

Abstract
We previously reported the phenotype of depletion of polo-like kinase 1 (Plk1) using RNA interference (RNAi) and showed that p53 is stabilized in Plk1-depleted cancer cells. In this study, we further analyzed the Plk1 depletion-induced phenotype in both cancer cells and primary cells. The vector-based RNAi approach was used to evaluate the role of the p53 pathway in Plk1 depletion-induced apoptosis in cancer cells with different p53 backgrounds. Although DNA damage and cell death can occur independently of p53, p53-deficient cancer cells were much more sensitive to Plk1 depletion than cancer cells with functional p53. Next, the lentivirus-based RNAi approach was used to generate a series of Plk1 hypomorphs. In HeLa cells, two weak hypomorphs showed only slight G2/M arrest, a medium hypomorph arrested with 4N DNA content, followed later by apoptosis, and a strong Plk1 hypomorph underwent serious mitotic catastrophe. In well-synchronized HeLa cells, a medium level of Plk1 depletion caused a 2-h delay of mitotic progression, and a high degree of Plk1 depletion significantly delayed mitotic entry and completely blocked cells at mitosis. In striking contrast, normal hTERT-RPE1 and MCF10A cells were much less sensitive to Plk1 depletion than HeLa cells; no apparent cell proliferation defect or cell cycle arrest was observed after Plk1 depletion in these cells. Therefore, these data further support suggestions that Plk1 may be a feasible cancer therapy target.
The polo-like kinase (Plk) family has emerged as a vital regulator in many aspects of cell cycle progression (2). The known functions of polo kinases include activation of Cdc2, chromosome segregation, centrosome maturation, bipolar spindle formation, regulation of anaphase-promoting complex, and execution of cytokinesis (2). This family has at least three members in mammals, namely, Plk1, -2, and -3. Both the mammalian Plk1 and Plk3 can complement a temperature-sensitive mutation of budding yeast Cdc5, indicating that Plk1 and Plk3 have overlapping functions in mitosis (17, 21).
To investigate the function of Plk1 in mammalian cells in more detail, we along with others recently investigated the phenotype of cultured cancer cells after Plk1 depletion by use of RNA interference (RNAi) (8, 18, 19, 28, 31, 34). We first observed that Plk1 depletion in randomly growing HeLa cells led to partial mitotic arrest, as indicated by the stabilization of cyclin B, and thus elevated Cdc2 protein kinase activity, suggesting that Plk1 is essential for the activation of anaphase-promoting complex, whose activation leads to the degradation of cyclin B (15). We also observed that Plk1 depletion significantly reduced centrosome hyperamplification in hydroxyurea-treated U2OS cells, indicating its potential involvement in centrosome duplication. Phenotype analysis based on the vector-based RNAi approach revealed the essential role of Plk1 for cell proliferation and viability. Plk1 depletion in HeLa cells over an extended time induced apoptosis, as supported by the appearance of subgenomic DNA in fluorescence-activated cell sorting (FACS) profiles, the activation of caspase 3, and the formation of fragmented nuclei. Finally, we showed that p53 was stabilized in Plk1-depleted HeLa cells and that inhibition of ATM strongly potentiated the lethality of Plk1 depletion (19). A more detailed analysis of the Plk1/p53 interaction reported recently showed that Plk1 physically interacts with p53 directly and inhibits its transactivation activity as well as its proapoptotic function (1).
A close correlation between mammalian Plk1 expression and carcinogenesis was recently documented, and overexpression of Plk1 was observed in various human tumors (5, 33). For example, among patients with melanomas, elevated Plk1 expression was detected in the melanomas, and patients with moderate Plk1 expression survived significantly longer than those with high Plk1 levels (30). Similarly, non-small cell lung cancer patients whose tumors showed moderate Plk1 expression survived significantly longer than those patients with high levels of Plk1 transcripts (35). Analysis of patients with head and neck squamous cell carcinomas, oropharyngeal carcinomas, ovarian and endometrial carcinomas, and esophageal and gastric cancer revealed a close correlation between lower survival rates and higher Plk1 expression levels (13, 14, 33). Thus, it was proposed that Plk1 could serve as a novel diagnostic marker for several types of cancers (5, 33). Moreover, constitutive expression of Plk1 in NIH 3T3 cells causes oncogenic focus formation and induces tumor growth in nude mice (25). Therefore, inhibition of Plk1 function may be an important approach for cancer therapy. In line with this notion, it was recently shown that injection of plasmids containing short hairpin RNA (shRNA) to target Plk1 significantly inhibited tumor growth in nude mice carrying xenograft tumors (27), and Plk1 small interfering RNA also inhibited colony formation in soft agar and tumorgenesis in an HT1080 xenograft model in a dose-dependent manner (8).
Because inhibition of cell proliferation and reactivation of apoptosis are basic principles in anticancer therapy, it was of interest to test the effect of Plk1 depletion in primary cells. In this communication, lentivirus-based RNAi was utilized to deplete Plk1 in normal human cells, and the phenotype after Plk1 depletion in these cells was analyzed.
MATERIALS AND METHODS
Lentiviral plasmid preparation and viral vector production.
Four lentivirus-based RNAi transfer plasmids (pLKO.1-Plk1) were constructed as described previously (29). The targeting sequences of human Plk1 (GenBank accession no. NM_005030) are GGGCGGCTTTGCCAAGTGCTT, AGATTGTGCCTAAGTCTCTGC, AACGACTTCGTGTTCGTGGTG, CAACCAAAGTCGAATATGACG, and AGATCACCCTCCTTAAATATT, corresponding to the coding regions of positions 183 to 203, 245 to 265, 367 to 387, 593 to 613, and 1424 to 1444, respectively. The targeting sequence of human p53 tumor suppressor (GenBank accession no. NM_000546) is TGTTCCGAGAGCTGAATGAGG, corresponding to the coding region of position 1019 to 1039. To generate lentivirus vectors, 293T cells in 10-cm culture dishes were cotransfected with 4 μg of pLKO.1-Plk1 or pLKO.1-p53, 4 μg of pHR′-CMV-ΔR8.20vpr, and 2 μg of pHR′-CMV-VSV-G (where CMV is cytomegalovirus and VSV-G is protein G of vesicular stomatitis virus) using GenePorter reagent. Viruses were harvested every 12 h from 24 h until 72 h posttransfection. After filtration through a 0.45-μm-pore-size filter, viruses were concentrated at 167,000 rpm for 90 min in an SW27 rotor. The viral pellets were resuspended in 500 μl of TNE buffer (50 mM Tris-HCl, pH 7.8, 130 mM NaCl, 1 mM EDTA) and incubated overnight at 4°C. Infections were carried out in the presence of 10 μg/ml of polybrene and 10 mM HEPES. Following transduction for 1 day, cells were selected with 1 μg/ml puromycin for at least one additional day. After removal of the floating cells, the remaining attached cells were analyzed.
Cell culture and synchronization.
HeLa, Saos2, H1299, and MCF7 cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin at 37°C in 8% CO2. hTERT-RPE1 cells were maintained in F12-DMEM (50/50) plus 10% fetal bovine serum. MCF10A cells were maintained in F12-DMEM (50/50) supplemented with 5% horse serum, 20 ng/ml epidermal growth factor, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, and 10 μg/ml insulin. To synchronize HeLa and hTERT-RPE1 cells, cells were treated with 2.5 mM thymidine for 16 h, released for 8 h, and then treated again with thymidine for 16 h. After two washes with phosphate-buffered saline (PBS), cells were cultured for different times as indicated in each experiment and harvested. Based on our experience, HeLa cells accumulate at M phase after a 10-h release into normal medium, whereas the majority of hTERT-RPE1 cells peak at mitosis after 8 h of release.
DNA transfections.
For phenotype analysis of Plk1 depletion in most cancer cells, cells were cotransfected with pBS/U6-Plk1 and pBabe-puro at a ratio of 9:1 using GenePorter reagents. After a 2-day selection of transfection-positive cells with 2 μg/ml puromycin, floating cells were washed away with PBS, and the attached cells were incubated until harvesting for phenotype analysis. For phenotype analysis of Plk1 depletion in Saos2 cells, cells were transfected with pBS/U6-Plk1 only, and puromycin selection was not used.
Immunoblotting.
Cells were lysed in TBSN buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1.5 mM EDTA, 5 mM EGTA, 0.5% Nonidet P-40, 0.5 mM Na3VO4) supplemented with phosphatase and proteinase inhibitors (20 mM p-nitrophenyl phosphate, 1 mM pefabloc, 10 μg/ml pepstatin A, 10 μg/ml leupeptin, 5 μg/ml aprotinin), and the lysates were clarified by centrifugation at 15,000 × g for 30 min. Cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and proteins of interest were detected by Western blotting using the antibodies indicated (see figures) in the each specific experiment.
Immunofluorescence staining.
Cells growing on coverslips were fixed with paraformaldehyde and permeabilized with methanol. After three washes with 0.1% Triton X-100-PBS, cells were incubated with the antibodies indicated in each specific experiment. Finally, DNA was stained with DAPI (4′,6′-diamidino-2-phenylinodole). Rabbit phospho-H3 antibody (Upstate Cells Signaling) was used at a 1:100 dilution, mouse α-tubulin antibody (Sigma) was diluted 1:2,000, and rabbit active caspase 3 (Cell Signaling Technology) and rabbit phospho-H2AX (Trevigen) antibodies were diluted 1:1,000 and 1:100, respectively.
RESULTS
Involvement of p53 in Plk1 depletion-induced apoptosis in cancer cells.
To further evaluate the involvement of p53 in apoptosis induced by Plk1 depletion, vector-based RNAi was used to deplete Plk1 in Saos2 cells, which are p53 deficient. The vector pBS/U6-Plk1 was constructed as described previously, and plasmid pBS/U6-Plk1-1st half (sense strand) was used as a control vector (19). Plasmids were transfected into Saos2 cells, the cells were cultured for 48 h, lysates were prepared, and standard Western blotting was performed. The level of Plk1 was efficiently reduced by small interfering RNA (Fig. 1A), whereas the level of Erk2 was unchanged. Since Plk1 can be efficiently depleted in Saos2 cells, we next determined whether Plk1 depletion influences the proliferation of these cells. Although transfection with the control vector did not affect the growth rate, transfection with pBS/U6-Plk1 strongly inhibited cell proliferation (Fig. 1B, upper panel). At the same time, Plk1 depletion significantly decreased the viability of these cells (Fig. 1B, lower panel).
FIG. 1.

p53-deficient cancer cells are very sensitive to Plk1 depletion. (A) Saos2 cells were transfected with pBS/U6-Plk1 or a control vector (19). Two days posttransfection, cells were harvested, and cell lysates were subjected to direct Western blotting using anti-Plk1 antibody. The same filter was stripped and reprobed with anti-Erk2 antibody. (B) Saos2 cells were Plk1-depleted as in panel A and harvested at different posttransfection times as indicated, and cell proliferation (upper panel) or viability (lower panel) was monitored. (C) Following transfection with pBS/U6-Plk1, Saos2 cells were harvested and subjected to FACS analysis at 3 days or 4 days posttransfection as indicated. The positions of G1, G2/M, and sub-G1 populations are labeled. (D) Caspase 3 activation occurs in Plk1-depleted Saos2 cells. After transfection with pBS/U6-Plk1 for 60 h, Saos2 cells were subjected to immunofluorescence staining with active-caspase 3 antibody and DNA was stained with DAPI. (E to I) Plk1 depletion-induced apoptosis can be p53 dependent or independent in cancer cells. HeLa, H1299, or MCF7 cells were cotransfected with pBS/U6-Plk1 and pBabe-puro at a ratio of 9:1. One day posttransfection, cells were treated with puromycin for two additional days to select the transfection-positive cells. After the floating cells were washed away, the remaining attached cells were harvested and subjected to Western blotting using antibodies as indicated (E), anti-phospho-H2AX antibody immunofluorescence staining (F), anti-active-caspase 3 immunofluorescence staining (G), FACS (H), or viability assay (I). For panels F and G, cells were transfected with pBS/U6-GFP-Plk1 vector, which expresses green fluorescent protein (GFP) independent of shRNA. Bars, 20 μm.
We further analyzed the effect of Plk1 depletion on cell cycle progression using FACS. As shown in Fig. 1C, transfection with control vector did not significantly affect the cell cycle profiles, whereas Plk1 depletion induced an obvious increase in the percentage of cells with 4N DNA content. At 3 days posttransfection, approximately 40% of Plk1-depleted cells had a 4N DNA content compared to 10% of control vector-transfected cells. Moreover, cells with sub-G1 DNA content also increased dramatically after Plk1 depletion, suggesting that Plk1 depletion induces apoptosis in Saos2 cells.
We next examined if Plk1 depletion in Saos2 cells resulted in caspase activation. As shown in Fig. 1D, no active caspase 3 staining was observed in control cells. In Plk1-depleted cells, a proportion of cells showed positive active caspase 3 staining at 60 h posttransfection. Careful inspection of the active caspase 3-positive cells revealed that DNA staining of these cells was abnormal compared to control cells. The nuclei in cells with active caspase 3 were either relatively small (Fig. 1D, frames 2 and 3), fragmented (Fig. 1D, frame 4) or almost completely absent (Fig. 1D, bottom frame). Thus, caspase 3 activation in Saos2 cells appeared to correlate with reduced DNA content.
We continued to analyze the potential involvement of p53 in Plk1 depletion-induced apoptosis by comparing the sensitivity of cell lines with different p53 backgrounds to Plk1 depletion. For that purpose, three cancer cell lines were utilized: HeLa cells, whose p53 is significantly downregulated due to overexpression of human papillomavirus E6 (24), p53-null H1299 cells, and MCF7 cells, which are p53 wild type. These cell lines were cotransfected with pBS/U6-Plk1 and pBabe-puro at a ratio of 9:1 for 1 day. Cells were then treated with puromycin for two additional days to select the transfection-positive cells, and the remaining attached cells were analyzed. Western blotting indicated that Plk1 depletion was efficient for all three cell lines (Fig. 1E), and anti-phospho-H2AX and active caspase 3 immunofluorescence staining suggested that DNA damage and apoptosis occurred for all three cell lines after Plk1 depletion (Fig. 1F and G). However, FACS analysis showed that H1299 cells were much more sensitive than MCF7 cells to Plk1 depletion, and both HeLa cells and MCF7 cells had much higher survival rates than did H1299 cells after Plk1 depletion (Fig. 1H and I).
Creation of a series of Plk1 hypomorphs using lentivirus-based RNAi in HeLa cells.
We wanted to extend the Plk1 depletion-induced phenotype analysis to normal primary cells. Toward that end, we turned our attention to the lentivirus-based RNAi system, due to very low transfection efficiency of primary cells with plasmid DNA. As described in Materials and Methods, four Plk1 mRNA targeting sites (nucleotides 183, 245, 367, and 1424 relative to the start codon) were chosen to prepare lentivirus vectors, with the hope of generating a series of Plk1 hypomorphs. To test the efficiency of lentivirus vectors to deplete Plk1, we first used these viruses to infect HeLa cells. One day postinfection, cells were treated with puromycin to select the infection-positive cells for two additional days. After the floating cells were washed away, the remaining attached cells were analyzed by Western blotting. To compare the degrees of Plk1 depletion, the results from different exposure times are presented (Fig. 2A). Even after a 2-min exposure, Plk1 was still undetectable after lentivirus-based RNAi targeting site 1424, indicating that this vector can lead to the strongest Plk1 hypomorph among the four sites we chose. A medium level of Plk1 depletion was observed with lentivirus targeting site 245, whereas Plk1 depletion was detected only under much shorter exposure conditions with lentivirus targeting sites 183 and 367, suggesting that these two vectors are weak hypomorphs, with the vector targeting site 367 having a better Plk1 depletion efficiency than the vector targeting site 183.
FIG. 2.

Lentivirus-based RNAi was used to create a series of Plk1 hypomorphs. (A) Lentiviruses targeting four different positions of Plk1 mRNA (nucleotides 183, 245, 367, and 1424 relative to the starting codon) were prepared as described in Materials and Methods and used to infect HeLa cells. One day postinfection, cells were treated with puromycin for two additional days to select the infection-positive cells. After the floating cells were washed away, the remaining attached cells were harvested and subjected to Western blotting with anti-Plk1 antibody using different exposure times as indicated on the left or anti-Erk2 antibody. (B and C) HeLa cells were infected with lentiviruses as in panel A, harvested at different postinfection times as indicated, and analyzed with FACS (B) or anti-phospho-H3 immunofluorescence staining (C). Phospho-H3 immunofluorescence staining was obtained 3 days postinfection. (D) HeLa cells stably expressing green fluorescent protein (GFP)-tubulin were infected with lentiviruses as in panel A, and multinucleated cells were counted 3 days postinfection.
We then compared the phenotype of Plk1 depletion using these different lentivirus vectors. In the FACS profiles shown in Fig. 2B, the strong hypomorph targeting site 1424 caused a significant increase of cell populations with either 4N or sub-G1 DNA content at 2 days postinfection, and very few cells were still alive at 3 days postinfection, suggesting the essential role of Plk1 for cancer cell survival. The medium hypomorph targeting site 245 caused obvious cell cycle arrest at G2/M at 2 days postinfection, followed by apoptosis of a subpopulation at 3 days postinfection. The weakest hypomorph targeting site 183 did not show any obvious change of cell cycle profiles within 3 days postinfection; however, minor G2/M arrest and apoptosis were observed in the weak hypomorph targeting site 367.
We continued to compare the phenotype of lentivirus-based RNAi targeting different regions of Plk1 by staining phospho-H3, a mitosis marker. Three days postinfection, Plk1 depletion with the medium hypomorph targeting site 245 led to about 20% phospho-H3 positive cells, whereas only minor increases of phospho-H3-positive cells were observed for the two weak hypomorphs targeting sites 183 and 367 (Fig. 2C). HeLa cells stably expressing green fluorescent protein-tubulin were also subjected to Plk1 depletion using lentivirus, and multinucleated cells were counted; 35% of cells infected by the medium hypomorph targeting site 245 were multinucleated at 3 days postinfection (Fig. 2D).
Plk1 depletion in well-synchronized HeLa cells.
Considering the essential role of Plk1 during mitosis, we next depleted Plk1 using lentivirus-based RNAi in a well-synchronized culture. As outlined in Fig. 3A, HeLa cells were first infected with lentivirus targeting site 245 and then subjected to a double thymidine block. Upon release into fresh medium for different times, cells were costained with phospho-H3 and tubulin antibodies. Control virus-infected cells started to enter mitosis after 8 h, reached a peak of phospho-H3 staining after 10 h, and exited mitosis at 12 h of release. Although cells infected with lentivirus targeting site 245 entered mitosis with the same kinetics, they remained at mitosis after 12 h; however, they were eventually able to exit from mitosis after 14 h of release (Fig. 3B). In line with the phospho-H3 staining pattern, a 2-h shift in the percentage of cells with a midbody structure was also observed. While most of control virus-infected cells reached telophase/cytokinesis at 12 h of release, the maximum number of cells with a midbody structure was not detected until 14 h of release after Plk1 depletion targeting site 245 (Fig. 3C). After 12 h of release from the block, control virus-infected cells were almost phospho-H3 staining negative, and a significant number of cells were in telophase/cytokinesis with midbody structure formation. In contrast, most Plk1-depleted cells were still phospho-H3 staining positive and showed no midbody structure formation (Fig. 3D). At 16 h of release from the block, control virus-infected cells were already in G1 phase, with the typical microtubule network pattern, whereas Plk1-depleted cells were at different stages of cytokinesis (Fig. 3E).
FIG. 3.

Lentivirus-based RNAi targeting position 245 of Plk1 caused mitotic delay in HeLa cells. (A) The protocol used to deplete Plk1 in well-synchronized HeLa cells. (B and C) HeLa cells were infected with lentivirus as in panel A. After release from the double thymidine block, cells were analyzed with phospho-H3 and tubulin staining at different times. (D) Representative images of cells after 12 h of release from the block. (E) Representative images of cells after 16 h of release from the block. Scale bar, 20 μm.
Next, we depleted Plk1 by lentivirus-based RNAi targeting site 1424 in well-synchronized HeLa cells (Fig. 4). Compared to the control virus infected-cells, strong Plk1 depletion in HeLa cells by lentivirus targeting site 1424 significantly delayed mitotic entry. The Plk1-depleted HeLa cells did eventually enter mitosis, but they remained arrested at mitosis afterwards (Fig. 4B). As with the medium hypomorph targeting site 245, we also analyzed mitotic exit in cells infected with the strong Plk1 hypomorph. HeLa cells were depleted of Plk1 with the protocol indicated in the legend of Fig. 4A and stained with α-tubulin antibody. Cells at telophase/cytokinesis were counted based on the tubulin staining pattern, and the percentage of cells at telophase/cytokinesis was plotted versus the time of release. Control virus-infected cells efficiently exited mitosis after 12 h of release, whereas very few Plk1-depleted cells were observed at telophase/cytokinesis over the entire releasing period (Fig. 4C). Figure 4D shows representative cells infected with the strong Plk1 hypomorph 16 h after release from the double thymidine block. These cells, with condensed chromosomes, were phospho-H3 staining positive. Careful inspection of the tubulin staining pattern indicated that they had monopolar rather than bipolar spindles (Fig. 4D and E). To further examine the involvement of Plk1 in mitotic entry, we depleted Plk1 in a modified protocol as shown in Fig. 4F. Upon release from the double thymidine block for different times, cells were stained with phospho-H3 antibody. Under this condition, only half of the Plk1-depleted cells were able to enter mitosis (Fig. 4G).
FIG. 4.

Lentivirus-based RNAi targeting position 1424 of Plk1 caused mitotic arrest in HeLa cells. (A) The protocol used to deplete Plk1 in well-synchronized HeLa cells. (B and C) HeLa cells were infected with lentivirus targeting position 1424 as in panel A. After release from the double thymidine block, cells were subjected to phospho-H3 (B) or α-tubulin (C) immunofluorescence staining at different times. (D) The representative images of Plk1-depleted HeLa cells after a 16-h release from the double thymidine block. The cell at the left bottom corner of the upper panel is presented again in higher magnification in the bottom panel to visualize the monopolar spindle. Scale bars, 20 μm (top panel) and 5 μm (bottom panel). (E) Histograms quantifying the results. (F) The modified protocol used to deplete Plk1 in well-synchronized HeLa cells. (G) HeLa cells were infected with lentivirus targeting position 1424 as in panel F. After release from the double thymidine block, cells were subjected to phospho-H3 immunofluorescence staining at different times.
Phenotypic analysis of Plk1 depletion in two nontransformed cells.
Next, we extended the phenotype analysis of Plk1 loss-of-function to normal cells by using lentivirus targeting Plk1 site 1424 to infect hTERT-RPE1 cells. HTERT-RPE1 is a human retinal pigment epithelial cell line that stably expresses human telomerase reverse transcriptase (hTERT). This cell line is an attractive replacement for primary and transformed cell lines and is ideal for studying the long-term biochemical and physiological aspects of cell growth. Due to increased telomere length caused by overexpression of hTERT, these cells continue to divide at the rate of young primary cells, even after 300 population doublings (3). Importantly, hTERT-RPE1 cells maintain a primary cell phenotype, albeit dividing indefinitely (10). First, hTERT-RPE1 cells were infected with lentivirus targeting site 1424 of Plk1 and then harvested for Western blotting. As shown in Fig. 5A, the level of Plk1 was significantly reduced in hTERT-RPE1 cells after infection for 2 days with lentivirus targeting site 1424. Moreover, the Plk1 level remained fairly low even at 12 days postinfection. Next, hTERT-RPE1 cells were infected with lentivirus targeting site 1424, and cell proliferation curves were obtained after different times of incubation. Only a minor effect was observed for proliferation of hTERT-RPE1 cells (Fig. 5B), although Plk1 depletion led to serious growth inhibition in HeLa cells. Moreover, in contrast to the case in HeLa cells, where Plk1 depletion caused significant cell cycle arrest with 4N DNA content, followed by cell death, no apparent defects in FACS profiles were detected in hTERT-RPE1 cells after Plk1 depletion (Fig. 5C).
FIG. 5.

Plk1 depletion in hTERT-RPE1 cells. hTERT-RPE1 cells were infected with lentivirus targeting position 1424 of Plk1 and harvested at different times. (A) Western blotting of hTERT-RPE1 cells at 2 days or 12 days postinfection using the antibodies indicated on the left. Cells were treated with 200 ng/ml of nocodazole for 10 h before harvesting. (B) Growth curves of hTERT-RPE1 cells after Plk1 depletion using lentivirus-based RNAi targeting position 1424 of Plk1. (C) FACS profiles of hTERT-RPE1 cells after Plk1 depletion for the times indicated. (D to F) Plk1 was depleted in well-synchronized hTERT-RPE1 cells using the protocol shown in panel D, and cells were harvested at different releasing times as indicated and analyzed by Western blotting (E) or stained with phospho-H3 antibody (F). (G) Plk1-depleted hTERRT-RPE1 cells were stained with anti-α-tubulin antibody and analyzed by immunofluorescence microscopy. Bar, 20 μm.
We further examined the phenotype of Plk1 depletion in well-synchronized hTERT-RPE1 cells after lentivirus infection targeting site 1424 (Fig. 5D). Upon release from the double thymidine block, hTERT-RPE1 cells were harvested after 8 h, and Western blotting indicated efficient Plk1 depletion under this condition (Fig. 5E). We found that hTERT-RPE1 cells reached the peak of mitosis at 8 h after release (Fig. 5F), 2 h earlier than HeLa cells. In striking contrast to the strong mitotic block we observed in HeLa cells, Plk1 depletion in hTERT-RPE1 cells caused only a minor effect on the kinetics and percentage of these cells to enter and exit from mitosis, compared to the control virus-infected cells. One apparent phenotype we did observe after Plk1 depletion in hTERT-RPE1 cells is a change in cell morphology. Normal hTERT-RPE1 cells are elongated, whereas Plk1-depleted hTERT-RPE1 cells have a shorter and flatter morphology (Fig. 5G).
We also investigated the phenotype of Plk1 depletion in another nontransformed cell line, MCF10A, which is an immortal cell line that arose spontaneously without viral or chemical intervention, from mortal human diploid breast epithelial cells (26). At 1 day postinfection with lentivirus targeting position 1424 of Plk1, MCF10A cells were incubated in fresh medium in the presence of puromycin to select the infection-positive cells. At 3 days postinfection, floating cells were washed away, and the remaining attached cells were incubated in the presence of puromycin until harvest. Lentivirus-based RNAi can efficiently deplete Plk1 in MCF10A cells, as shown by the Western blotting results (Fig. 6A). Moreover, the Plk1 level remained low at 12 days postinfection. To examine the effect of Plk1 depletion on cell proliferation of MCF10A cells, Plk1-depleted cells were reseeded on the plates at 12 days postinfection, and cell proliferation was analyzed for the following 5 days. Proliferation of MCF10A cells was not affected by Plk1 depletion (Fig. 6B). FACS profiles of Plk1-depleted MCF10A cells indicated that no apparent cell cycle arrest was observed in these cells, compared to control cells (Fig. 6C). In line with this, Plk1-depleted MCF10A cells did not show an increase of phospho-H3 staining and responded to nocodazole treatment similarly to control cells (Fig. 6D). Proliferation of Plk1-depleted MCF10A cells was also confirmed by immunofluorescence staining with Ki67 (Fig. 6E), an antibody raised against a nuclear protein that stains nonquiescent cells (7). Similar to the experiments with HeLa and hTERT-RPE1 cells, Plk1-depleted MCF10A cells were next synchronized with the double thymidine block, harvested at different releasing times, and stained with phospho-H3 antibody to follow mitotic entry and progression. Plk1-depleted MCF10A cells showed almost identical kinetics of entry into and exit from mitosis compared to control cells (Fig. 6F). Plk1 depletion did not affect chromosome condensation during prophase, bipolar spindle formation during metaphase, sister chromatid separation during anaphase, or cytokinesis of MCF10A cells (Fig. 6G).
FIG. 6.

Plk1 depletion in MCF10A cells. At 1 day postinfection with lentivirus targeting position 1424 of Plk1, MCF10A cells were incubated in fresh medium in the presence of puromycin to select the infection-positive cells. At 3 days postinfection, floating cells were washed away, and the remaining attached cells were incubated in the presence of puromycin until harvest. (A) Western blotting of MCF10A cells at 3 days or 12 days postinfection using the antibodies indicated on the left. Cells were treated with 200 ng/ml of nocodazole for 10 h before harvesting. (B) Growth curves of MCF10A cells after Plk1 was depleted at 12 days postinfection. MCF10A cells were reseeded on the plates, and cell proliferation was examined for the following 5 days. (C) FACS profiles of MCF10A cells after Plk1 depletion for the times indicated. (D) At 12 days postinfection, MCF10A cells were treated with 100 ng/ml of nocodazole (Noc) for 12 h and analyzed with phospho-H3 staining. (E) Ki67 antibody staining of MCF10A cells at 12 days postinfection. Bars, 20 μm. (F) At 12 days postinfection, MCF10A cells were subjected to a double thymidine block, harvested at different releasing times, and stained with phospho-H3 antibody. (G) Representative images of Plk1-depleted MCF10A cells during mitosis. At 12 days postinfection, cells were costained with anti-α-tubulin and phospho-H3 antibodies, and analyzed by immunofluorescence microscopy. Bar, 5 μm.
Codepletion of Plk1 and p53 in MCF10A cells.
Loss-of-function mutation of the p53 tumor suppressor is one major defect in most cancer cells. Therefore, we wanted to analyze the potential involvement of p53 in Plk1-depleted normal cells. For that purpose, the lentivirus-based RNAi approach was first used to generate a p53-depleted MCF10A cell line. As a series of control experiments, the phenotype of p53 depletion alone in MCF10A cells was analyzed. MCF10A cells were infected with lentivirus targeting p53, and after 1 day, cells were incubated in fresh medium in the presence of puromycin for 2 days to select for infection-positive cells. At 3 days postinfection, floating cells were washed away, and the remaining attached cells were either further incubated in the presence of puromycin to obtain a p53-deficient MCF10A cell line or harvested and analyzed by an anti-p53 Western blot (Fig. 7A, top panel). The p53-deficient MCF10A cells were subsequently infected with lentivirus targeting Plk1, harvested 2 days postinfection, and analyzed with anti-Plk1 antibody (Fig. 7A, bottom panel). At 2 days postinfection with lentivirus targeting Plk1, the p53-deficient MCF10A cells were reseeded on new plates, and cell proliferation was examined for 4 days. Depletion of p53 alone did not affect proliferation of MCF10A cells, whereas the depletion of Plk1 significantly inhibited cell growth of p53-deficient MCF10A cells (Fig. 7B). FACS analysis did not reveal any defects in p53-deficient MCF10A cells, whereas obvious G2/M arrest and serious cell death were observed after Plk1 depletion in these cells (Fig. 7C). To further distinguish whether Plk1/p53 codepletion caused G2 or M phase arrest in MCF10A cells, we stained these cells with phospho-H3 antibodies after nocodazole treatment (Fig. 7D and E). Nocodazole treatment blocked control cells at mitosis as expected; however, a much lower percentage of Plk1/p53-codepleted MCF10A cells was phospho-H3 positive, indicating that these cells were not arrested at mitosis but that Plk1/p53 codepletion led to G2 arrest (Fig. 7E). The positive Ki67 staining of these cells suggests they had not exited from the cell cycle to enter a G0-like state (Fig. 7F). At 4 days postinfection with lentiviruses targeting Plk1 and p53, MCF10A cells were also analyzed with active caspase 3 and α-tubulin staining. Compared to control and p53− cells, Plk1/p53 codepletion caused the disappearance of microtubule fiber structure, DNA fragmentation, and activation of caspase 3 (Fig. 7G). Finally, 1 day after infection with lentivirus targeting Plk1, p53-deficient MCF10A cells were subjected to a double thymidine block, harvested at different releasing times, and stained with phospho-H3 antibody. Depletion of p53 alone did not affect mitotic entry and progression of MCF10A cells, whereas additional Plk1 depletion in p53-depleted cells led to obvious G2 block (Fig. 7H). Compared to control cells, a significant population of Plk1/p53-codepleted MCF10A cells never entered mitosis.
FIG. 7.

Codepletion of Plk1 and p53 causes G2/M arrest and cell death in MCF10A cells. (A) Generation of p53-deficient MCF10A cell line. MCF10A cells were infected with lentivirus targeting p53, and after 1 day, cells were incubated in fresh medium in the presence of puromycin for 2 days to select for infection-positive cells. At 3 days postinfection, floating cells were washed away by PBS, and the remaining attached cells were either left to incubate in the presence of puromycin to obtain a p53-deficient MCF10A cell line or harvested and analyzed by anti-p53 Western blotting (top panel). Cells were treated with VP16 for 3 h before harvesting. The p53-deficient MCF10A cells were further infected with lentivirus targeting Plk1, harvested 2 days postinfection, and analyzed with an anti-Plk1 antibody (bottom panel). (B) At 2 days postinfection with lentivirus targeting Plk1, the p53-deficient MCF10A cells were reseeded on new plates, and cell proliferation was examined for 4 days. (C) FACS profiles of p53-deficient MCF10A cells after Plk1 depletion for 4 days. (D and E) At 3 days postinfection with lentiviruses targeting Plk1 and p53, MCF10A cells were treated with 200 ng/ml of nocodazole for 12 h and analyzed with phospho-H3 staining. Bar, 10 μm. (F) Ki67 antibody staining of MCF10A cells after a 3-day codepletion of Plk1 and p53. Bar, 20 μm. (G) At 4 days postinfection with lentiviruses targeting Plk1 and p53, MCF10A cells were analyzed with active caspase 3 and α-tubulin staining. Bar, 10 μm. (H) One day after infection with lentivirus targeting Plk1, p53-deficient MCF10A cells were subjected to a double thymidine block, harvested at different releasing times, and stained with phospho-H3 antibody.
Next, we wanted to examine the potential defects of Plk1/p53-codepleted MCF10A cells during mitotic progression. Toward that end, p53- or Plk1-depleted MCF10A cells were synchronized with the double thymidine block protocol, infected with lentiviruses targeting Plk1 or p53 during the 8-h interval, and released for different times (Fig. 8A). Phospho-H3 staining indicated that this shorter version of second virus infection led to mitotic entry of MCF10A cells, but mitotic progression was significantly delayed (Fig. 8B and C). Careful inspection of Plk1/p53-codepleted MCF10A cells during mitosis reveals the following defects: multipolar spindle formation during prophase, misalignment of condensed chromosomes during metaphase, and chromosome lagging during anaphase (Fig. 8D).
FIG. 8.

Codepletion of Plk1 and p53 leads to delay of mitotic progression in MCF10A cells. (A) Protocols used to generate Plk1/p53-codepleted-MCF10A cells. (B) MCF10A cells were subjected to Plk1/p53 codepletion as shown in panel A, harvested at different releasing times, and stained with phospho-H3 antibody. (C) Representative images of MCF10A cells as described in panel A after release from the block for 14 h. Bar, 20 μm. (D) Mitotic defects observed in Plk1/p53-codepleted-MCF10A cells. Lagging chromosomes are indicated by the arrowheads. Bar, 5 μm.
To gain additional insights about the mechanism of the effects of p53 depletion on the sensitivity of MCF10A cells to Plk1 inhibition, the p53- and Plk1-deficient MCF10A cell lines were further examined for the protein levels of Chk1, Chk2, and Plk3 (Fig. 9A). Neither p53 nor Plk1 depletion caused significant changes in these proteins, indicating that these proteins are unlikely to be responsible for the apoptosis observed in p53/Plk1-codepleted MCF10A cells. Next, three cancer cell lines with different p53 backgrounds were depleted of p53, and the effect on the level of Plk1 was determined (Fig. 9B). At 4 days postinfection, p53 depletion led to downregulation of Plk1 in both MCF7 and HeLa cells, whereas no obvious change was observed in H1299 cells. In MCF10A cells, the level of Plk1 was not affected at 4 days postinfection; however, downregulation of Plk1 was observed at 21 days postinfection (Fig. 9C).
FIG. 9.
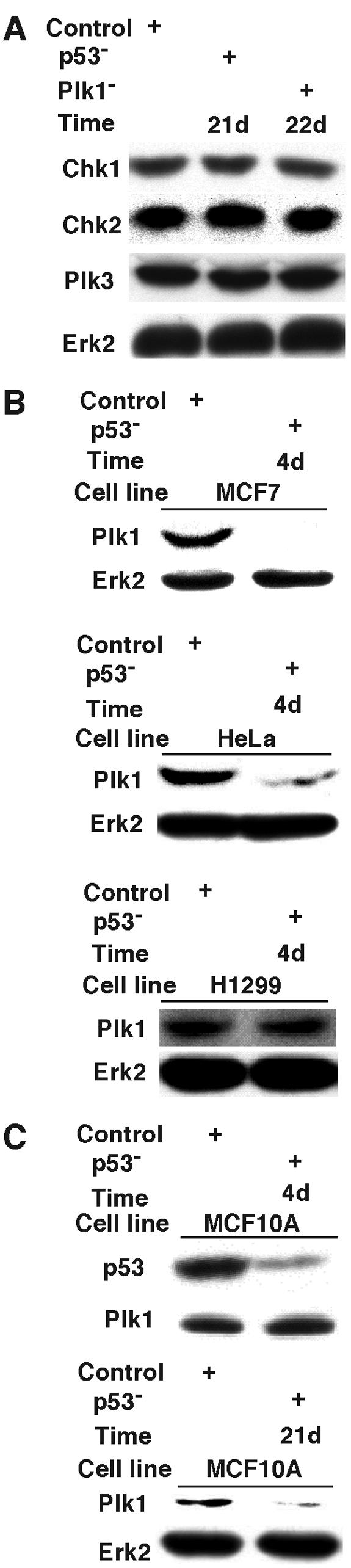
The effects of p53 depletion on several cell cycle-related proteins. (A) MCF10A cells were depleted of p53 or Plk1 as described in the legend of Fig. 7, harvested at 21 or 22 days postinfection, and analyzed by Chk1, Chk2, Plk3, and Erk2 Western blotting. (B) MCF7, HeLa, or H1299 cells were depleted of p53 as described in the legend of Fig. 7A, harvested at 4 days postinfection, and subjected to Western blotting using antibodies as indicated. (C) MCF10A cells were depleted of p53 as described in the legend of Fig. 7A, harvested at 4 or 21 days postinfection, and analyzed with anti-Plk1 Western blotting.
To rule out off-target effects of RNAi, we depleted Plk1 by targeting a different site that also generates a strong Plk1 hypomorph. For that purpose, HeLa cells were infected with lentiviruses targeting either site 593 or site 1424. Western blotting indicated that the depletion efficiencies of these two lentiviruses were comparable (Fig. 10A). Lentivirus-based RNAi targeting site 593 also can efficiently deplete Plk1 in MCF10A cells (Fig. 10B). Similar to depletion of Plk1 by targeting site 1424, proliferation of MCF10A cells was not affected by depletion of Plk1 by targeting 593 (Fig. 10C). FACS profiles of Plk1-depleted MCF10A cells indicated no apparent cell cycle arrest in comparison to control cells. In contrast, severe apoptosis was observed by depletion of Plk1 by targeting site 593 in HeLa cells (Fig. 10D). MCF10A cells depleted of Plk1 by targeting site 593 were also synchronized with the double thymidine block, harvested at different releasing times, and stained with phospho-H3 antibody to follow mitotic entry and progression. These cells showed normal kinetics of entry into and exit from mitosis (Fig. 10E).
FIG. 10.
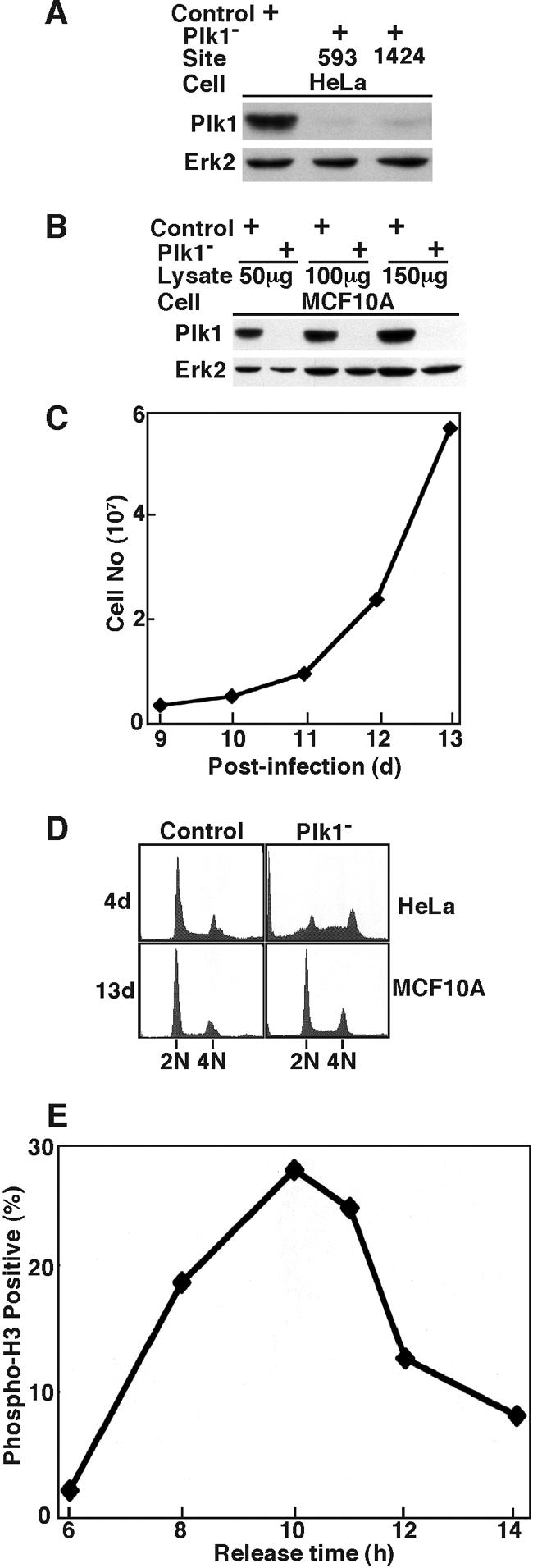
Plk1 depletion in MCF10A cells by targeting site 593. At 1 day postinfection with lentiviruses targeting position 593 or 1424 of Plk1, HeLa or MCF10A cells were incubated in fresh medium in the presence of puromycin to select the infection-positive cells. At 3 days postinfection, floating cells were washed away, and the remaining attached cells were incubated in the presence of puromycin until harvest. (A) Western blotting of HeLa cells at 3 days postinfection by lentiviruses targeting the positions indicated. (B) Western blotting of MCF10A cells at 3 days postinfection by lentivirus targeting site 593 of Plk1. Cells were treated with 200 ng/ml of nocodazole for 10 h before harvesting. (C) Growth curve of MCF10A cells after Plk1 was depleted by targeting position 593 at 8 days postinfection. MCF10A cells were reseeded on the plates, and cell proliferation was examined for the following 5 days. (D) FACS profiles of HeLa (top panels) and MCF10A (bottom panels) cells after Plk1 depletion by targeting position 593 for the times indicated. (E) At 8 days postinfection by targeting position 593 of Plk1, MCF10A cells were subjected to a double thymidine block, harvested at different releasing times, and stained with phospho-H3 antibody.
DISCUSSION
Analysis of the Drosophila polo1 mutant a decade ago revealed the phenotype of monopolar spindles, indicating a role for Plk in centrosome assembly and separation (32). Additional data obtained from two strong polo hypomorphs (polo9 and polo10) demonstrate an absolute requirement for polo in the metaphase-anaphase transition and also provide further insight into the requirement for Plk in organization of microtubule-nucleating centers (4). Although a complete proliferation defect of diploid tissues is observed in the two strong hypomorphs, polo9 and polo10, the weakly hypomorphic polo1 mutant is able to progress through multiple cell cycles. In the case of mitotic entry, Plx1 in Xenopus was proposed as a trigger kinase to activate Cdc2/cyclin B complex via phosphorylation and activation of Cdc25C (16). It was shown that partial (∼90%) inhibition of Plx1 by antibody microinjection only delays M phase entry (22), whereas complete immunodepletion of Plx1 from prophase extracts totally blocks mitotic entry (23). Using the vector-based RNAi approach to target Plk1, we recently showed that partial Plk1 knockdown leads to mitotic arrest and that complete Plk1 depletion blocks cells at G2 phase in well-synchronized cultures (20). Thus, in various systems different phenotypes are observed based on different degrees of polo deficiency. Using the lentivirus-based RNAi approach, we were able to generate a series of Plk1 hypomorphs in mammalian cells, due to intrinsic differences between individual shRNAs (Fig. 2). Since different levels of Plk1 activity may be required for different phases of mitosis, these hypomorphs provide the opportunity to study different functions of Plk1 in many aspects of mitosis in mammalian cells.
In our studies using lentivirus delivery of shRNA into HeLa cells, we achieve very efficient infection: most of the cells are puromycin resistant at 24 h and are expressing shRNA. Cells infected with the two weak Plk1 hypomorphs targeting sites 183 and 367 were viable and cell lines were obtained, but a greater percentage of cells had a G2 DNA content at 3 days postinfection. Cells infected with the medium Plk1 hypomorph targeting site 245 were viable for about 1 week, whereas very few HeLa cells survived 3 days after lentivirus infection targeting site 1424. Because of efficient infection and expression of shRNA, we were also able to synchronize cells with a double thymidine block immediately after infection. As shown here, targeting site 245 did not affect entry into mitosis, but targeting site 1424 significantly delayed mitotic entry (Fig. 3 and 4). Mitotic defects as determined by phospho-H3 staining and formation of multinucleated cells were detected after targeting site 245 as early as 2 days postinfection.
We show here that the proliferation of two apparently normal cell lines after severe Plk1 depletion is indistinguishable from their controls. This is in sharp contrast to the observation with cancer cell lines, which undergo cell death rapidly following similar levels of depletion. In view of the many published reports that M phase polo-like kinases in yeast and flies are required for mitosis, it is surprising that Plk1-depleted normal cells have no detectable proliferation phenotype. However, these cells are not completely devoid of Plk1, but appear to express less than 10% that of control normal cells. Thus, there may be sufficient activity to support mitotic events. It seems unlikely that such a low abundance of Plk1 would be sufficient to support normal proliferation events, however, and suggests that mammalian cells have an activity that complements Plk1. Loss of Plk1 may be compensated by other homologs of polo kinase in primary cells. Whether Plk3 can perform similar functions as Plk1 during mitosis in primary cells is a question that remains untested.
Targeting Plk1 expression in normal human fibroblasts was attempted previously with antisense oligodeoxynucleotides. Although the authors stated that proliferation of normal human skin amniocytes was not affected after treatment with antisense oligodeoxynucleotides, neither Western blotting nor immunofluorescence staining data were presented to indicate that Plk1 was actually depleted under their conditions (6). In fact, a separate report describing the use of direct transfection of synthetic double-stranded RNA to deplete Plk1 in both cancer and primary cells clearly demonstrated that transfection of double-stranded RNA into human mammary epithelial cells was much less efficient than into cancer cells (28). The lentivirus-based RNAi approach does not appear to discriminate between cancer cells and normal cells with regard to vector delivery efficiency, and efficient Plk1 depletion was achieved in both cases under the same conditions (Fig. 5 and 6). The fact that significant depletion of Plk1 did not affect the proliferation of either hTERT-RPE1 or MCF10A cells strongly suggests that Plk1 might be a good cancer therapy target, since obvious mitotic catastrophe was induced after Plk1 depletion in cancer cells. Our data are consistent with those obtained with ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, which induces mitotic arrest followed by apoptosis in tumor cells but not in normal cells (9). Moreover, these data support the interpretation that the effect of ON01910 did not result from inhibition of a kinase other than Plk1, although that possibility cannot be excluded.
Due to the expression of the papillomavirus E6 protein, expression of the p53 tumor suppressor protein is significantly compromised in HeLa cells (24). However, these cells do have an intact p53 gene, and a functional p53 protein is induced under some circumstances. For example, the malfunction of telomeres due to inhibition of the telomeric-repeat binding factor 2 leads to the stabilization of p53 and subsequent apoptosis, possibly due to activation of a DNA damage checkpoint (11). Similarly, we observed stabilization of p53 in Plk1-depleted HeLa cells, a process in which ATM is clearly involved (19). We show here that Plk1 depletion induces cancer cell death in a p53-independent fashion. HeLa and p53 wild-type MCF7 cells survive somewhat longer than p53-null H1299 cells, demonstrating that p53 may delay, but not prevent, cell death in tumor cells after Plk1 depletion.
A more detailed analysis of the Plk1/p53 connection showed that Plk1 physically binds to p53 in cultured cells. Mapping experiments indicated that the DNA-binding domain of p53 is responsible for Plk1 association, and the interaction between Plk1 and the DNA-binding domain of p53 may prevent its ability to bind DNA and, thus, inhibit its transactivating and proapoptotic functions (1). Considering the fact that most mutations of p53 in tumor cells occur in the DNA-binding domain, it is intriguing to speculate on the potential significance of Plk1 interaction with p53 in tumorgenesis.
A genome-wide approach was used to identify the p53-regulated genes involved in genotoxic stress-induced apoptosis (12). Surprisingly, these authors found that p53 regulates gene expression primarily through transcriptional repression rather than activation, and selective blockade of p53-dependent gene repression resulted in the reduction of 5-fluorouracil-induced apoptosis. Further analysis demonstrated that p53 suppressed the activity of the Plk1 promoter without direct binding to the promoter region. It was proposed that p53 might repress Plk1 through an indirect mechanism by recruiting a repressor complex via p21 (12). Therefore, p53 and Plk1 mutually and negatively regulate each other.
We also depleted p53 in MCF10A cells and showed that their growth rate and morphology appeared normal. p53 is ordinarily not abundant in the absence of DNA damage in normal cells. Thus, the depletion of p53 in MCF10A cells would not be expected to cause any immediate phenotypic changes. However, upon depletion of Plk1 by targeting site 1424, p53-depleted MCF10A cells were arrested with a 4N DNA content and exhibited a subgenomic DNA content, consistent with induction of apoptosis events. Thus, under these conditions p53-depleted MCF10A cells partially mimic cancer cells. In view of the results reported by Kho et al. (12), the absence of p53 in otherwise apparently normal cells may influence the outcome following Plk1 depletion as the result of failure to repress gene products that promote cell death. Depletion of p53 may eventually result in deleterious mutations over time, however, and contribute to sensitivity to Plk1 depletion in otherwise normal cells. In agreement with this notion, p53 depletion in the short term did not affect the level of Plk1 in MCF10A cells, whereas p53 depletion in the long term did cause downregulation of Plk1 (Fig. 9). Taken together, these data suggest new approaches for the study of Plk1 function in cell regulation and survival, as well as emphasizing the potential of Plk1 as a target in cancer therapy.
Acknowledgments
We thank Eleanor Erikson for critical reading of the manuscript. We thank Carl Novina and Phillip Sharp for providing lentivirus vectors.
This work is supported by National Institutes of Health Grant GM59172. R.L.E. is the John F. Drum American Cancer Society Research Professor. X.L. is a recipient of the Howard Temin Award from the National Cancer Institute (K01 CA114401).
REFERENCES
- 1.Ando, K., T. Ozaki, H. Yamamoto, K. Furuya, M. Hosoda, S. Hayashi, M. Fukuzawa, and A. Nakagawara. 2004. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 279:25549-25561. [DOI] [PubMed] [Google Scholar]
- 2.Barr, F. A., H. H. W. Sillje, and E. A. Nigg. 2004. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell. Biol. 5:429-440. [DOI] [PubMed] [Google Scholar]
- 3.Bodnar, A. G., M. Ouellette, M. Frolkis, S. E. Holt, C. P. Chiu, G. B. Morin, C. B. Harley, J. W. Shay, S. Lichtsteiner, and W. E. Wright. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279:349-352. [DOI] [PubMed] [Google Scholar]
- 4.Donaldson, M. M., A. A. Tavares, H. Ohkura, P. Deak, and D. M. Glover. 2001. Metaphase arrest with centromere separation in polo mutants of Drosophila. J. Cell Biol. 153:663-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckerdt, F., J. Yuan, and K. Strebhardt. 2005. Polo-like kinases and oncogenesis. Oncogene 24:267-276. [DOI] [PubMed] [Google Scholar]
- 6.Elez, R., A. Piiper, B. Kronenberger, M. Kock, M. Brendel, E. Hermann, U. Pliquett, E. Neumann, and S. Zeuzem. 2003. Tumor regression by combination antisense therapy against Plk1 and Bcl-2. Oncogene 22:69-80. [DOI] [PubMed] [Google Scholar]
- 7.Gerdes, J., H. Lemke, H. Baisch, H. H. Wacker, U. Schwab, and H. Stein. 1984. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 133:1710-1715. [PubMed] [Google Scholar]
- 8.Guan, R., P. Tapang, J. D. Leverson, D. Albert, V. L. Giranda, and Y. Luo. 2005. Small Interfering RNA-mediated polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res. 65:2698-2704. [DOI] [PubMed] [Google Scholar]
- 9.Gumireddy, K., M. V. Reddy, S. C. Cosenza, R. B. Nathan, S. J. Baker, N. Papathi, J. Jiang, J. Holland, and E. P. Reddy. 2005. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 7:275-286. [DOI] [PubMed] [Google Scholar]
- 10.Jiang, X. R., G. Jimenez, E. Chang, M. Frolkis, B. Kusler, M. Sage, M. Beeche, A. G. Bodnar, G. M. Wahl, T. D. Tlsty, and C. P. Chiu. 1999. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet. 21:111-114. [DOI] [PubMed] [Google Scholar]
- 11.Karlseder, J., D. Broccoli, Y. Dai, S. Hardy, and T. de Lange. 1999. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283:1321-1325. [DOI] [PubMed] [Google Scholar]
- 12.Kho, P. S., Z. Wang, L. Zhuang, Y. Li, J. L. Chew, H. H. Ng, E. T. Liu, and Q. Yu. 2004. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J. Biol. Chem. 279:21183-21192. [DOI] [PubMed] [Google Scholar]
- 13.Knecht, R., R. Elez, M. Oechler, C. Solbach, C. von Ilberg, and K. Strebhardt. 1999. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. 59:2794-2797. [PubMed] [Google Scholar]
- 14.Knecht, R., C. Oberhauser, and K. Strebhardt. 2000. PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int. J. Cancer 89:535-536. [PubMed] [Google Scholar]
- 15.Kotani, S., S. Tugendreich, M. Fujii, P. Jorgensen, N. Watanabe, C. Hoog, P. Hieter, and K. Todokoro. 1998. PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell 1:371-380. [DOI] [PubMed] [Google Scholar]
- 16.Kumagai, A., and W. Dunphy. 1996. Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science 273:1377-1380. [DOI] [PubMed] [Google Scholar]
- 17.Lee, K. S., and R. L. Erikson. 1997. Plk is a functional homolog of Saccharomyces cerevisiae Cdc5, and elevated Plk activity induces multiple septation structures. Mol. Cell. Biol. 17:3408-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu, X., and R. L. Erikson. 2002. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc. Natl. Acad. Sci. USA 99:8672-8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, X., and R. L. Erikson. 2003. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA 100:5789-5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, X., C. Lin, M. Lei, S. Yan, T. Zhou, and R. L. Erikson. 2005. CCT chaperonin complex is required for the biogenesis of functional Plk1. Mol. Cell. Biol. 25:4993-5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang, B., H. Pan, L. Lu, J. Li, P. Stambrook, B. Li, and W. Dai. 1997. Human Prk is a conserved protein serine/threonine kinase involved in regulating M phase functions. J. Biol. Chem. 272:28646-28651. [DOI] [PubMed] [Google Scholar]
- 22.Qian, Y. W., E. Erikson, C. Li, and J. L. Maller. 1998. Activated polo-like kinase Plx1 is required at multiple points during mitosis in Xenopus laevis. Mol. Cell. Biol. 18:4262-4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian, Y. W., E. Erikson, F. E. Taieb, and J. L. Maller. 2001. The polo-like kinase Plx1 is required for activation of the phosphatase Cdc25C and cyclin B-Cdc2 in Xenopus oocytes. Mol. Biol. Cell 12:1791-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheffner, M., B. A. Werness, J. M. Huibregtse, A. J. Levine, and P. M. Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129-1136. [DOI] [PubMed] [Google Scholar]
- 25.Smith, M. R., M. L. Wilson, R. Hamanaka, D. Chase, H. Kung, D. L. Longo, and D. K. Ferris. 1997. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem. Biophys. Res. Commun. 234:397-405. [DOI] [PubMed] [Google Scholar]
- 26.Soule, H. D., T. M. Maloney, S. R. Wolman, W. D. Peterson, R. Brenz, C. M. McGrath, J. Russo, R. J. Pauley, R. F. Jones, and S. C. Brooks. 1990. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF10. Cancer Res. 50:6075-6086. [PubMed] [Google Scholar]
- 27.Spankuch, B., Y. Matthess, R. Knecht, B. Zimmer, M. Kaufmann, and K. Strebhardt. 2004. Cancer inhibition in nude mice after systemic application of U6 promoter-driven short hairpin RNAs against PLK1. J. Natl. Cancer Inst. 96:862-872. [DOI] [PubMed] [Google Scholar]
- 28.Spankuch-Schmitt, B., J. Bereiter-Hahn, M. Kaufmann, and K. Strebhardt. 2002. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J. Natl. Cancer Inst. 94:1863-1877. [DOI] [PubMed] [Google Scholar]
- 29.Stewart, S. A., D. M. Dykxhoorn, D. Palliser, H. Mizuno, E. Y. Yu, D. S. An, D. M. Sabatini, I. S. Chen, W. C. Hahn, P. A. Sharp, R. A. Weinberg, and C. D. Novina. 2003. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9:493-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strebhardt, K., L. Kneisel, C. Linhart, A. Bernd, and R. Kaufmann. 2000. Prognostic value of polo-like kinase expression in melanomas. JAMA 283:479-480. [DOI] [PubMed] [Google Scholar]
- 31.Sumara, I., J. F. Gimenez-Abian, D. Gerlich, T. Hirota, C. Kraft, C. De La Torre, J. Ellenberg, and J. M. Peters. 2004. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr. Biol. 14:1712-1722. [DOI] [PubMed] [Google Scholar]
- 32.Sunkel, C. E., and D. M. Glover. 1988. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci. 89:25-38. [DOI] [PubMed] [Google Scholar]
- 33.Takai, N., R. Hamanaka, J. Yoshimatsu, and I. Miyakawa. 2005. Polo-like kinases (Plks) and cancer. Oncogene 24:287-291. [DOI] [PubMed] [Google Scholar]
- 34.Van Vugt, M. A., B. C. Van De Weerdt, G. Vader, H. Janssen, J. Calafat, R. Klompmakerf, R. M. Wolthuis, and R. H. Medema. 2004. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for APC/Cdc20 activation and initiation of cytokinesis. J. Biol. Chem. 279:36841-36854. [DOI] [PubMed] [Google Scholar]
- 35.Wolf, G., R. Elez, A. Doermer, U. Holtrich, H. Ackermann, H. J. Stutte, H. M. Altmannsberger, H. Rubsamen-Waigmann, and K. Strebhardt. 1997. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 14:543-549. [DOI] [PubMed] [Google Scholar]