

Abstract
Myosin VI is an unconventional motor protein, and its mutation is responsible for the familiar conditions sensorineural deafness and hypertrophic cardiomyopathy. Myosin VI is found to play a key role in the protein trafficking and homeostasis of the Golgi complex. However, very little is known about how myosin VI is regulated and whether myosin VI has a function in the DNA damage response. Here, we found that myosin VI is regulated by DNA damage in a p53-dependent manner and possesses a novel function in the p53-dependent prosurvival pathway. Specifically, we show that myosin VI is induced by p53 and DNA damage in a p53-dependent manner. We found that p53 directly binds to, and activates, the promoter of the myosin VI gene. We also show that the intracellular localization of myosin VI is substantially altered by p53 and DNA damage in a p53-dependent manner such that the pool of myosin VI in endocytic vesicles, membrane ruffles, and cytosol migrates to the Golgi complex, perinuclear membrane, and nucleus. Furthermore, we show that knockdown of myosin VI attenuates activation of p53 and impairs Golgi complex integrity, which makes myosin VI-deficient cells susceptible to apoptosis upon DNA damage. Taken together, we found a novel function for p53 in the maintenance of Golgi complex integrity and for myosin VI in the p53-dependent prosurvival pathway.
p53, a tumor suppressor protein, exerts its tumor suppression by regulating a plethora of p53 target genes (22). Based on their biological functions, these target genes can be classified into the following categories based on their products: cell cycle arrest, such as p21; apoptosis, such as Bax and Fas; DNA repair, such as XPB and DDB2; and antiangiogenesis and antimetastasis, such as VEGF and maspin. Induction of these genes is necessary for the host to repair or eliminate damaged cells and to prevent cellular transformation during exposure to genotoxic stresses (14, 22, 28, 43). Interestingly, recent evidence also indicates that p53 up-regulates a group of target genes that are prosurvival, such as those coding for DDR1, COX2, and HB-EGF (16, 20, 34). Presumably, induction of these prosurvival genes is necessary for the tissue homeostasis once the stress signals are subsided (22).
Myosins are the primary motor protein that use the energy derived from ATP to move cargos along actin filaments (11, 18, 23). Myosin VI, a member of the myosin superfamily, is an unconventional actin-based motor protein. One of the unique features of myosin VI is that myosin VI moves toward the minus end of actin filaments and therefore moves away from the plasma membrane into the cell and away from the surface of internal organelles such as the Golgi complex (11, 18, 23). Due to its localization in endocytic vesicles, membrane ruffles, cytosol, the Golgi complex, and perinuclear membrane, the motor activity of myosin VI is required for several physiological functions of the cell, such as protein secretion (9, 44), endocytosis (2, 3, 8, 10, 44), cell migration (19), spindle orientation (37), and spermatogenesis (24, 27).
In addition to its motor function, myosin VI is found to play a role in the maintenance of integrity of cellular organelles. For example, as part of the Golgi complex, myosin VI is necessary for the proper maintenance of Golgi complex morphology such that loss of myosin VI leads to a reduction of up to 40% of the organelle (44). Myosin VI is also found to be necessary for the maintenance of the normal stereocilial structure of the inner ear hair cell. Loss of myosin VI leads to the fusion of stereocilia and subsequently results in loss of hearing observed in humans and the Snell's waltzer mouse deficient in the myosin VI gene (1, 4, 31, 39). Furthermore, myosin VI-deficient mice exhibit a decrease in synapse number, abnormally short dendritic spines, and profound astrogliosis in hippocampus (35).
Although myosin VI is expressed in several organelles with distinct physiological functions, as outlined above, very little is known about how myosin VI is regulated within the cell and whether myosin VI has a function in the DNA damage response (11). Alternative splicing of the myosin VI gene produces four myosin VI isoforms (8), some of which have targeted expression in various organelles of the cell. Phosphorylation of myosin VI has also been shown to regulate its cellular localization (9, 32), but the responsible kinase(s) has not been characterized. In this report, we show that p53 regulates myosin VI in two major pathways: transactivation of myosin VI gene expression and intracellular localization of myosin VI protein such that the pool of myosin VI in the endocytic vesicles, membrane ruffles, and cytosol migrates to the Golgi complex, perinuclear membrane, and nucleus. Furthermore, we found a novel function for myosin VI in the p53-dependent prosurvival pathway and for p53 in the maintenance of Golgi complex integrity.
MATERIALS AND METHODS
Cell culture.
Tetracycline-inducible H1299 cell lines that express wild-type p53, p53(ΔAD1), p53(ΔPRD), p53(ΔBD), p53(AD1−), or p53(R249S) were cultured as previously reported (49). Dually inducible H1299 cell lines that express wild-type p53 under the ecdysone-inducible system and mutant p53(R273H) under the tetracycline-inducible system or that express wild-type p53 under the ecdysone-inducible system and wild-type p73β under the tetracycline-inducible system, were cultured as previously reported (45, 46, 49). RKO, LS174T, and HeLa cells were purchased from the American Type Culture Collection and cultured as instructed.
siRNA knockdown of myosin VI.
To generate a cell line in which myosin VI is stably knocked down, a U6 promoter-based small interfering RNA (siRNA) expression vector, pBabe-U6, was used (29). The following oligonucleotides were used: forward primer, 5′-tagaggtcccGATATTAGAGAAAAACTTCttcaagagaGAAGTTTTTCTCTAATATCtttttg-3′; and reverse primer, 5′-gatccaaaaaGATATTAGAGAAAAACTTCtctcttgaaGAAGTTTTTCTCTAATATCggacc-3′. Upon annealing, DNA oligonucleotides were cloned into XhoI and BamHI sites. The resulting construct, designated pBabe-U6-myo6, was transfected into RKO cells, and siRNA expression cell lines were selected with 0.6 μg/ml of puromycin. Western blot analysis was used to measure the level of myosin VI in RKO cells. To generate inducible myosin VI-knockdown cell lines, an H1 promoter-based tetracycline-inducible siRNA expression vector, pBabe-H1, was used (41). The following oligonucleotides were used: forward, 5′-gatccccGATATTAGAGAAAAACTTCttcaagagaGAAGTTTTTCTCTAATATCtttttggaaa-3′; and reverse, 5′-agcttttccaaaaaGATATTAGAGAAAAACTTCtctcttgaaGAAGTTTTTCTCTAATATCggg-3′. Upon annealing, DNA oligonucleotides were cloned into BglII and HindIII sites. The resulting construct, designated pBabe-H1-myo6, was transfected into RKO cells in which a tetracycline repressor is expressed by pcDNA6. siRNA-expressing RKO cell lines were selected with 0.6 μg/ml of puromycin. Western blot analysis was used to measure the level of myosin VI in RKO cells with or without tetracycline.
Northern blot analysis.
Northern blots were prepared by using 10 μg of total RNA isolated from cells induced to express p53 or treated with the DNA-damaging agent camptothecin. The myosin VI probe was prepared with a cDNA fragment of the myosin VI gene isolated from H1299 cells. The p21 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) probes were prepared as previously described (48).
Western blot analysis.
Whole cells were harvested by scrapping and extracted with 2× sodium dodecyl sulfate sample buffer. Proteins were separated by 7.5% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and probed with indicated antibodies followed by ECL enhanced chemiluminescence detection. The following antibodies were used in this study: anti-myosin VI (KA-15; Sigma), anti-p53 (FL-393 and Pab1801; Santa Cruz), anti-p21 (C-19; Santa Cruz), anti-Mdm2 (Ab-2 and SMP-14; Oncogene), anti-Fas (FL-335; Santa Cruz), anti-14-3-3σ (Ab-1; Oncogene), anti-Bax (Ab-2 from Oncogene and P-19 from Santa Cruz), anti-phospho-histone H2AX (Upstate), antiactin (Sigma), antihemagglutinin (anti-HA) (Berkeley Antibody Company), anti-GM130 (Transduction Laboratories), anti-TGN p230 (Transduction Laboratories), anti-PARP (Pharmingen), anti-caspase 8 (BioSource), and anti-cyclin B1 (H-433; Santa Cruz). Anti-GBF and anti-p115 were provided by E. Sztul's laboratory (The University of Alabama at Birmingham).
Luciferase assay.
A 795-bp DNA fragment containing the myosin VI promoter was PCR amplified using genomic DNAs from MCF7 cells with the forward primer 5′-TGAGGGTACCGGGGCTCTAAATAAAGT-3′ and the reverse primer 5′-AATTAAGCTTGCGGTGAGAAGTGATGAGGG-3′. The PCR product, Myo6-766, was cloned into pGEM-T-Easy vector and confirmed by DNA sequencing. After digesting with KpnI and HindIII, Myo6-766 was cloned into pGL2-Basic vector. The resulting luciferase reporter was designated as pGL2-Myo6-766. Using pGL2-Myo6-766 as a template, several mutated constructs were generated by PCR using the above reverse primer and one of the following forward primers: Myo6-571 (5′-GGTACCAACCGCCACGCAGCCTCG-3′), Myo6-371 (5′-GGTACCAAAGAGGCCGTT-3′), Myo6-mut 371 (5′-GGTACCAAAGAGGCCGTTTCGCCATGGGAAGAAAAATAACTTCCGGATT-3′), Myo6-del-348 (5′-GGTACCAAAGAGGCCGTTTCGCCATGGGAAAGGGGCAGCCCCCCTCTGCGCTGCAGG-3′), Myo6-321 (5′-GGTACCGGCAGCCCCCCTCT-3′), Myo6-171 (5′-GGTACCCCCGCCCCTTCG-3′), and Myo6-71 (5′-GGTACCCGAGCAGGAAGCCAG-3′). To generate various internal deletions in the GC-rich region, deletion constructs were generated by PCR using the above forward primer and a primer that spans the deleted region as indicated. These PCR products were confirmed and then cloned into pGL2-Basic vector. To measure whether the myosin VI promoter is responsive to p53, 0.5 μg of pGL2-Myo6-766 or one of its derivatives was cotransfected with 2 μg of pcDNA3 control vector or a vector expressing wild-type p53 or mutant p53(R249S) into p53-null H1299 cells by the calcium phosphate method. Twenty nanograms of pRL-CMV was also cotransfected as an internal control. A dual-luciferase assay was performed 24 h after transfection as instructed (Promega).
ChIP assay.
The chromatin immunoprecipitation (ChIP) assay was performed as previously described (29). The fragment containing the p53-responsive element in the myosin VI promoter was detected with a forward primer (5′-Myo6, 5′-GCAAAAGAGGCCGTTTCGCCATGGG-3′) and a reverse primer (3′-Myo6, 5′-GTCACGGCTACGGAGGCCCGAAGG-3′). The fragment containing the p53-responsive element in the p21 promoter was detected with a forward primer (5′-p21, 5′-CAGGCTGTGGCTCTGATTGG-3′) and a reverse primer (3′-p21, 5′-TTCAGAGTAACAGGCTAAGG-3′).
Immunofluorescence microscopy.
Cells were grown on four-well chamber slides and treated as indicated. After being washed with phosphate-buffered saline, cells were fixed with 3% formaldehyde for 45 min, permeabilized with 0.5% Triton X-100 for 5 min, blocked with 1% bovine serum albumin for 1 h, and then incubated with primary antibody/antibodies for 1 to 2 h followed by incubation with fluorescein isothiocyanate (FITC)-, Texas red-, and/or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch and Molecular Probes). Cells were also stained with 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma) to visualize nuclei and then mounted with a solution containing 0.1% of purified protein derivative (Sigma) and 80% glycerol in phosphate-buffered saline. Intracellular localization of proteins was analyzed by immunofluorescence microscopy.
DNA histogram analysis.
Various cells were seeded at 2 × 105 per 90-mm plate and then uninduced or induced to express various proteins for 36 h or myosin VI siRNA for 3 days followed by treatment with 1 μg/ml of doxorubicin for 16 h. Both floating cells in the medium and live cells on the plates were collected, and DNA histogram analysis was performed as described previously (12).
RESULTS
The myosin VI gene is a novel p53 target gene.
Upon exposure to various extrinsic and intrinsic stresses, p53 is activated in cells (21, 28, 43), which then regulates an array of target genes that mediate the function of p53 as a tumor suppressor (14, 22). To identify novel p53 target genes, we performed a cDNA subtraction assay and found many known target genes induced by p53, such as those coding for p21, MDM2, and PIG3. We also found the gene coding for myosin VI, a potential target gene induced by p53. To confirm the cDNA subtraction assay, Northern blot analysis was performed. We showed that myosin VI was highly induced by p53 in a time-dependent manner (Fig. 1A, upper panel). As a control, the expression of p21 was examined and found to be induced by p53 (Fig. 1A, bottom panel). We also examined the induction of myosin VI by several p53 mutants (Fig. 1B). As expected, p53(R249S), a tumor-derived mutant, was incapable of inducing myosin VI and p21 (Fig. 1B, compare lanes 11 and 12, upper and lower panels). In addition, p53(AD1−), a double-point mutant at codons 22 and 23 that inactivates the first activation domain in p53, was also unable to induce myosin VI (Fig. 1B). In contrast, myosin VI was weakly induced by p53(ΔAD1), which lacks the first activation domain in residues 1 to 42, p53(ΔPRD), which lacks the proline-rich domain in residues 62 to 91, and p53(ΔBD), which lacks the C-terminal basic domain in residues 364 to 393 (Fig. 1B). These data are consistent with a previous report that these p53 mutants have a reduced transcriptional activity (49).
FIG. 1.

Myosin VI is a novel target gene of p53. (A) Myosin VI is induced by p53 in a time-dependent manner. (B) Induction of myosin VI by wild-type and various mutated forms of p53. (C) Myosin VI is induced by DNA damage in a p53-dependent manner. RKO and LS174T were either untreated (−) or treated (+) with 300 nM camptothecin for 24 h. Northern blots were probed with 32P-labeled cDNA for myosin VI, p21, and/or GAPDH.
DNA damage stabilizes and activates p53, leading to induction of p53 target genes (14, 22). If myosin VI is a true p53 target, it would be induced by DNA damage in cells that contain an endogenous wild-type p53 gene. To this end, we tested two cell lines untreated or treated with camptothecin, an inhibitor of topoisomerase 1 which can induce double-strand breaks (33). We found that myosin VI was induced in camptothecin-treated RKO and LS174T cells (Fig. 1C, lanes 1 to 4). These cell lines contain wild-type p53. As a positive control, the expression of p21 was measured and found to be induced as expected (Fig. 1C, p21 panel).
Next, we determined whether an increase in myosin VI transcripts correlates with an increase in myosin VI protein. Endogenous wild-type p53 was activated in RKO cells by treatment with doxorubicin or camptothecin. RKO-p53-KD, an RKO derivative in which endogenous p53 is silenced by RNA interference (RNAi), was similarly treated with doxorubicin or camptothecin. As expected, p53 was stabilized and p21 was concomitantly induced upon treatment with doxorubicin or camptothecin in RKO but not RKO-p53-KD cells (Fig. 2A, p53 and p21 panels). Similarly, we found that the level of myosin VI was increased in RKO cells by treatment with either doxorubicin or camptothecin (Fig. 2A). However, the level of myosin VI was not significantly increased in RKO-p53-KD cells upon DNA damage (Fig. 2A, myosin VI panel). We also determined the induction of myosin VI in RKO cells when treated with doxorubicin for various times. We found that like p21, myosin VI was induced in RKO cells by DNA damage in a time-dependent manner (Fig. 2B).
FIG. 2.
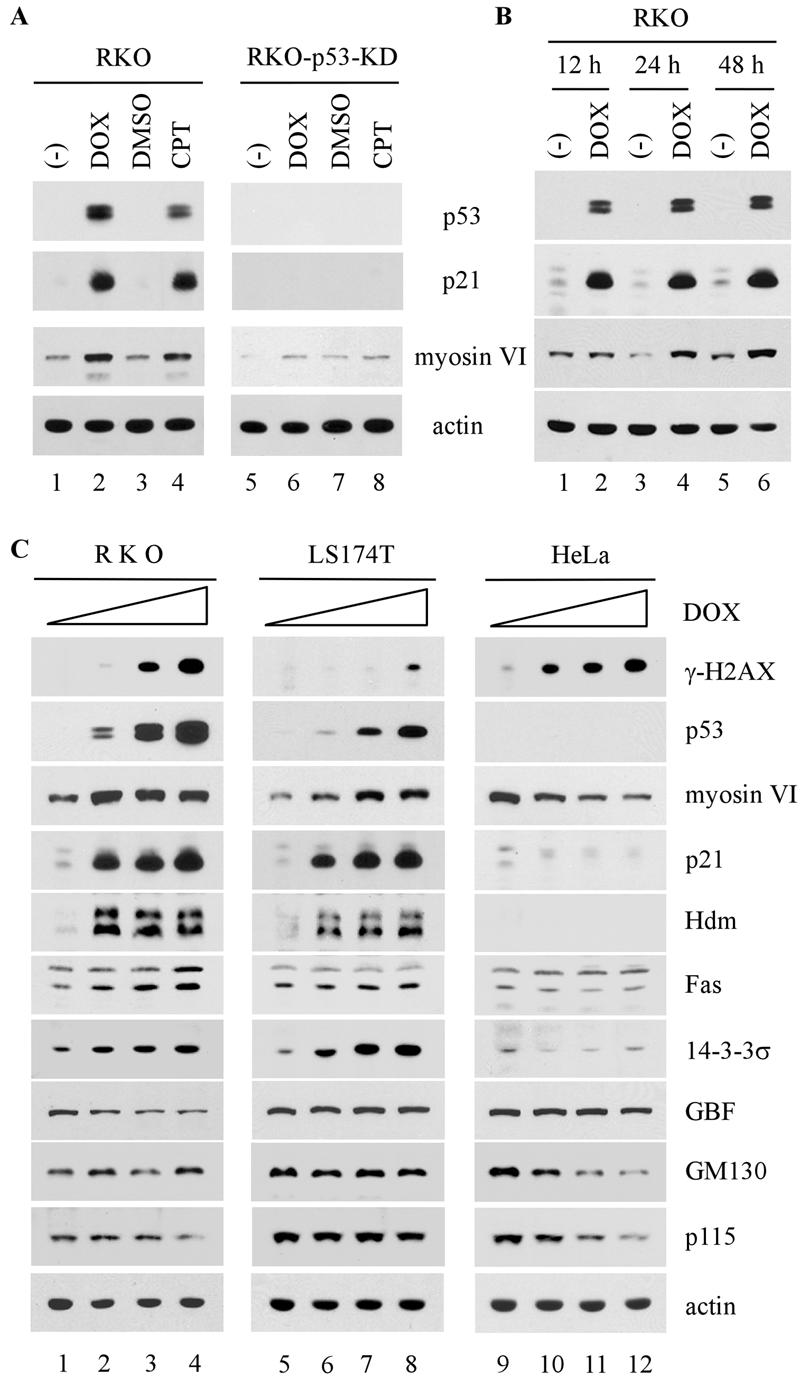
Level of myosin VI, but not GBF, GM130 and p115, is increased by DNA damage in a p53-dependent manner. (A) The induction of myosin VI by DNA damage is dependent on p53. RKO and RKO-p53-KD were untreated (−) or treated with dimethyl sulfoxide (DMSO), 0.2 μg/ml of doxorubicin (DOX), or 200 nM camptothecin (CPT). (B) The level of myosin VI protein is increased by DNA damage in a time-dependent manner. RKO cells were untreated (−) or treated with 0.2 μg/ml of DMSO, 0.2 μg/ml of doxorubicin, or 100 nM camptothecin for 12, 24, and 48 h. (C) The level of myosin VI, but not other Golgi proteins, is increased by DNA damage in a p53-dependent manner. RKO, LS174T, and HeLa cells were treated with 0, 0.1, 0.3, or 0.5 μg/ml of doxorubicin. Whole-cell extracts were analyzed by Western blotting with antibodies as indicated. Actin was analyzed as a loading control.
Since myosin VI is known to be localized in the Golgi complex, we wanted to determine whether other Golgi proteins are regulated by p53. To test this, the induction of myosin VI and other Golgi proteins, including GBF, GM130, and p115, was assayed in RKO, LS174T, and HeLa cells upon treatment with doxorubicin. As shown in Fig. 2C, the level of phosphorylated histone H2AX (γ-H2AX) in all three cell lines was increased upon treatment with doxorubicin in a dose-dependent manner, indicating that double-strand DNA breaks were generated (17). As expected, the level of p53 was increased in wild-type p53-containing RKO and LS174T cells, but not in p53-null-like HeLa cells (Fig. 2C, p53 panel). We also showed that as a p53 target gene, the myosin VI, p21, Hdm2, Fas, and 14-3-3σ genes were induced by activated p53 in RKO and LS174T cells, but not in HeLa cells (Fig. 2C). However, unlike myosin VI, GBF, GM130, and p115 were not increased by activated p53 (Fig. 2C). Taken together, we showed that myosin VI, a novel p53 target, is specifically up-regulated by DNA damage in a p53-dependent manner.
If myosin VI is transcriptionally regulated by p53, one or more p53 responsive elements should exist in the myosin VI gene (14, 22). To test this, we analyzed the genomic locus containing myosin VI and found one potential p53 binding site (GAACAAGAACtccCGGCTTGTCC) (Fig. 3A, box A) and a stretch of GC-rich region (Fig. 3A, box B) located within the promoter of the myosin VI gene between nucleotides (nt) −347 and −71, with +1 as the putative transcription start site. The potential p53 binding site contains three mismatches in the noncritical positions of the consensus p53-binding site (15). The GC-rich region is also shown to be recognized by p53 (5). To determine whether p53 transactivates myosin VI via the potential p53 binding site or the GC-rich region, a 795-bp DNA fragment, which contains both putative p53-responsive elements, was cloned into the promoterless luciferase reporter vector, pGL2. The resulting vector was designated Myo6-766 (Fig. 3A). We also generated several deletion or point mutants of the myosin VI promoter (Fig. 3A). Each of these luciferase reporters was cotransfected into H1299 cells with either a pcDNA3 control vector or a vector that expresses wild-type or mutant p53. We found that the luciferase activity for Myo6-766 was markedly increased by wild-type p53, but not by mutant p53(R249S) (Fig. 3B). Interestingly, deletion or mutation of the potential p53 binding site had no effect on the transactivation of the myosin VI promoter by p53 (Fig. 3A and B). In contrast, the GC-rich region located between nt −171 and −71 was required since p53 was unable to increase the luciferase activity for myo6-71 reporter (Fig. 3A and B). To demonstrate the requirement of the GC-rich region for p53 induction of myosin VI, we generated 11 additional promoter constructs that still contain box A but lack a part of or the entire GC-rich region (Fig. 3C). We found that the 3′ half of the GC-rich region was required for the response of the myosin VI promoter to p53 as the promoter with deletion of nt −120 to −71 was inert to p53 (Fig. 3D). Upon further dissection of the 3′ half of the GC-rich region, we found that nucleotides from −80 to −71 (GGGATCTGTC) were required for the promoter activity, whereas nucleotides from −120 to −110 (GACTCGGCGG) were partially necessary (Fig. 3E). Nevertheless, both of the half-sites conform somewhat to the consensus sequence as determined by the p53MH algorithm (25).
FIG. 3.

Myosin VI is a direct target of the p53 transcription factor. (A) Schematic presentation of the human myosin VI gene locus and luciferase (Luc) reporter constructs. Box A represents a potential p53-binding site from nt −347 to −325, whereas box B is a GC-rich region from nt −171 to −72. (B) The GC-rich region in the myosin VI promoter is responsive to wild-type (wt) p53, but not mutant (mut) p53. The luciferase assay was performed as described in Materials and Methods. con, control. (C) Schematic presentation of 11 additional myosin VI promoter/reporter constructs with deletion in the GC-rich region. (D and E) The GC-rich region is required for p53 transactivation of the myosin VI promoter. (F and G) Schematic representation of the myosin VI and p21 promoters with the locations of the transcription start site, p53-responsive elements, and primers used for ChIP assays. (H) p53 binds directly to the myosin VI promoter. The ChIP assay was performed as described in Materials and Methods. (I) Wild-type p53, but not mutant p53(R249S), binds directly to the myosin VI promoter. α-p53 and α-419, anti-p53 and anti-419 antibodies, respectively.
To determine whether wild-type p53 induces myosin VI by directly binding to the myosin VI promoter, we performed a ChIP assay with a pair of primers that amplify the GC-rich region (Fig. 3F). As a positive control, the interaction of p53 with the responsive element 1 (p53RE-1) in the p21 promoter was determined (Fig. 3G). The p53-DNA complexes were immunoprecipitated with anti-p53 antibody. We found that the captured DNA fragment with the GC-rich region was markedly increased upon inducible expression of p53 (Fig. 3H and I, compare lanes 3 and 4). Similarly, p53 bound to the responsive element 1 (p53RE-1) in the p21 promoter (Fig. 3H, compare lanes 1 and 2). As a negative control, no DNA fragment was enriched by the control antibody Pab419 (Fig. 3I, compare lanes 5 and 6). In addition, mutant p53(R249S) was incapable of binding to the GC-rich region in the myosin VI promoter (Fig. 3I, lanes 7 to 12).
The intracellular localization of myosin VI is altered by wild-type p53, but not mutant p53 or wild-type p73β.
The intracellular localization dictates the function of myosin VI in the homeostasis of the Golgi complex, intracellular protein trafficking, endocytosis, and cell migration (11, 18, 23). In order to determine what role myosin VI plays in the p53 pathway, we examined the cellular localization of myosin VI in the presence or absence of p53. To test this, two previously characterized H1299-derived cell lines, H24-208 and H24-112, were used (45, 46). H1299 is a p53-null and non-small-cell lung carcinoma cell line. H24-208 is a dually inducible cell line, which expresses Flag-tagged wild-type p53 by the ecdysone-inducible system and HA-tagged mutant p53(R273H) by the tetracycline-inducible system (45). H24-112 is also a dually inducible cell line, which expresses HA-tagged wild-type p53 by the ecdysone-inducible system and HA-tagged wild-type p73β by the tetracycline-inducible system (46). First, we checked the inducibility of various proteins upon induction. In H24-208 cells, Flag-tagged wild-type p53 was expressed upon treatment with the ecdysone analog ponasterone A; HA-tagged mutant p53 was expressed upon withdrawal of tetracycline; and both proteins were expressed upon treatment with ponasterone and withdrawal of tetracycline (Fig. 4A, the top panel). In H24-112 cells, HA-tagged wild-type p53 was expressed upon treatment with ponasterone, HA-tagged wild-type p73β was expressed upon withdrawal of tetracycline, and both proteins were expressed upon treatment with ponasterone and withdrawal of tetracycline (Fig. 4B, the top panel). Second, we examined the activity of p53 and p73 to induce myosin VI expression. In H24-208 cells, myosin VI and p21 were induced by wild-type, but not mutant, p53, as expected (Fig. 4A, myosin VI and p21 panels). In addition, we showed that as a dominant-negative mutant, p53(R273H) inhibited the induction of p21 and myosin VI by wild-type p53 (Fig. 4A, myosin VI and p21 panels). The level of GBF, a Golgi protein, was not affected by both wild-type and mutant p53 (Fig. 4A, GBF panel). In H24-112 cells, p21 was induced by both wild-type p53 and p73β (Fig. 4B, p21 panel). Interestingly, myosin VI was induced by wild-type p53, but not wild-type p73β (Fig. 4B, myosin VI panel). This is not entirely surprising since p53 and p73 are known to regulate both common and distinct target genes (22). The level of p115, a Golgi protein, was not affected by both wild-type p53 and p73β (Fig. 4B, p115 panel).
FIG.4.

Intracellular localization of myosin VI is altered by wild-type p53, but not mutant p53 or wild-type p73β. (A) Myosin VI is induced by wild-type (wt) p53, but not mutant (mut) p53(R273H). H24-208 cells were uninduced (control), induced to express wild-type p53, induced to express mutant p53(R273H), or induced to express both for 48 h. The expression level of wild-type p53, mutant p53, myosin VI, p21, and GBF was quantified by Western blot analysis. Actin was analyzed as a loading control. (B) Myosin VI is induced by wild-type p53, but not wild-type p73β. H24-112 cells were uninduced (control), induced to express wild-type p73β, induced to express wild-type p53, or induced to express both for 48 h. The expression level of p73β, p53, myosin VI, p21, and p115 was quantified by Western blot analysis. (C) The intracellular localization of myosin VI is altered by wild-type p53, which is inhibited by mutant p53(R273H). Immunofluorescence staining was performed as described in Materials and Methods. p53 was stained green, whereas myosin VI was stained red. Nuclei were stained blue. con, control. (D) The intracellular localization of myosin VI is altered by wild-type p53, but not wild-type p73β. (E) Colocalization of myosin VI with trans-Golgi marker TGN p230. (F) Cell cycle arrest is not sufficient to alter cellular localization of myosin VI. H1299 cells were uninduced or induced to express p53, p73β, or both. The cell cycle profile was determined by DNA histogram analysis 36 h following induction of p53, p73, or both.
Next, the intracellular localization of myosin VI was examined in these cells upon expression of wild-type p53, mutant p53(R273H), and/or wild-type p73β. In H24-208 cells, both wild-type and mutant p53 were detected in the nucleus upon induction (Fig. 4C, p53 column). We also found that in general, the intensity of myosin VI staining was much stronger in the presence of wild-type p53 than that in the absence of p53 or in the presence of mutant p53(R273H) (data not shown). This is consistent with the above data that myosin VI is induced by wild-type p53. Interestingly, we found that the myosin VI localization was markedly altered in the presence of wild-type p53 such that the pool of myosin VI in the intracellular vesicles, membrane ruffles, and cytosol migrates to the Golgi complex, perinuclear membrane, and nucleus (Fig. 4C, wild-type p53 panel). In contrast, mutant p53(R273H) had no effect (Fig. 4C, mutant p53 panel). In addition, the effect of wild-type p53 on myosin VI localization was attenuated by mutant p53 (Fig. 4C, wild-type p53 and mutant p53 panel). In H24-112 cells, we also found that the myosin VI localization was similarly altered by wild-type p53, whereas p73β had no effect (Fig. 4D). When both wild-type p53 and p73β were expressed, wild-type p53 was still capable of altering the localization of myosin VI (Fig. 4D). To make certain that myosin VI is located in the trans-Golgi, we examined the colocalization of myosin VI and TGN p230, a well-defined trans-Golgi marker (6). As shown in Fig. 4E, myosin VI was colocalized with TGN p230 and the level of myosin VI in trans-Golgi was markedly increased by p53, but not p73β. To rule out the possibility that the effect of p53 on myosin VI is an indirect outcome of cell cycle arrest by p53, we examined the cell cycle profile in the presence of p53, p73, or both. We found that under the same condition, p73β had an effect on the cell cycle similar to that of p53 (Fig. 4F) and yet did not affect myosin VI relocalization (Fig. 4D to E).
To examine whether endogenous p53, when activated upon DNA damage, is capable of altering the intracellular localization of myosin VI, both parental RKO cells (Fig. 5A) and RKO-p53-KD cells in which endogenous p53 is knocked down by siRNA (Fig. 5B) were mock treated or treated with doxorubicin. These cells were then stained with anti-p53, anti-myosin VI, and DAPI. We found that p53 was accumulated upon DNA damage in parental RKO but not RKO-p53-KD cells (Fig. 5A and B, p53 column). Furthermore, like that in the H1299 cells (Fig. 4C and D), myosin VI localization was altered by DNA damage in parental RKO cells (Fig. 5A, myosin VI column), but not in RKO-p53-KD cells (Fig. 5B, myosin VI column), demonstrating that the DNA damage-induced alteration of myosin VI localization is p53 dependent. To make certain that some fraction of myosin VI is located in the trans-Golgi, we examined the colocalization of myosin VI and trans-Golgi TGN p230. As shown in Fig. 5C, myosin VI was colocalized with TGN p230 and the level of myosin VI in the trans-Golgi was markedly increased in RKO cells upon treatment with nutlin and camptothecin. Nutlin is a small molecular inhibitor of Mdm2 that binds to Mdm2, blocks the interaction of p53 with Mdm2, and leads to p53 activation without causing other cellular stresses (40, 42). Therefore, the enhanced expression and accumulation of myosin VI in the trans-Golgi by Nutlin reconfirmed the notion that p53 directly induces myosin VI expression and alters its subcellular localization.
FIG. 5.

(A and B) Intracellular localization of myosin VI is altered in RKO cells by DNA damage in a p53-dependent manner. Parental RKO and RKO-p53-KD cells were mock treated or treated with doxorubicin (DOX). These cells were then stained with anti-p53, anti-myosin VI, and DAPI. (C) Colocalization of myosin VI with trans-Golgi marker TGN p230. Parental RKO cells were mock treated or treated with nutlin or camptothecin (CPT).
To further confirm that myosin VI nuclear localization is increased by p53, whole-cell extracts and nuclear extracts were prepared from H1299 cells, which were uninduced or induced to express p53. We showed that upon induction of p53, myosin VI, but not GM130, a Golgi marker (6), was markedly increased (Fig. 6), consistent with the above data (Fig. 4A and B). In addition, we showed that the nuclear fraction of myosin VI was also substantially increased (Fig. 6). We would like to mention that only a minute fraction of GM130 was detected in the nuclear extracts, suggesting that the level of myosin VI in the nuclear extracts reflects the extent of the nuclear localization of myosin VI.
FIG. 6.
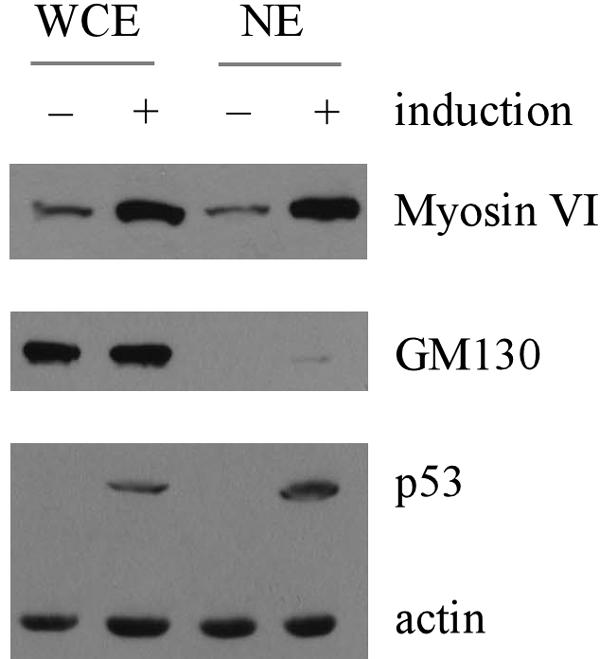
Nuclear localization of myosin VI is increased by p53. Whole-cell extracts (WCE) or nuclear extracts (NE) were prepared from H1299 cells uninduced or induced to express p53. The level of myosin VI, cis-Golgi marker GM130, p53, and actin was quantified by Western blot analysis.
Myosin VI knockdown attenuates the activation of p53 and inhibits myosin VI-mediated maintenance of Golgi complex integrity.
To determine the role of myosin VI in the p53 pathway, we generated stable RKO cell lines in which endogenous myosin VI is silenced by siRNA. Interestingly, we observed that upon DNA damage, the accumulation of p53 and induction of p21 were impaired in an RKO cell line in which endogenous myosin VI is >80% knocked down compared to the parental RKO cell line or an RKO cell line in which myosin VI is only partially knocked down (data not shown). To further test this, we generated RKO cell lines in which myosin VI is inducibly knocked down by siRNA using the tetracycline-inducible system. One representative cell line, RKO-myo6-KD-87, was chosen for further characterization (Fig. 7A). Upon induction of myosin VI siRNA, the level of myosin VI was markedly reduced (Fig. 7A, compare lanes 1 and 2). To determine the effect of myosin VI on p53 activation, RKO cells were treated with doxorubicin for 0.5, 2, or 4 h with or without myosin VI knockdown. We found that phosphorylated histone H2AX (γ-H2AX) was increased in a time-dependent manner, indicating that double-strand DNA breaks were generated upon treatment with doxorubicin. Again, we found that the accumulation of p53 upon DNA damage was attenuated in RKO cells with myosin VI knockdown (Fig. 7A, compare lanes 3 with 4, 7 with 8, and 11 with 12). It should be noted that in RKO cells, a short-term exposure to doxorubicin (within 4 h) did not lead to an increase of myosin VI (Fig. 7A, compare lanes 1 with 3, 5 with 7, and 9 with 11), whereas long-term exposure (12 h or longer) did lead to a marked increase (Fig. 2B, compare lanes 1, 3, and 5 with 2, 4, and 6, respectively).
FIG. 7.

Myosin VI knockdown attenuates the activation of p53 and impairs myosin VI-mediated maintenance of Golgi complex integrity. (A) Myosin VI is required for efficient stabilization of p53 by DNA damage. Myo6-KD-87, in which myosin VI is inducibly knocked down by siRNA, was uninduced (−) or induced (+) to express siRNA against myosin VI for 3 days followed by treatment with 0 or 5 μg/ml of doxorubicin for 0.5, 2, or 4 h. The expression level of myosin VI, p53, and γ-H2AX was quantified by Western blot analysis. Actin was analyzed as an internal loading control. (B) Intracellular localization of p53 and myosin VI in Myo6-KD-87 RKO cells. RKO cells were uninduced (top panel), treated with 1.0 μg/ml of doxorubicin (DOX) for 16 h (second panel), induced to express siRNA against myosin VI (third panel), or induced to express siRNA against myosin VI and treated with 1.0 μg/ml of doxorubicin (bottom panel). Immunofluorescence staining was performed as described in Materials and Methods. p53 was stained green, whereas myosin VI was stained red. Nuclei were stained blue. (C) Intracellular localization of myosin VI and Golgi marker protein TGN p230 in Myo6-KD-87 RKO cells. The experiments were performed similarly to those in panel B. TGN p230 was stained green, whereas myosin VI was stained red. Nuclei were stained blue.
In the experiments above, we showed that in addition to the enhanced expression of myosin VI, p53 also alters the intracellular localization of myosin VI (Fig. 4C and D, above). Thus, we wanted to examine the intracellular localization of myosin VI in RKO-myo6-KD-87 cells (Fig. 7B). We found that myosin VI was distributed normally in the Golgi complex, intracellular vesicles, membrane ruffles and cytosol under the control condition (Fig. 7B, the top panel), which is similar to that in the parental RKO cells (Fig. 5A). Upon treatment with doxorubicin, p53 was accumulated in nucleus and the level of myosin VI increased. In addition, the concentration of myosin VI in the Golgi complex and nucleus was detectably increased (Fig. 7B, second panel). Upon myosin VI knockdown, the myosin VI signal in the intracellular organelles, including the Golgi complex, was substantially reduced (Fig. 7B, third panel). When RKO cells were subjected to both myosin VI knockdown and treatment with doxorubicin, we found that p53 was still accumulated in the nucleus, albeit more weakly than that in RKO cells without myosin VI knockdown (Fig. 7B, p53 column, compare the second panel with the bottom panel). We also found that the intensity of myosin VI staining was markedly reduced as expected (Fig. 7B, bottom panel). However, we found that the accumulation of the intracellular myosin VI in the Golgi complex and nucleus following doxorubicin treatment was markedly reduced in myosin VI-knockdown RKO cells (Fig. 7B, compare the second panel with the bottom panel).
To determine whether the decreased accumulation of myosin VI in the Golgi complex has any effect on the integrity of the organelle upon DNA damage, we analyzed the expression of TGN p230, a trans-Golgi protein (6). We showed that under the control condition, myosin VI was colocalized with TGN p230 in the Golgi complex, although the majority of myosin VI was expressed in the perinuclear membrane and cytosol (Fig. 7C, the top panel). When cells were treated with doxorubicin, the level of myosin VI was substantially increased as expected and myosin VI was still colocalized with TGN p230 in the Golgi complex (Fig. 7C, second panel). When cells were induced to knock down myosin VI, the level of myosin VI was substantially reduced, as expected (Fig. 7C, third panel). Similarly, when cells were induced to knock down myosin VI and treated with doxorubicin, the level of myosin VI was slightly increased due to stabilized p53 (Fig. 7C, bottom panel), but was still lower than that in RKO cells without myosin VI knockdown (Fig. 7C, myosin VI column, compare the second panel with the bottom panel). Most importantly, we found that the colocalization of myosin VI with TGN p230 in the Golgi complex was markedly reduced when myosin VI was knocked down (Fig. 7C, compare the top two panels with the bottom two panels). Furthermore, upon knockdown of myosin VI and/or DNA damage, the integrity of the Golgi complex was altered since the pattern of TGN p230 staining was scattered (Fig. 7C).
Previous studies have shown that when the integrity of the Golgi complex is compromised by cleavage of a Golgi protein, the cell becomes prone to stress-induced apoptosis (13). Thus, we reasoned that upon DNA damage, p53 is activated, which then regulates myosin VI to protect the integrity of the Golgi complex for cell survival. To test this, we examined the extent of cell death in two RKO cell lines in which myosin VI is inducibly knocked down. We showed that myosin VI knockdown alone did not induce apoptosis in Myo6-KD-87 RKO cells (Fig. 8A, compare myosin VI-KD with the control). Upon treatment with doxorubicin, approximately 43% of RKO cells underwent apoptosis (Fig. 8A, doxorubicin panel). Interestingly, we found that the susceptibility to undergo DNA damage-induced apoptosis was significantly increased in RKO cells upon myosin VI knockdown (Fig. 8A, compare doxorubicin panel with doxorubicin plus myosin VI-KD). Similar results were detected in another myosin VI-knockdown RKO cell line, Myo6-KD-3 (data not shown). We also analyzed the apoptotic response of RKO cells to nutlin, an inhibitor of Mdm2. We found that upon myosin VI knockdown, the nutlin-induced apoptosis was substantially increased in RKO cells.
FIG. 8.

Myosin VI knockdown accelerates DNA damage-induced apoptosis. (A) Myo6-KD-87 cells were uninduced (control), induced to express siRNA against myosin VI (myosin VI-KD), treated with 1.0 μg/ml of DOX (DOX), or induced to express siRNA against myosin VI and treated with 1.0 μg/ml of doxorubicin (DOX + Myosin VI-KD). DNA histogram analysis was performed as described in Materials and Methods. The percentage of sub-G1 (M1) cells represents apoptotic cells, whereas M2, M3, and M4 represent cells in G0-G1, S, and G2-M, respectively. (B) DNA damage-induced apoptotic response is enhanced by myosin VI knockdown. Both Myo6-KD-87 and Myo6-KD-3 cells, which were uninduced or induced to express siRNA against myosin VI, were untreated or treated with 1.0 μg per ml of doxorubicin. The expression level of myosin VI, p53, p21, PARP, caspase 8, and actin was quantified by Western blot analysis. (C) Knockdown of myosin VI leads to impaired phosphorylation of p53 via ATM and increased expression of Fas, a target of p53. The experiment was performed as in panel B.
To determine the mechanism by whether myosin VI knockdown predisposes a cell to DNA damage-induced apoptosis, we examined the cleavage of caspase 8 and PARP. Cleavage of caspase leads to caspase activation, whereas PARP is an essential enzyme for cell survival and its cleavage by caspase is a hallmark of apoptosis (26). We found that both caspase 8 and PARP were cleaved in RKO cells upon treatment with doxorubicin (Fig. 8B, caspase 8 and PARP panels, lanes 3 and 7). However, the extent of caspase 8 and PARP cleavage was increased in myosin VI-knockdown RKO cells (Fig. 8B, caspase 8 and PARP panels, compare lanes 3 with 4 and 7 with 8). We also examined the level of p21 and determined whether the weakened accumulation of p53 in myosin VI-knockdown cells leads to weak induction of p21. We found that the level of p21 was markedly lower in RKO cells with myosin VI knockdown than that without myosin VI knockdown (Fig. 8C, p21 panel, compare lanes 3 with 4 and 7 with 8).
To further determine the underlying mechanism by which lack of myosin VI alters the prosurvival function of the p53 pathway, we examined the ATM-p53 pathway and Fas, which is known to be stored in the Golgi complex and is an effector of p53-dependent apoptosis (7, 36). We showed that knockdown of myosin VI lead to a decreased activation of ATM and its subsequent phosphorylation of p53 (Fig. 8C). In addition, we showed that the level of Fas was slightly increased by myosin VI knockdown, probably due to alteration of the Golgi structure and thus release of Fas from the Golgi reservoir. Interestingly, the level of Fas in the myosin VI-knockdown cells was further increased by activated p53 upon DNA damage. Thus, there is a feedback loop in which p53 induces myosin VI, which then maintains the Golgi structure for cell survival upon DNA damage.
DISCUSSION
Myosin VI is known to be ubiquitously expressed in a variety of tissues and at different stages of development (11, 18, 23). However, very little is known about how myosin VI is regulated, and, especially, whether myosin VI is subject to transcriptional regulation is not certain. Here, we showed that myosin VI is induced by p53 and DNA damage in a p53-dependent manner. We also showed that p53 binds directly to the promoter of the myosin VI gene and transcriptionally regulates myosin VI gene expression. Interestingly, we found that myosin VI is a unique target of p53, although some p53 target genes are also regulated by other p53 family proteins (22). Furthermore, we found that the p53-responsive element in the myosin VI promoter appears to be a GC-rich element, which is different from the consensus p53-binding site. This is not surprising since one of the first identified p53-responsive elements was found to be GC-rich (5). Thus, for the first time, we found that myosin VI can be transcriptionally regulated by p53 and stress signals.
In this study, we also found that p53 regulates the intracellular localization of myosin VI. This is particularly significant given that the intracellular localization dictates the function of myosin VI (11, 18, 23). For example, myosin VI is involved in moving endocytic vesicles out of actin-rich regions when expressed in uncoated endocytic vesicles in epithelial cells, protein secretion and maintenance of Golgi complex integrity when expressed in the Golgi complex, cell migration when expressed in membrane ruffles at the leading edge, and maintaining the hearing capability of the host when expressed in inner ear stereocilia. We showed that upon expression of p53 or in response to DNA damage in cells carrying a functional p53 pathway, myosin VI is relocated from endocytic vesicles, membrane ruffles, and cytosol to the Golgi complex, perinuclear membrane, and nucleus. Since loss of myosin VI leads to increased DNA damage-induced apoptotic response, we conclude that in response to stress stimuli, p53 has a novel function in the maintenance of Golgi complex integrity via myosin VI and that myosin VI has a novel prosurvival function in the DNA damage response pathway.
It is well established that when cells are exposed to various stress signals, p53 stability is increased by several mechanisms, including phosphorylation and acetylation (30, 38). In this study, we showed that upon DNA damage, p53 stabilization is compromised in myosin VI-knockdown cells. As an essential organelle of the secretory pathway, the Golgi complex regulates the posttranslational modification and the destination of every protein synthesized in the endoplasmic reticulum. It is possible that as a protein expressed in the Golgi complex, myosin VI knockdown alters the structure of the Golgi complex and subsequently the normal function of the secretory pathway (11, 18, 23), which then leads to reduced stability of p53. Since myosin VI is also relocalized to the nucleus upon DNA damage, the possibility exists that nuclear myosin VI may regulate p53 stability. Indeed, we found that myosin VI knockdown results in decreased activation of ATM, which is known to phosphorylate p53 and thus leads to reduced stability and activation of p53. How myosin VI serves as a DNA damage sensor for ATM activation merits further investigation, which is beyond the scope of the current study.
Myosin VI has been shown to be overexpressed in advanced ovarian carcinomas compared to normal tissues (47). As a result, myosin VI knockdown impedes cell spreading and migration of ovarian carcinoma cells in vitro and dissemination of ovarian carcinoma cells propagated in nude mice (47). In this study, we found that myosin VI is a mediator of the p53 prosurvival pathway and myosin VI knockdown increases the susceptibility of a cell to DNA damage-induced apoptosis, at least in part through release of Fas death receptor from the Golgi reservoir and inhibition of the p53 prosurvival pathway. Thus, blocking the prosurvival function of p53, such as through myosin VI, can be explored as a potential sensitizer of tumor cells to chemo- or radiotherapies.
Acknowledgments
We thank E. Sztul's laboratory for providing Golgi marker antibodies and Albert Tousson for assistance with immunofluorescence microscopy. We also thank W. Yan, K. Harms, and S. Helton for providing technical advice.
This work is supported in part by NIH grant CA076069.
REFERENCES
- 1.Ahmed, Z. M., R. J. Morell, S. Riazuddin, A. Gropman, S. Shaukat, M. M. Ahmad, S. A. Mohiddin, L. Fananapazir, R. C. Caruso, T. Husnain, S. N. Khan, A. J. Griffith, T. B. Friedman, and E. R. Wilcox. 2003. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 72:1315-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aschenbrenner, L., T. Lee, and T. Hasson. 2003. Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol. Biol. Cell 14:2728-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aschenbrenner, L., S. N. Naccache, and T. Hasson. 2004. Uncoated endocytic vesicles require the unconventional myosin, Myo6, for rapid transport through actin barriers. Mol. Biol. Cell 15:2253-2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avraham, K. B., T. Hasson, K. P. Steel, D. M. Kingsley, L. B. Russell, M. S. Mooseker, N. G. Copeland, and N. A. Jenkins. 1995. The mouse Snell's waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat. Genet. 11:369-375. [DOI] [PubMed] [Google Scholar]
- 5.Bargonetti, J., I. Reynisdottir, P. N. Friedman, and C. Prives. 1992. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev. 6:1886-1898. [DOI] [PubMed] [Google Scholar]
- 6.Barr, F. A., and B. Short. 2003. Golgins in the structure and dynamics of the Golgi apparatus. Curr. Opin. Cell Biol. 15:405-413. [DOI] [PubMed] [Google Scholar]
- 7.Bennett, M., K. Macdonald, S. W. Chan, J. P. Luzio, R. Simari, and P. Weissberg. 1998. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science 282:290-293. [DOI] [PubMed] [Google Scholar]
- 8.Buss, F., S. D. Arden, M. Lindsay, J. P. Luzio, and J. Kendrick-Jones. 2001. Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J. 20:3676-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buss, F., J. Kendrick-Jones, C. Lionne, A. E. Knight, G. P. Cote, and J. P. Luzio. 1998. The localization of myosin VI at the Golgi complex and leading edge of fibroblasts and its phosphorylation and recruitment into membrane ruffles of A431 cells after growth factor stimulation. J. Cell Biol. 143:1535-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buss, F., J. P. Luzio, and J. Kendrick-Jones. 2001. Myosin VI, a new force in clathrin mediated endocytosis. FEBS Lett. 508:295-299. [DOI] [PubMed] [Google Scholar]
- 11.Buss, F., G. Spudich, and J. Kendrick-Jones. 2004. Myosin VI: cellular functions and motor properties. Annu. Rev. Cell Dev. Biol. 20:649-676. [DOI] [PubMed] [Google Scholar]
- 12.Chen, X., L. J. Ko, L. Jayaraman, and C. Prives. 1996. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 10:2438-2451. [DOI] [PubMed] [Google Scholar]
- 13.Chiu, R., L. Novikov, S. Mukherjee, and D. Shields. 2002. A caspase cleavage fragment of p115 induces fragmentation of the Golgi apparatus and apoptosis. J. Cell Biol. 159:637-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.el-Deiry, W. S. 1998. Regulation of p53 downstream genes. Semin. Cancer Biol. 8:345-357. [DOI] [PubMed] [Google Scholar]
- 15.el-Deiry, W. S., S. E. Kern, J. A. Pietenpol, K. W. Kinzler, and B. Vogelstein. 1992. Definition of a consensus binding site for p53. Nat. Genet. 1:45-49. [DOI] [PubMed] [Google Scholar]
- 16.Fang, L., G. Li, G. Liu, S. W. Lee, and S. A. Aaronson. 2001. p53 induction of heparin-binding EGF-like growth factor counteracts p53 growth suppression through activation of MAPK and PI3K/Akt signaling cascades. EMBO J. 20:1931-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Capetillo, O., A. Lee, M. Nussenzweig, and A. Nussenzweig. 2004. H2AX: the histone guardian of the genome. DNA Repair 3:959-967. [DOI] [PubMed] [Google Scholar]
- 18.Frank, D. J., T. Noguchi, and K. G. Miller. 2004. Myosin VI: a structural role in actin organization important for protein and organelle localization and trafficking. Curr. Opin. Cell Biol. 16:189-194. [DOI] [PubMed] [Google Scholar]
- 19.Geisbrecht, E. R., and D. J. Montell. 2002. Myosin VI is required for E-cadherin-mediated border cell migration. Nat. Cell Biol. 4:616-620. [DOI] [PubMed] [Google Scholar]
- 20.Han, J. A., J. I. Kim, P. P. Ongusaha, D. H. Hwang, L. R. Ballou, A. Mahale, S. A. Aaronson, and S. W. Lee. 2002. P53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO J. 21:5635-5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen, R., and M. Oren. 1997. p53; from inductive signal to cellular effect. Curr. Opin. Genet. Dev. 7:46-51. [DOI] [PubMed] [Google Scholar]
- 22.Harms, K., S. Nozell, and X. Chen. 2004. The common and distinct target genes of the p53 family transcription factors. Cell Mol. Life Sci. 61:822-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasson, T. 2003. Myosin VI: two distinct roles in endocytosis. J. Cell Sci. 116:3453-3461. [DOI] [PubMed] [Google Scholar]
- 24.Hicks, J. L., W. M. Deng, A. D. Rogat, K. G. Miller, and M. Bownes. 1999. Class VI unconventional myosin is required for spermatogenesis in Drosophila. Mol. Biol. Cell 10:4341-4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoh, J., S. Jin, T. Parrado, J. Edington, A. J. Levine, and J. Ott. 2002. The p53MH algorithm and its application in detecting p53-responsive genes. Proc. Natl. Acad. Sci. USA 99:8467-8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang, X., and X. Wang. 2004. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 73:87-106. [DOI] [PubMed] [Google Scholar]
- 27.Kelleher, J. F., M. A. Mandell, G. Moulder, K. L. Hill, S. W. L'Hernault, R. Barstead, and M. A. Titus. 2000. Myosin VI is required for asymmetric segregation of cellular components during C. elegans spermatogenesis. Curr. Biol. 10:1489-1496. [DOI] [PubMed] [Google Scholar]
- 28.Ko, L. J., and C. Prives. 1996. p53: puzzle and paradigm. Genes Dev. 10:1054-1072. [DOI] [PubMed] [Google Scholar]
- 29.Liu, G., T. Xia, and X. Chen. 2003. The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J. Biol. Chem. 278:17557-17565. [DOI] [PubMed] [Google Scholar]
- 30.Meek, D. W. 2004. The p53 response to DNA damage. DNA Repair 3:1049-1056. [DOI] [PubMed] [Google Scholar]
- 31.Melchionda, S., N. Ahituv, L. Bisceglia, T. Sobe, F. Glaser, R. Rabionet, M. L. Arbones, A. Notarangelo, E. Di Iorio, M. Carella, L. Zelante, X. Estivill, K. B. Avraham, and P. Gasparini. 2001. MYO6, the human homologue of the gene responsible for deafness in Snell's waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 69:635-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris, C. A., A. L. Wells, Z. Yang, L. Q. Chen, C. V. Baldacchino, and H. L. Sweeney. 2003. Calcium functionally uncouples the heads of myosin VI. J. Biol. Chem. 278:23324-23330. [DOI] [PubMed] [Google Scholar]
- 33.Nelson, W. G., and M. B. Kastan. 1994. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol. Cell. Biol. 14:1815-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ongusaha, P. P., J. I. Kim, L. Fang, T. W. Wong, G. D. Yancopoulos, S. A. Aaronson, and S. W. Lee. 2003. p53 induction and activation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation through a positive feedback loop. EMBO J. 22:1289-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osterweil, E., D. G. Wells, and M. S. Mooseker. 2005. A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J. Cell Biol. 168:329-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owen-Schaub, L. B., W. Zhang, J. C. Cusack, L. S. Angelo, S. M. Santee, T. Fujiwara, J. A. Roth, A. B. Deisseroth, W.-W. Zhang, E. Kruzel, and R. Radinsky. 1995. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol. Cell. Biol. 15:3032-3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petritsch, C., G. Tavosanis, C. W. Turck, L. Y. Jan, and Y. N. Jan. 2003. The Drosophila myosin VI Jaguar is required for basal protein targeting and correct spindle orientation in mitotic neuroblasts. Dev. Cell 4:273-281. [DOI] [PubMed] [Google Scholar]
- 38.Prives, C., and J. L. Manley. 2001. Why is p53 acetylated? Cell 107:815-818. [DOI] [PubMed] [Google Scholar]
- 39.Self, T., T. Sobe, N. G. Copeland, N. A. Jenkins, K. B. Avraham, and K. P. Steel. 1999. Role of myosin VI in the differentiation of cochlear hair cells. Dev. Biol. 214:331-341. [DOI] [PubMed] [Google Scholar]
- 40.Thompson, T., C. Tovar, H. Yang, D. Carvajal, B. T. Vu, Q. Xu, G. M. Wahl, D. C. Heimbrook, and L. T. Vassilev. 2004. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J. Biol. Chem. 279:53015-53022. [DOI] [PubMed] [Google Scholar]
- 41.van de Wetering, M., I. Oving, V. Muncan, M. T. Pon Fong, H. Brantjes, D. van Leenen, F. C. Holstege, T. R. Brummelkamp, R. Agami, and H. Clevers. 2003. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 4:609-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vassilev, L. T., B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi, and E. A. Liu. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844-848. [DOI] [PubMed] [Google Scholar]
- 43.Vogelstein, B., D. Lane, and A. J. Levine. 2000. Surfing the p53 network. Nature 408:307-310. [DOI] [PubMed] [Google Scholar]
- 44.Warner, C. L., A. Stewart, J. P. Luzio, K. P. Steel, R. T. Libby, J. Kendrick-Jones, and F. Buss. 2003. Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell's waltzer mice. EMBO J. 22:569-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willis, A., E. J. Jung, T. Wakefield, and X. Chen. 2004. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 23:2330-2338. [DOI] [PubMed] [Google Scholar]
- 46.Willis, A. C., T. Pipes, J. Zhu, and X. Chen. 2003. p73 can suppress the proliferation of cells that express mutant p53. Oncogene 22:5481-5495. [DOI] [PubMed] [Google Scholar]
- 47.Yoshida, H., W. Cheng, J. Hung, D. Montell, E. Geisbrecht, D. Rosen, J. Liu, and H. Naora. 2004. Lessons from border cell migration in the Drosophila ovary: a role for myosin VI in dissemination of human ovarian cancer. Proc. Natl. Acad. Sci. USA 101:8144-8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu, J., J. Jiang, W. Zhou, and X. Chen. 1998. The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 58:5061-5065. [PubMed] [Google Scholar]
- 49.Zhu, J., S. Zhang, J. Jiang, and X. Chen. 2000. Definition of the p53 functional domains necessary for inducing apoptosis. J. Biol. Chem. 275:39927-39934. [DOI] [PubMed] [Google Scholar]