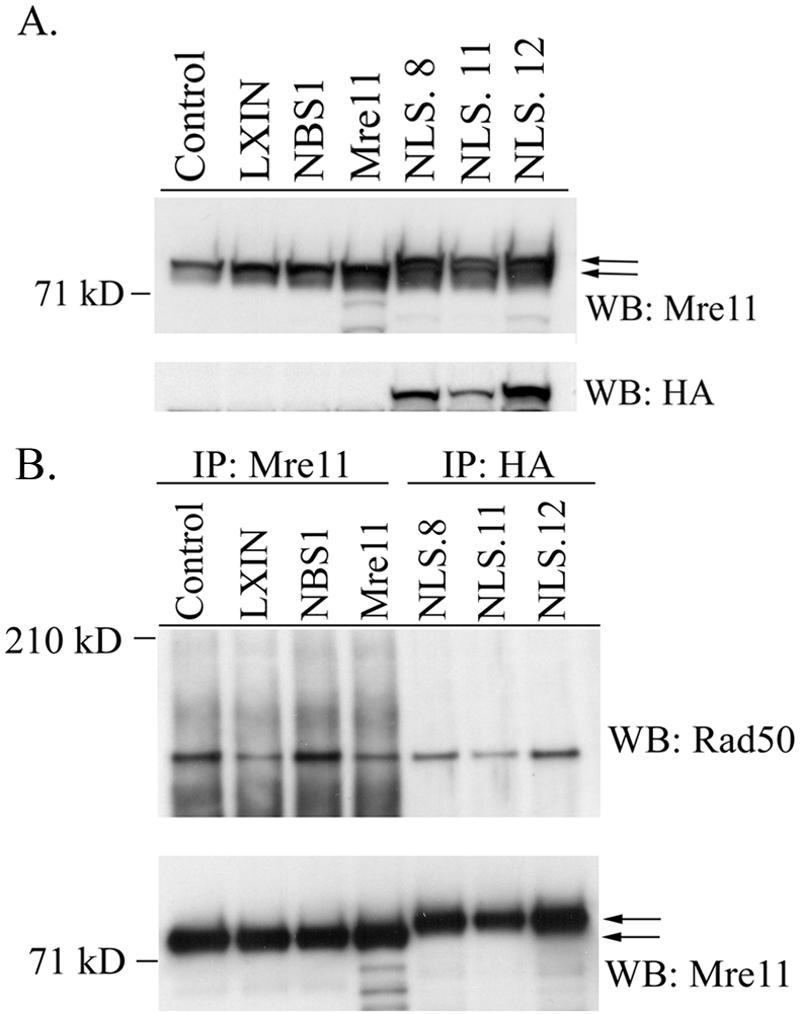
Expression of the Mre11-NLS transgene in NBS cells and interaction with Rad50. (A) Western blot analysis of GM0637 fibroblasts or NBS-ILB1 cells stably transduced with the pLXIN retroviral vector alone (LXIN), wild-type nibrin (NBS1), wild-type Mre11 (Mre11), or an Mre11-NLS transgene with two artificial NLS sequences and an HA tag attached to the Mre11 C terminus. Individual clones of NBS cells expressing the Mre11-NLS transgene were isolated (NLS.8, NLS.11, and NLS.12). A total of 25 μg of total cellular protein per lane was separated on a 7% sodium dodecyl sulfate (SDS)-polyacrylamide gel. The Western blot (WB) was probed with monoclonal antibodies specific for Mre11 or the HA tag. (B) Mre11 was immunoprecipitated (IP) from 250 μg of total cellular protein isolated from the above cell lines using a polyclonal anti-Mre11 antibody or a monoclonal anti-HA antibody. Immunoprecipitated proteins were separated on a 3 to 8% SDS-polyacrylamide gel, and the immunoblot was probed with a polyclonal Rad50 antisera or a monoclonal anti-Mre11 antibody. The arrows indicate the positions of endogenous Mre11 protein and the higher-molecular-weight Mre11-NLS protein.
To determine whether the Mre11-NLS protein could interact with endogenous Rad50 in NBS-ILB1 cells, Mre11-NLS was immunoprecipitated with anti-HA antibodies, and Western blots were probed with Rad50 antisera. As shown in Fig. 1B, Rad50 was coimmunoprecipitated with Mre11-NLS protein, indicating that the two proteins were complexed in NBS-ILB1 cells. The amount of Rad50 detected in NLS.8, NLS.11, and NLS.12 correlated with the levels of Mre11-NLS protein expressed in each clone. The Mre11-NLS protein appeared to complex with slightly less Rad50 than endogenous Mre11 in normal control cells or NBS-ILB1 cells expressing a wild-type nibrin transgene.
The Mre11-NLS protein localized to the nucleus of NBS-ILB1 cells, as determined by Western blot analysis of cytoplasmic and nuclear proteins and by immunofluorescence. In NBS-ILB1 cells infected with vector alone, Mre11 was localized primarily in the cytoplasm, but expression of wild-type nibrin in NBS cells resulted in nuclear localization of Mre11 (Fig. 2A). The Mre11-NLS protein was also localized to the nucleus of NBS-ILB1 cells, although approximately half of the transgenic protein remained in the cytoplasm, along with the endogenous Mre11 protein in these cells. By comparison, Mre11 was localized only in the cytoplasm of NBS cells infected with a wild-type Mre11 transgene lacking NLS sequences. Similar results were observed by immunofluorescence, detecting the Mre11-NLS protein with the anti-HA antibody (Fig. 2B). Thus, expression of an Mre11-NLS transgene in NBS cells resulted in substantial nuclear expression of Mre11/Rad50 in the absence of nibrin.
FIG. 2.

Subcellular localization of the Mre11-NLS protein in NBS cells. (A) Cytoplasmic and nuclear proteins were isolated from GM0637 control fibroblasts or NBS-ILB1 cells stably transduced with the pLXIN retroviral vector alone (LXIN), wild-type nibrin (NBS1), wild-type Mre11 (Mre11), or the Mre11-NLS transgene (NLS.8, NLS.11, and NLS.12). A total of 105 cell equivalents of protein per lane were electrophoresed on 7% SDS-polyacrylamide gels. Western blots (WB) were probed with a monoclonal anti-Mre11 antibody, a polyclonal antisera specific for the cytoplasmic heat shock protein (Hsp90), or a monoclonal antibody specific for the telomeric protein, TRF2. The arrows indicate the positions of endogenous Mre11 protein and the higher-molecular-weight Mre11-NLS protein. (B) Immunofluorescence analysis was performed on NBS-ILB1 cells stably transduced with the pLXIN retroviral vector alone, wild-type nibrin (NBS1), or the Mre11-NLS transgene (NLS.8). Fibroblasts were stained for nibrin and Mre11 expression using a polyclonal anti-nibrin antibody (red) and a monoclonal antibody to Mre11 (green). The Mre11-NLS protein was specifically detected using a monoclonal antibody specific for the HA tag (HA, green). Immunofluorescence was detected by confocal microscopy at 488 and 568 nm. Individual fields were Z-planed and stacked. Magnification, ×600.
Atm activation and function are not stimulated by Mre11-NLS/Rad50.
To determine whether Mre11/Rad50 alone could stimulate the kinetics of Atm activation, as measured by serine 1981 autophosphorylation, NBS cells expressing Mre11-NLS protein were exposed to 3 Gy of ionizing radiation and harvested 15 min later. Atm protein was immunoprecipitated, and serine 1981 phosphorylation was analyzed by Western blotting (Fig. 3A). As observed previously (5), Atm activation under these conditions was barely detectable in NBS-ILB1 cells infected with vector alone but was restored to robust levels in NBS cells complemented with wild-type nibrin or the C terminus of nibrin. In contrast, all three clonal cell lines expressing the Mre11-NLS protein failed to stimulate Atm autophosphorylation after irradiation. The levels of S1981 phosphorylation observed in the Mre11-NLS clones were comparable to NBS cells infected with a wild-type Mre11 cDNA lacking an NLS sequence, nibrin lacking the C-terminal 100 amino acids, or the empty LXIN vector. Consistent with deficient Atm activation in the Mre11-NLS cell lines, phosphorylation of the downstream Atm targets Chk2 threonine 68 and p53 serine 15 was reduced in the Mre11-NLS clones relative to wild-type nibrin and NbFR5 complemented cells (Fig. 3B).
FIG. 3.

Atm activation and function after irradiation in NBS cells expressing the Mre11-NLS protein. (A) Atm autophosphorylation at serine 1981 was assessed in NBS-ILB1 cells stably transduced with the pLXIN vector alone (LXIN), wild-type nibrin (NBS1), nibrin lacking the C-terminal 100 aa (Nb652), a C-terminal 300-aa fragment of nibrin (NbFR5), wild-type Mre11 (Mre11), or the Mre11-NLS transgene (NLS.8, NLS.11, and NLS.12). Cells were unirradiated or exposed to 3 Gy of ionizing radiation (IR), and total cellular lysates were harvested after 15 min. Atm was immunoprecipitated (IP) from 800 μg of protein using a polyclonal anti-Atm antibody and protein was separated on 3 to 8% SDS-polyacrylamide gels. Western blots (WB) were probed with a monoclonal phosphoserine 1981 antibody or a polyclonal Atm antibody. (B) Chk2 threonine 68 phosphorylation and p53 serine 15 phosphorylation by Atm were assessed in the above cells, as well as GM0637 control fibroblasts, 15 min after exposure to 0 or 3 Gy of ionizing radiation. A total of 20 μg of total cellular protein per lane was separated on 7% SDS-polyacrylamide gels. Immunoblots were probed with a phosphothreonine 68 antisera or a monoclonal anti-Chk2 antibody. For p53, the immunoblots were probed with phosphoserine 15 antisera or a monoclonal p53 antibody.
Mre11-NLS complements Atm activation and function in ATLD cells.
The C terminus of Mre11 encodes one of the two DNA-binding domains of the Mre11 protein (9). To rule out the trivial possibility that the attachment of the HA tag and NLS sequences to the C terminus of Mre11 interfered with the function of the protein, we tested the ability of the Mre11-NLS protein to complement ATLD cells, that lack full-length Mre11. A primary ATLD fibroblast cell line was infected with the empty pLXIN vector or pLXIN encoding wild-type Mre11 or Mre11-NLS. ATLD cells express low levels of endogenous nibrin protein that is stabilized in the presence of exogenous Mre11 (38). Therefore, exogenously expressed Mre11-NLS protein can potentially enter the nucleus by two routes, by binding nibrin and translocating to the nucleus via nibrin, as well as via the added C-terminal NLS sequences. Figure 4A shows that ATLD cells infected with a wild-type Mre11 cDNA or the Mre11-NLS cDNA expressed Mre11 at levels comparable to a control cell line. Immunoprecipitation with an anti-nibrin antisera demonstrated that the Mre11-NLS transgene could complex with nibrin and Rad50 comparable to the wild-type Mre11 cDNA (Fig. 4B and data not shown). In contrast to NBS cells, however, expression of the Mre11-NLS protein in ATLD cells was sufficient to stimulate Atm autophosphorylation at 15 min after exposure to 3 Gy of ionizing radiation, similar to levels observed with a wild-type Mre11 transgene (Fig. 4C). This stimulatory effect on Atm activation resulted in phosphorylation of nibrin, Chk2, and p53 in the complemented cells compared to cells infected with vector alone (Fig. 4B and C). Thus, the Mre11-NLS transgene was functional with regard to Atm activation.
FIG. 4.

Atm activation and function after irradiation in ATLD cells expressing the Mre11-NLS protein. (A) Expression of the Mre11 NLS transgene in ATLD cells was assessed by Western blot analysis. Total cellular lysates were isolated from GM0637 control fibroblasts and ATLD D6809 cells stably transduced with the pLXIN retroviral vector (ATLD LXIN), wild-type Mre11 (ATLD Mre11), or the Mre11-NLS transgene (ATLD NLS). Then, 25 μg of protein per lane was electrophoresed on a 7% SDS-polyacrylamide gel and Western blotted (WB). The immunoblot was probed with a monoclonal anti-Mre11 antibody or a polyclonal antibody specific for Hsp90, as a loading control. The arrows indicate the endogenous Mre11 protein and the higher-molecular-weight Mre11-NLS protein. (B) Nibrin phosphorylation in the above cell lines was analyzed 15 min after exposure to 0 or 3 Gy of ionizing radiation (IR). Nibrin was immunoprecipitated (IP) from 250 μg of total cellular protein using a polyclonal antinibrin antibody and proteins were separated on a 3 to 8% SDS-polyacrylamide gel. Western blots were probed with a phosphoserine 343 monoclonal antibody or a polyclonal antibody specific for nibrin. To assess Mre11-NLS complex formation with nibrin, the blot was reprobed with a monoclonal anti-Mre11 antibody. (C) Atm autophosphorylation at serine 1981 was assessed in the above cells lines 15 min after exposure to 0 or 3 Gy of irradiation. Next, 25 μg of total cellular protein per lane was electrophoresed on a 3 to 8% SDS-polyacrylamide gel. The Western blot was probed with a monoclonal phosphoserine 1981 antibody or a polyclonal anti-Atm antisera. Chk2 threonine 68 phosphorylation was analyzed by probing a similar immunoblot with phosphothreonine 68 antisera or a monoclonal anti-Chk2 antibody. To detect p53 serine 15 phosphorylation, immunoblots were probed with phosphoserine 15 antisera or a monoclonal p53 antibody. (D) Irradiation-induced foci were examined 6 h after 12 Gy of irradiation in the above ATLD fibroblasts. Mre11 or Mre11-NLS foci were detected with Mre11 antisera (red) and γ-H2AX foci were detected with a monoclonal γ-H2AX antibody (green). Immunofluorescence was collected by using confocal microscopy at 488 and 568 nm. Individual fields were Z-planed and stacked. Magnification, ×1,000.
The Mre11-NLS transgene localizes to chromatin.
The MRN complex associates with chromatin, even in the absence of DNA damage (27, 31, 45). To ensure that the Mre11-NLS protein localized to chromatin in the absence of nibrin, NBS-ILB1 cells infected with vector alone, wild-type nibrin, or the Mre11-NLS transgene were treated with or without 3 Gy of ionizing radiation, and 30 min later cells were fractionated. Proteins isolated in the various cellular fractions were examined by Western blot analysis. As shown in Fig. 5A, Mre11 was localized primarily in the cytoplasmic and nuclear soluble fractions in NBS-ILB1 cells infected with the pLXIN vector alone. The presence of Mre11 in the nuclear soluble fraction is likely due to contamination of this fraction with cytoplasmic proteins, as evidenced by reprobing the blot with antibodies to Hsp90, a cytoplasmic protein. Very little Mre11 protein was localized to the nuclear pellet, or chromatin-bound, fraction from LXIN cells, a finding consistent with the inability of Mre11 to translocate to the nucleus in the absence of nibrin. Expression of wild-type nibrin in NBS cells resulted in a decrease of Mre11 in the cytoplasmic fraction and a proportional increase in the amount of Mre11 detected in nuclear fractions, particularly the chromatin bound nuclear pellet (Fig. 5B). Likewise, Mre11-NLS protein was detected in the nuclear fractions, including the nuclear pellet, in NBS cells expressing the Mre11-NLS transgene. Thus, the Mre11-NLS protein appeared to be able to associate with chromatin in the absence of nibrin.
FIG. 5.

Localization of the Mre11-NLS protein to chromatin in NBS cells. NBS-ILB1 cells stably transduced with the pLXIN retroviral vector alone (LXIN), wild-type nibrin (NBS1), or the Mre11-NLS transgene (Mre11-NLS) were unirradiated or exposed to 3 Gy of ionizing radiation. After 30 min cells were harvested and fractionated into cytoplasmic (lanes C), nuclear soluble (lanes NS), nuclear 125 mM NaCl wash (lanes 125), nuclear 250 mM NaCl wash (lanes 250), and the insoluble nuclear pellet (lanes NP). For comparison, whole-cell lysate was included (lanes WC). A total of 105 cell equivalents of protein per lane was separated on 3 to 8% SDS-polyacrylamide gels and Western blotted (WB). (A) Immunoblots of pLXIN cellular fractions were probed with an anti-Mre11 monoclonal antibody to assess the localization of Mre11 in the absence of full-length nibrin. The blot was reprobed with anti-Hsp90 antisera as a control for cytoplasmic proteins or with a monoclonal TRF2 antibody as a control for nuclear proteins. (B) Localization of Mre11 in cellular fractions from LXIN, NBS1, and Mre11-NLS expressing cells was detected by probing immunoblots with a monoclonal Mre11 antibody.
Relocalization of Mre11/Rad50 to the sites of DNA DSBs in chromatin appears to be mediated in part by nibrin (3, 6, 40, 45). To determine whether the Mre11-NLS protein could relocalize to the sites of DNA damage, we examined formation of irradiation-induced foci in ATLD cells where the Mre11-NLS protein was complexed with nibrin. ATLD fibroblasts complemented with vector alone, wild-type Mre11, or the Mre11-NLS transgene were exposed to 12 Gy of irradiation, and 6 h later cells were stained with antibodies specific for γ-H2AX, to indicate sites of DNA damage, and Mre11. As shown in Fig. 4D, the Mre11-NLS protein formed foci that overlapped with γ-H2AX foci, a finding comparable to ATLD cells expressing wild-type Mre11. Similar Mre11 foci were not observed in ATLD cells infected with vector alone. Thus, the Mre11-NLS protein was capable of relocalizing to sites of DNA damage in cells where nibrin is present.
Atm activation is not stimulated by nibrin lacking the Atm binding domain.
The results presented above indicated that nuclear expression of Mre11/Rad50 in the absence of nibrin was insufficient to stimulate Atm autophosphorylation after irradiation, despite the fact that the complex localized to chromatin. Thus, nibrin appears to plays a more active role in stimulating Atm autophosphorylation beyond simply directing the translocation of Mre11/Rad50 to the nucleus. Recent studies have shown that the C-terminal 20 amino acids of nibrin bind Atm and that deletion of this region is sufficient to disrupt nibrin-Atm interaction and the recruitment of Atm to the sites of DNA damage (15, 32, 44). However, contradictory results obtained in different systems have failed to resolve whether interaction between nibrin and Atm is necessary to stimulate Atm activation at early times after DNA damage (15, 44).
To test the contribution of nibrin-Atm interaction to the stimulation of Atm activation in our in vivo system, we introduced a nonsense mutation (K735X) in nibrin that deleted the C-terminal 19 amino acids. The K735X mutation was introduced into a full-length nibrin cDNA, as well as NbFR5, and the nibrin ΔAtm transgenes were expressed in NBS-ILB1 fibroblasts. Immunoprecipitation with nibrin antisera revealed that the NBS1 ΔAtm and NbFR5 ΔAtm transgenes were expressed at similar levels as the parental full-length nibrin and NbFR5 transgenes in NBS cells (Fig. 6A). Immunoblot analysis with Mre11 and Rad50 antisera demonstrated that both NBS1 ΔAtm and NbFR5 ΔAtm proteins complexed with Mre11 and Rad50. The amount of Mre11/Rad50 coimmunoprecipitated with the Atm binding domain mutants was slightly less than the parent protein but, given the proximity of the Mre11 and Atm binding domains in nibrin, this result was not surprising. As expected, the Mre11/Rad50/nibrin ΔAtm complex localized to the nucleus on Western blots of nuclear and cytoplasmic extracts (data not shown).
FIG. 6.

Expression of nibrin ΔAtm transgenes in NBS cells and Atm activation after irradiation. (A) Expression and MRN complex formation was analyzed in GM0637 control fibroblasts and NBS-ILB1 cells stably transduced with the pLXIN retroviral vector alone (LXIN), wild-type nibrin (NBS1), nibrin lacking the C-terminal Atm binding domain (NBS1 ΔAtm), a 300-aa C-terminal fragment of nibrin (NbFR5), and NbFR5 lacking the C-terminal Atm binding domain (NbFR5 ΔAtm). Nibrin was immunoprecipitated (IP) from 250 μg of total cellular protein using polyclonal nibrin antisera and proteins were separated on a 3 to 8% SDS-polyacrylamide gel. The Western blot (WB) was probed with an anti-Mre11 monoclonal antibody, a polyclonal Rad50 antibody, or a monoclonal nibrin antibody. (B) Atm autophosphorylation at serine 1981 was assessed in the above cells 15 min after treatment with 0 or 3 Gy of ionizing radiation (IR). Atm was immunoprecipitated from 800 μg of total cellular protein using a polyclonal Atm antibody and proteins were separated on 3 to 8% SDS-polyacrylamide gels. Immunoblots were probed with a monoclonal phosphoserine 1981 antibody or a polyclonal Atm antibody. To detect Chk2 and p53 phosphorylation, 20 μg of total cellular protein isolated from the above cells 15 min after 0 or 3 Gy of irradiation was separated on 7% SDS-polyacrylamide gels. Immunoblots were probed with phosphothreonine 68 antisera or a Chk2 monoclonal antibody. For p53, Western blots were probed with phosphoserine 15 antisera or a monoclonal p53 antibody.
The ability of the nibrin ΔΑtm proteins to stimulate the kinetics of Atm activation was assessed by exposing cells to 3 Gy of ionizing radiation and harvesting cells 15 min later. Atm protein was immunoprecipitated and serine 1981 phosphorylation was analyzed by Western blotting. As shown in Fig. 6B, NBS-ILB1 cells expressing wild-type nibrin or the NbFR5 transgenes stimulated robust Atm autophosphorylation early after irradiation, whereas Atm activation was dramatically reduced in NBS cells expressing NBS1 ΔΑtm and NbFR5 ΔΑtm. There appeared to be some residual ability of the NBS1 ΔΑtm protein to stimulate Atm activation compared to NBS cells expressing the NbFR5 ΔΑtm transgene or vector alone.
The reduction in Atm activation in the nibrin binding site mutants was reflected in decreased phosphorylation of downstream Atm targets (Fig. 6B). As expected, phosphorylation of Chk2 threonine 68 was reduced in NBS-ILB1 cells expressing the nibrin ΔAtm proteins, since this target is phosphorylated by Atm at the sites of DNA DSBs. More importantly, however, Atm phosphorylation of p53 serine 15 was also reduced in the nibrin binding site mutants. Since p53 serine 15 phosphorylation takes place in the nucleoplasm, this result confirms that nibrin-Atm interaction is necessary for stimulation of Atm activation after irradiation.
DISCUSSION
Studies using NBS cells complemented with various nibrin transgenes have revealed a role for the nibrin C terminus in the activation of the Atm protein kinase (5, 18). The contribution of nibrin to Atm activation is not an absolute requirement, but rather nibrin stimulates the kinetics of Atm activation. This stimulatory effect is evident primarily at early time points after low doses of irradiation (5, 18, 20). The recent finding that Atm activation is deficient using in vitro reactions lacking recombinant nibrin and in mouse cells conditionally null for nibrin expression raises the possibility that nibrin is actually required for Atm activation and that the quantitative effect on Atm activation seen in NBS cells is due to the hypomorphic nature of the 657del5 mutation in these cells (12, 24, 28).
The mechanism by which nibrin stimulates Atm autophosphorylation was addressed in the current study. Since the nibrin C-terminal 100 aa contains binding domains for Mre11 and Atm, we sought to determine whether the role of nibrin in Atm activation is to translocate Mre11/Rad50 to nucleus, or to interact with Atm. Using an Mre11 transgene with artificial NLS sequences attached to the C terminus, we were able to translocate Mre11/Rad50 to the nucleus in the absence of nibrin. Although the Mre11-NLS transgene was not as efficient at directing Mre11/Rad50 nuclear localization as intact nibrin, Mre11-NLS localized to chromatin and complemented Atm activation in ATLD cells. Despite this, Mre11-NLS/Rad50 failed to stimulate Atm after irradiation. These results indicate that whereas the role of nibrin in translocating Mre11 and Rad50 to the nucleus is necessary for normal Atm activation kinetics, it is not sufficient to restore this function.
What else is nibrin doing then? In vitro studies have demonstrated that nibrin stimulates the enzymatic activities of the Mre11/Rad50 complex (22, 34). Clearly, Mre11/Rad50 enzymatic activity is required for Atm activation, since ATLD cells complemented with a nuclease-deficient mutant of Mre11 fail to activate Atm after neocarzinostatin treatment and a Rad50 ATPase mutant fails to activate Atm in vitro (24, 41). These results suggest that DNA DSBs must be processed by Mre11/Rad50 in order to activate the DNA DSB response, perhaps through the induction of chromatin alterations or a single-stranded DNA intermediate.
An equally or more important function for nibrin appears to be the ability of nibrin to bind Atm. The C-terminal 20 amino acids of nibrin have been shown to interact with the HEAT repeats of Atm in yeast, Xenopus laevis, and mammals (15, 32, 44). This interaction is required for recruitment of Atm to DNA damage. Nibrin transgenes lacking the Atm binding domain fail to recruit Atm to DNA, and mouse cells conditionally null for nibrin expression also fail to recruit Atm to DNA after irradiation (12, 15).
The literature is less clear about the contribution of nibrin-Atm interaction to activating Atm. Falck et al. (15) reported that NBS cells expressing nibrin lacking the Atm binding region had indistinguishable Atm serine 1981 phosphorylation compared to cells expressing wild-type nibrin. However, downstream Atm targets, including Chk2, nibrin, and Smc1, were not phosphorylated in the Atm binding mutants. The authors of that study propose a model where Atm is activated in the nucleoplasm after DNA damage but is then recruited to sites of DNA DSBs where it phosphorylates downstream targets that are also recruited to DNA DSBs. This model is appealing because it is consistent with the rapid kinetics by which Atm becomes activated after DNA damage, an event argued to favor activation of Atm in a diffusible state (1). In contrast to these findings, however, the nibrin Atm binding domain was required for recruiting Atm to DNA and for subsequent activation of Atm in studies using Xenopus egg extracts (44). In fact, You et al. (44) were able to demonstrate the association of unphosphorylated Atm with double-stranded DNA, which became phosphorylated at serine 1981 within 5 to 10 min.
The results obtained here expressing nibrin transgenes lacking the Atm binding domain in NBS cells support the conclusions of You et al. and studies carried out with purified recombinant proteins (24, 44). In contrast to Falck et al. (15), we observed that Atm activation was dramatically reduced in the absence of the nibrin-Atm interaction domain. This decrease in Atm activation was reflected in reduced phosphorylation of p53 serine 15, which is not dependent upon recruitment of Atm to DNA DSBs, clearly delineating the role of nibrin in facilitating Atm activation from recruitment of Atm to DNA DSBs by nibrin. The K735X nonsense mutation used here to delete the Atm binding domain of nibrin is adjacent to the A734X mutation made by Falck et al. (J. Falck and S. Jackson, personal communication) and is unlikely to explain the difference in our results. However, Falck et al. have now performed additional experiments using a different Atm phospho-serine 1981 antibody and have obtained results consistent with our findings (J. Falck and S. Jackson, personal communication). Thus, there now is a consensus on this issue, with all studies indicating that Atm activation is stimulated by nibrin-Atm interaction.
Although we found that interaction between the nibrin C terminus and Atm had a profound effect on Atm activation, we consistently observed that the NBS1 ΔΑtm transgene had some residual ability to stimulate Atm activation after irradiation, compared to the NbFr5 ΔΑtm transgene that contains only the C terminus of nibrin. These findings suggest that other regions of nibrin can also contribute to Atm activation. The obvious candidate for this interaction would be via the nibrin FHA and BRCT domains. We, and others, have previously reported that mutations in the nibrin FHA and BRCT domains interfere with Atm phosphorylation of nibrin (6, 45). The nibrin FHA/BRCT domains are also required for accumulation of nibrin at the sites of DNA damage via interaction with MDC1, which may enhance activation of recruited Atm (6, 17, 27, 40, 45).
In the context of the overall DNA DSB response from DNA damage to the activation of downstream effectors, nibrin appears to play multiple roles. To sense DNA damage nibrin plays a role by translocating Mre11/Rad50 to the nucleus, stimulating their enzyme activity, and binding Atm, leading to its activation. In transducing the damage signal to downstream targets, nibrin functions by directing the accumulation of Atm at the sites of DNA damage where some Atm targets are phosphorylated. MRN may also act as an adaptor for some phosphorylation events, increasing the avidity of Atm for its targets (5, 23). Lastly, nibrin contributes to the effector phase of the DNA damage response through its role in S-phase checkpoint activation and by stimulating the DNA repair enzymatic functions of Mre11/Rad50.
Acknowledgments
We thank Tony DeMaggio and Domenico Delia for providing antibodies and Eric Olson for nucleotide sequencing. We also thank Jacob Falck and Steve Jackson, for helpful discussions and sharing of unpublished results.
This study was supported by grants from the A-T Medical Research Foundation (to K.C.) and a grant from the National Cancer Institute (CA57569; to P.C.).
REFERENCES
- 1.Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499-506. [DOI] [PubMed] [Google Scholar]
- 2.Banin, S., L. Moyal, S. Shieh, Y. Taya, C. W. Anderson, L. Chessa, N. I. Smorodinsky, C. Prives, Y. Reiss, Y. Shiloh, and Y. Ziv. 1998. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281:1674-1677. [DOI] [PubMed] [Google Scholar]
- 3.Carney, J. P., R. S. Maser, H. Olivares, E. M. Davis, M. Le Beau, J. R. Yates III, L. Hays, W. F. Morgan, and J. H. Petrini. 1998. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93:477-486. [DOI] [PubMed] [Google Scholar]
- 4.Carson, C. T., R. A. Schwartz, T. H. Stracker, C. E. Lilley, D. V. Lee, and M. D. Weitzman. 2003. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 22:6610-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerosaletti, K., and P. Concannon. 2004. Independent roles for nibrin and Mre11-Rad50 in the activation and function of Atm. J. Biol. Chem. 279:38813-38819. [DOI] [PubMed] [Google Scholar]
- 6.Cerosaletti, K. M., and P. Concannon. 2003. Nibrin forkhead-associated domain and breast cancer C-terminal domain are both required for nuclear focus formation and phosphorylation. J. Biol. Chem. 278:21944-21951. [DOI] [PubMed] [Google Scholar]
- 7.Cerosaletti, K. M., A. Desai-Mehta, T. C. Yeo, M. Kraakman-Van Der Zwet, M. Z. Zdzienicka, and P. Concannon. 2000. Retroviral expression of the NBS1 gene in cultured Nijmegen breakage syndrome cells restores normal radiation sensitivity and nuclear focus formation. Mutagenesis 15:281-286. [DOI] [PubMed] [Google Scholar]
- 8.Cortez, D., Y. Wang, J. Qin, and S. J. Elledge. 1999. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 286:1162-1166. [DOI] [PubMed] [Google Scholar]
- 9.D'Amours, D., and S. P. Jackson. 2002. The Mre11 complex: at the crossroads of DNA repair and checkpoint signaling. Nat. Rev. Mol. Cell. Biol. 3:317-327. [DOI] [PubMed] [Google Scholar]
- 10.de Jager, M., J. van Noort, D. C. van Gent, C. Dekker, R. Kanaar, and C. Wyman. 2001. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 8:1129-1135. [DOI] [PubMed] [Google Scholar]
- 11.Desai-Mehta, A., K. M. Cerosaletti, and P. Concannon. 2001. Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol. Cell. Biol. 21:2184-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Difilippantonio, S., A. Celeste, O. Fernandez-Capetillo, H. T. Chen, S. M. Reina, F. Van Laethem, Y. P. Yang, G. V. Petukhova, M. Eckhaus, L. Feigenbaum, K. Manova, M. Kruhlak, R. D. Camerini-Otero, S. Sharan, M. Nussenzweig, and A. Nussenzweig. 2005. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat. Cell Biol. 7:675-685. [DOI] [PubMed] [Google Scholar]
- 13.Dolganov, G. M., R. S. Maser, A. Novikov, L. Tosto, S. Chong, D. A. Bressan, and J. H. Petrini. 1996. Human Rad50 is physically associated with human Mre11: identification of a conserved multiprotein complex implicated in recombinational DNA repair. Mol. Cell. Biol. 16:4832-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong, Z., Q. Zhong, and P. L. Chen. 1999. The Nijmegen breakage syndrome protein is essential for Mre11 phosphorylation upon DNA damage. J. Biol. Chem. 274:19513-19516. [DOI] [PubMed] [Google Scholar]
- 15.Falck, J., J. Coates, and S. P. Jackson. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434:605-611. [DOI] [PubMed] [Google Scholar]
- 16.Gatei, M., D. Young, K. M. Cerosaletti, A. Desai-Mehta, K. Spring, S. Kozlov, M. F. Lavin, R. A. Gatti, P. Concannon, and K. Khanna. 2000. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 25:115-119. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg, M., M. Stucki, J. Falck, D. D'Amours, D. Rahman, D. Pappin, J. Bartek, and S. P. Jackson. 2003. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421:952-956. [DOI] [PubMed] [Google Scholar]
- 18.Horejsi, Z., J. Falck, C. J. Bakkenist, M. B. Kastan, J. Lukas, and J. Bartek. 2004. Distinct functional domains of Nbs1 modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene 23:3122-3127. [DOI] [PubMed] [Google Scholar]
- 19.Kim, S. T., B. Xu, and M. B. Kastan. 2002. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 16:560-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitagawa, R., C. J. Bakkenist, P. J. McKinnon, and M. B. Kastan. 2004. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 18:1423-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kraakman-van der Zwet, M., W. J. I. Overkamp, A. A. Friedl, B. Klein, G. W. C. T. Verhaugh, N. J. G. Jaspers, A. T. Midro, F. Eckardt-Schupp, P. H. M. Lohman, and M. Z. Zdzienicka. 1999. Immortalization and characterization of Nijmegen breakage syndrome fibroblasts. Mutat. Res. 434:17-27. [DOI] [PubMed] [Google Scholar]
- 22.Lee, J. H., R. Ghirlando, V. Bhaskara, M. R. Hoffmeyer, J. Gu, and T. T. Paull. 2003. Regulation of Mre11/Rad50 by Nbs1: effects on nucleotide-dependent DNA binding and association with ataxia-telangiectasia-like disorder mutant complexes. J. Biol. Chem. 278:45171-45181. [DOI] [PubMed] [Google Scholar]
- 23.Lee, J. H., and T. T. Paull. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93-96. [DOI] [PubMed] [Google Scholar]
- 24.Lee, J. H., and T. T. Paull. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308:551-554. [DOI] [PubMed] [Google Scholar]
- 25.Lim, D. S., S. T. Kim, B. Xu, R. S. Maser, J. Lin, J. H. Petrini, and M. B. Kastan. 2000. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 404:613-617. [DOI] [PubMed] [Google Scholar]
- 26.Lukas, C., J. Falck, J. Bartkova, J. Bartek, and J. Lukas. 2003. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 5:255-260. [DOI] [PubMed] [Google Scholar]
- 27.Lukas, C., F. Melander, M. Stucki, J. Falck, S. Bekker-Jensen, M. Goldberg, Y. Lerenthal, S. P. Jackson, J. Bartek, and J. Lukas. 2004. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 23:2674-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maser, R. S., R. Zinkel, and J. H. Petrini. 2001. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat. Genet. 27:417-421. [DOI] [PubMed] [Google Scholar]
- 29.Matsuoka, S., G. Rotman, A. Ogawa, Y. Shiloh, K. Tamai, and S. J. Elledge. 2000. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 97:10389-10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mendez, J., and B. Stillman. 2000. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 20:8602-8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirzoeva, O. K., and J. H. Petrini. 2003. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 1:207-218. [PubMed] [Google Scholar]
- 32.Nakada, D., K. Matsumoto, and K. Sugimoto. 2003. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 17:1957-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paull, T. T., and M. Gellert. 1998. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1:969-979. [DOI] [PubMed] [Google Scholar]
- 34.Paull, T. T., and M. Gellert. 1999. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 13:1276-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petrini, J. H. J., M. E. Walsh, C. DiMare, X.-N. Chen, J. R. Korenberg, and D. T. Weaver. 1995. Isolation and characterization of the human MRE11 homologue. Genomics 29:80-86. [DOI] [PubMed] [Google Scholar]
- 36.Rappold, I., K. Iwabuchi, T. Date, and J. Chen. 2001. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 153:613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savitsky, K., A. Bar-Shira, S. Gilad, G. Rotman, Y. Ziv, L. Vanagaite, D. A. Tagle, S. Smith, T. Uziel, S. Sfez, et al. 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268:1749-1753. [DOI] [PubMed] [Google Scholar]
- 38.Stewart, G. S., R. S. Maser, T. Stankovic, D. A. Bressan, M. I. Kaplan, N. G. Jaspers, A. Raams, P. J. Byrd, J. H. Petrini, and A. M. Taylor. 1999. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99:577-587. [DOI] [PubMed] [Google Scholar]
- 39.Taniguchi, T., I. Garcia-Higuera, B. Xu, P. R. Andreassen, R. C. Gregory, S. T. Kim, W. S. Lane, M. B. Kastan, and A. D. D'Andrea. 2002. Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell 109:459-472. [DOI] [PubMed] [Google Scholar]
- 40.Tauchi, H., J. Kobayashi, K. Morishima, S. Matsuura, A. Nakamura, T. Shiraishi, E. Ito, D. Masnada, D. Delia, and K. Komatsu. 2001. The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50/hMRE11/NBS1 complex DNA repair activity. J. Biol. Chem. 276:12-15. [DOI] [PubMed] [Google Scholar]
- 41.Uziel, T., Y. Lerenthal, L. Moyal, Y. Andegeko, L. Mittelman, and Y. Shiloh. 2003. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22:5612-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varon, R., C. Vissinga, M. Platzer, K. M. Cerosaletti, K. H. Chrzanowska, K. Saar, G. Beckmann, E. Seemanova, P. R. Cooper, N. J. Nowak, M. Stumm, C. M. R. Weemaes, R. A. Gatti, R. K. Wilson, M. Digweed, A. Rosenthal, K. Sperling, P. Concannon, and A. Reis. 1998. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93:467-476. [DOI] [PubMed] [Google Scholar]
- 43.Wu, X., V. Ranganathan, D. S. Weisman, W. F. Heine, D. N. Ciccone, T. B. O'Neill, K. E. Crick, K. A. Pierce, W. S. Lane, G. Rathbun, D. M. Livingston, and D. T. Weaver. 2000. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 405:477-482. [DOI] [PubMed] [Google Scholar]
- 44.You, Z., C. Chahwan, J. Bailis, T. Hunter, and P. Russell. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 25:5363-5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao, S., W. Renthal, and E. Y. Lee. 2002. Functional analysis of FHA and BRCT domains of NBS1 in chromatin association and DNA damage responses. Nucleic Acids Res. 30:4815-4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao, S., Y. C. Weng, S. S. Yuan, Y. T. Lin, H. C. Hsu, S. C. Lin, E. Gerbino, M. H. Song, M. Z. Zdzienicka, R. A. Gatti, J. W. Shay, Y. Ziv, Y. Shiloh, and E. Y. Lee. 2000. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature 405:473-477. [DOI] [PubMed] [Google Scholar]