

Abstract
Leishmaniasis is the second-most dreaded parasitic disease in the modern world, behind malaria. The lack of effective vaccines demand improved chemotherapy along with the development of lead compounds and newer targets. We report here that the pentacyclic triterpenoid, dihydrobetulinic acid (DHBA), is a novel lead compound for antileishmanial therapy. It acts by targeting DNA topoisomerases. DNA topoisomerase I and II activity was studied using relaxation and decatenation assays. Mechanistic studies were based on the decreased mobility of enzyme-bound DNA compared with free DNA and the differential mobility of nicked and supercoiled monomers in 1% agarose gel. Pulsed field gradient gel electrophoresis, confocal microscopy, and transmission electron microscopy were performed to assess cytotoxicity of the compound and ultrastructural damage of the parasite. Apoptosis was studied by the isolation of DNA from DHBA-treated parasites and subsequent electrophoresis in 1% agarose gel. DHBA inhibits growth of Leishmania donovani promastigotes and amastigotes with an IC50 of 2.6 and 4.1 μM respectively. The compound is a dual inhibitor of DNA topoisomerases that fails to induce DNA cleavage and acts by preventing the formation of enzyme-DNA binary complex, ultimately inducing apoptosis. Treatment of infected golden hamsters with the compound markedly reduces (> 92%) parasitic burden, both in spleen and liver. Interestingly, the 17-decarboxylated analogue, dihydrolupeol, does not inhibit DNA topoisomerase I and II, has no effect on parasitic growth, and also fails to induce apoptosis. DHBA is a potent antileishmanial agent that induces apoptosis by primarily targeting DNA topoisomerases. Therefore it is a strong candidate for use in designing new antileishmanial drugs.
INTRODUCTION
Kala-azar, or visceral leishmaniasis, is the most fatal form of leishmaniasis, affecting millions around the world. The toxicity of available drugs and the emergence of strains that are not responsive to drug therapy make the discovery of novel therapeutic agents imperative (1,2). In recent years, various new compounds with profound antileishmanial activity have been reported. Pilot study has demonstrated itraconazole to be a well-tolerated alternative treatment of mucocutaneous leishmaniasis (3) whereas the leaf extract of Kalanchoe pinnata was reported to reduce lesion size in cutaneous leishmaniasis (4). Various compounds, such as atovaquone (5), WR6026 (6), licochalcone A (7), ilmfosine (8), and formycin B (9) have been reported to inhibit Leishmania donovani infection, the causative agent of visceral leishmaniasis. Unfortunately, all such compounds are effective only at higher doses and none of them have been recognized as oral drugs. Compounds like ketoconazole, fluconazole, and terbinafine were found to be less effective than antimonial agents in reducing hepatic parasitic load (10). A plant-derived napthoquinone, lapachol, was reported to exert leishmanicidal activity in vitro with no in vivo therapeutic effect (11). The only effective new oral compound to date is alkyl phospholipid miltefosine, which is effective against even antimony unresponsive cases (12) and presently is under phase III clinical trials (13).
Search for antileishmanial agents have led scientists to newer therapeutic targets, which are essential for parasite growth and survival. DNA topoisomerases recently have emerged as principal therapeutic targets with a group of targeting agents having a broad spectrum of antiprotozoan activity (14). They are ubiquitous enzymes, involved in many vital cellular processes (for example, replication, transcription, chromosomal segregation, genome stability, and so on [15]) and are essential for cell survival. Studies by this laboratory have introduced potential antileishmanial agents that inhibit DNA topoisomerases (16–19). Camptothecin, an anti-tumor agent and a well-characterized topoisomerase I inhibitor, also is reported to have profound cytotoxic effect on Trypanosoma and Leishmania species (20). Betulinic acid, a pentacyclic triterpenoid with a wide spectrum of pharmacological properties, is reported to be a potent inhibitor of DNA topoisomerases I and II (21). Recently, we have reported that dihydrobetulinic acid (DHBA), a derivative of betulinic acid that exhibits anti-HIV activity (22,23), is another excellent inhibitor of DNA topoisomerase I (24) with the potential to become a lead therapeutic compound.
In the present study, we describe the antileishmanial activity of dihydrobetulinic acid (DHBA) (Figure 1A) on both L. donovani promastigotes and amastigotes. Dissecting the molecular mechanism, we show that similar to the parent compound (21), DHBA inhibits DNA topoisomerases I and II by preventing enzyme-DNA binary complex formation. The compound causes extensive DNA damage with the ultimate induction of apoptosis. Oral and intramuscular administrations of DHBA to hamsters with established infection markedly reduce (> 92%) the splenic and hepatic parasitic burden. Correlating DHBA-mediated cell death and topoisomerase inhibition with that of dihydrolupeol, the decarboxylated analogue of DHBA, we propose that DHBA could be a novel lead compound for antileishmanial therapy.
Figure 1.

Inhibition of catalytic activity of L. donovani DNA topoisomerases I and II by DHBA. A: Structure of DHBA. B: Simultaneous addition of topoisomerase I, DHBA, and DNA. Lane 1, supercoiled pGEM4Z DNA; lane 2, DNA with 2 units of purified L. donovani topoisomerase I and 4% DMSO; lanes 3 to 7, inhibition of catalytic activity with increasing concentration of DHBA. SM, supercoiled monomer; NM/RM nicked or relaxed monomer. C: Simultaneous addition of topoisomerase I, camptothecin, and DNA. Lanes 1 and 2, supercoiled pGEM4Z DNA; lanes 3 to 7, inhibition of catalytic activity with increasing concentration of camptothecin. D: Inhibition of catalytic activity of L. donovani DNA topoisomerase II by DHBA and etoposide. Lane 1, control kDNA; lane 2, kDNA with 2 units of purified L. donovani topoisomerase II; lanes 3 to 5, simultaneous addition of topoisomerase II, kDNA, and increasing concentration of etoposide; lanes 6 to 10, same as lanes 3 to 5 but with increasing concentration of DHBA. E: Effect of dihydrolupeol on catalytic activity of L. donovani DNA topoisomerase I. Lane 1, supercoiled pGEM4Z DNA; lane 2, DNA with 2 units of topoisomerase I; lanes 3 to 5, simultaneous addition of DNA, enzyme, and increasing concentrations of dihydrolupeol. F: Effect of dihydrolupeol on catalytic activity of L. donovani DNA topoisomerase II. Lane 1, kDNA; lane 2, kDNA with 2 units of topoisomerase II; lanes 3 to 5, simultaneous addition of DNA, enzyme, and increasing concentrations of dihydrolupeol.
MATERIALS AND METHODS
Isolation and Purification of DHBA
Betulinic acid, isolated from the leaves of Bacopa monniera as described (25), was subjected to hydrogenation (26) to create DHBA (see Figure 1A). DHBA was purified by chromatography over silica gel (60 to 120 mesh) and characterized by detailed spectral analysis of infrared, 1H nuclear magnetic resonance, 13C nuclear magnetic resonance, and mass spectroscopy. The final drug concentration was kept at 4% and 0.5% for all in vitro and cellular experiments, respectively.
Enzymes, DNA, and Chemicals
Kinetoplast DNA (kDNA) and DNA topoisomerase I and II were purified from Leishmania donovani strain UR6 promastigotes as described previously (27–29). Plasmids pHOT1 and pRYG were purchased from Topogen, Inc. (Columbus, OH, USA) and pGEM4Z was purchased from Promega (Madison, WI, USA). All other chemicals were purchased from Sigma Chemical (St. Louis, MO, USA).
Parasite Culture and Growth Conditions
Leishmania strain UR6 and AG83 promastigotes were grown at 22 °C in Rays modified media (30) and M 199 liquid media supplemented with 10% fetal calf serum.
DNA Topoisomerase I Assay
After treatment with topoisomerase I as described (18), the type I DNA topoisomerase was assayed by decreased mobility of relaxed isomers of supercoiled pGEM4Z DNA in an agarose gel. The standard topoisomerase assay mixture (25 μL) contained: 50 mM Tris HCl, pH 7.5, 5% glycerol, 50 mM KCl, 0.5 mM dithiothreitol (DTT), 10 mM MgCl2, 30 μg/mL bovine serum albumin, 0.5 μg pGEM4Z DNA, and 2 units of enzyme (1 unit is defined as the amount of enzyme required to convert 50% of 0.5 μg supercoiled DNA substrate to the relaxed form under standard assay conditions). Reactions were carried out at 37 °C for 30 min and then stopped by adding 1% sodium dodecyl sulfate, 10 mM EDTA, 0.25 mg/mL bromophenol blue, and 15% glycerol. Samples were electrophoresed in 1% agarose gel in Tris-acetate-EDTA buffer (40 mM Tris-acetate, 2mM EDTA, pH 8) at 1.5 V/cm for 14 to 16 h at room temperature. Gels were stained with ethidium bromide (0.5 μg/mL), destained with water, and photographed under UV illumination.
DNA Topoisomerase II Assay
The type II DNA topoisomerase was assayed by the decatenation of kDNA as described (19). The standard decatenation assay mixture (25 μL), containing 25 mM Tris HCl pH 7.9, 10 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 50 mM NaCl, 10% glycerol, and 1 μg kDNA, was incubated with 2 units of Leishmania topoisomerase II at 30 °C for 30 min. One unit is defined as the amount of enzyme required to release 50% of free minicircle DNA from 0.5 μg substrate kDNA. Decatenations were monitored in 1% agarose gel as described above.
Gel Retardation Assay
pHOT1 and pRYG DNA, containing high affinity topoisomerase I binding and topoisomerase II cleavage site respectively (31,32), were used as substrate DNA. Experiments were conducted as described (24). Substrate DNA was incubated with DNA topoisomerase I or II at 8 °C for 15 min, in the presence or absence of DHBA, in 50 μL binding buffer (24). After incubation, the reaction mixtures were electrophoresed in 1% agarose gel containing 0.5 μg/mL ethidium bromide at 4 °C as described above.
Analysis of DNA Cleavage
pHOT1 DNA and pRYG DNA were used as substrates for topoisomerase I and II mediated DNA cleavage, respectively. The standard reaction mixtures (50 μL) contained 50 mM Tris HCl, pH 7.5, 2mM MgCl2, 0.2 mM DTT, 0.5 mM EDTA, 30 μg/mL bovine serum albumin, and 20 μg/mL pHOT1 DNA (for topoisomerase I) or 20 μg/mL pRYG DNA (for topoisomerase II). Topoisomerase II assay mixtures also contained 2 mM ATP. Reactions were carried out with 50 units of enzymes at 37 °C for 30 min in presence and absence of drugs. The reactions were terminated by adding 1% sodium dodecyl sulfate, 150 μg/mL proteinase K and incubated for 1 h at 37 °C. DNA samples were electrophoresed in 1% agarose gel containing 0.5 μg/mL ethidium bromide as described above.
Drug Treatment of Intact Leishmania Parasites and Isolation of DNA
Exponentially growing AG83 promastigotes (2.5 × 106 cells/mL) were incubated at 22 °C for 15 min prior to the addition of DHBA and etoposide. Cells were further incubated at 22 °C for 30 min, washed, and lysed. DNA was extracted from the lysate, electrophoresed in 2% agarose gel, transferred into Hybond N membrane, and hybridized with 32P-labeled kDNA (19).
Cytotoxicity Assay
Cytotoxicity of the drugs was estimated by microscopic counting of the viable parasites by trypan blue exclusion method after treatment of L. donovani promastigotes with DHBA. Antileishmanial activity of DHBA on intracellular L. donovani amastigotes was carried out with thioglycollate-elicited peritoneal exudate as described (19). At least 400 macrophages were examined for each cover slip. Antileishmanial activity was determined by calculating the number of amastigotes per 100 macrophages.
Study of Dyskinetoplastidy by Confocal Microscopy
Smears of drug-treated L. donovani AG83 promastigotes (2.5 × 106 cells/mL) were assessed for dyskinetoplastic cells by ethidium bromide staining (0.1 μg/mL in 1× phosphate buffered saline containing 10% glycerol) under TCS-SP Leica confocal microscope system equipped with a krypton-argon mixed laser.
Study of Parasite Ultrastructure by TEM
For thin-section electron microscopy, cells were fixed with 6% glutaraldehyde in 0.125 M phosphate buffer (pH 7.2) for 14 to 16 h and then with 1% osmium tetroxide for 16 to 20 h at room temperature. The fixed cells were washed for 2 h in 0.5% uranyl acetate, dehydrated with increasing concentrations of ethanol, and embedded in Spurr medium at 70 °C for 48 h.
Sections were cut with a DuPont diamond knife in an LKB ultramicrotome, stained with uranyl acetate and lead citrate, and examined under JEOL 100 CX transmission electron microscope (33).
Pulsed Field Gradient Gel Electrophoresis of DHBA–Treated Promastigotes
Different concentrations of DHBA were added to AG83 promastigotes (2.5 × 106 cells/mL) and incubated at 22 °C up to 48 h. Cells were pelleted, lysed, and blocks prepared from low-melting agarose were subsequently digested with proteinase K (2 mg/mL) for 48 h at 55 °C as described (34). Electrophoresis was carried out for 24 h with an initial and final switch time of 60 and 120 s respectively (current, 6 V/cm; including angle, 120°) in 0.5X Tris-borate-EDTA buffer.
Analysis of Apoptosis by Agarose Gel Electrophoresis
DNA, from exponentially growing AG83 cells (2.5 × 106 cells/mL) treated with or without drugs, was isolated according to the protocol of Ward and others (35) and then electrophoresed on 1% agarose gel followed by ethidium bromide staining.
Determination of Antileishmanial Activity of the Compounds In Vivo
Male golden hamsters (6 wk, 60 to 75 g) with 2-wk-old established infection were administered 10 mg/kg body weight of DHBA both orally and by intramuscular injection for 6 wk before they were sacrificed. Control group received phosphate buffered saline (PBS). Splenic and liver parasitic loads were determined from impression smears after Giemsa staining and also from limiting dilution analysis of tissue homogenates. Results are expressed as the number of amastigotes/100 nuclei and the total parasitic load per organ, using the formula (36):
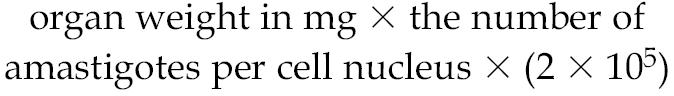 |
RESULTS
DHBA Inhibits DNA Topoisomerases I and II of L. donovani
DNA relaxation and kDNA decatenation assays were performed with 2 units of topoisomerases I and II with different concentrations of DHBA. Topoisomerase I activity was partially inhibited by 1 μM DHBA and completely at 2 μM (see Figure 1B). This inhibition was comparable to the effect of a potent antitumor agent, camptothecin (37) (see Figure 1C). During the decatenation assay, the topoisomerase II catalytic activity was completely inhibited by 5 μM DHBA. This inhibition was comparable to the effect of a well-studied topoisomerase II inhibitor, etoposide (38) (see Figure 1D). These results demonstrate that DHBA is a very potent dual inhibitor of L. donovani DNA topoisomerases. However, 50 μM of the decarboxylated form of DHBA, dihydrolupeol (17-CH3 instead of 17-CO2H) neither inhibited DNA topoisomerase I (see Figure 1E) nor topoisomerase II (see Figure 1F).
DHBA Inhibits Cleavable Complex Formation by Abrogating Topoisomerase-DNA Interaction
A topoisomerase reaction has 3 general mechanistic steps: 1) binding of the enzyme to the substrate DNA, 2) strand breakage and subsequent strand passage through the break leading to change in linking number, and 3) strand religation. Topoisomerase inhibition can be achieved by prevention of enzyme-DNA binary complex formation or by stabilization of enzyme-DNA putative reaction intermediate, that is, the cleavable complex. Gel retardation assays were performed to study the effect of DHBA on the initial topoisomerase–DNA binary complex formation. pHOT1 DNA, when incubated with 20 and 40 units of topoisomerase I at 8 °C for 30 min, showed retarded mobility indicating the formation of a protein-DNA complex that disappeared upon treatment with sodium dodecyl sulfate and proteinase K. When 40 units of topoisomerase I was preincubated with increasing concentrations of DHBA for 5 min, prior to the addition of pHOT1 DNA, no retardation was visible (Figure 2A). When the same experiment was carried out with pRYG DNA and topoisomerase II, similar results were obtained with the complete disappearance of the retarded band upon increasing DHBA concentration (see Figure 2B). These experiments clearly indicate that DHBA prevents the primary interaction between topoisomerases and substrate DNA.
Figure 2.

Mechanistic study of DHBA-mediated DNA topoisomerase inhibition. A: DHBA prevents topoisomerase I-DNA binary complex formation. pHOT1 DNA (lane 1) was incubated with 20 units (lane 2) and 40 units (lane 3) of topoisomerase I at 8 °C for 15 min and electrophoresed on a 7% nondenaturing polyacrylamide gel at 4 °C. Preincubation of 50 units of topoisomerase I with increasing concentration of DHBA prevented the formation of DNA-enzyme binary complex (lanes 5 to 7). B: DHBA prevents topoisomerase II-DNA binary complex formation. Preincubation of 50 units of topoisomerase II with increasing DHBA concentration (lanes 5 to 8). Lanes 2 and 3, incubation of DNA with 20 and 40 units of enzyme, respectively. C: DHBA does not induce topoisomerase I–mediated DNA cleavage. pHOT1 DNA (lane 1) was incubated with 50 units of enzyme alone (lane 2) and further treated with SDS/PK (lane 3). Lanes 4 to 6, with increasing concentrations of camptothecin, and lanes 7 to 9, with increasing concentration of DHBA. D: DHBA antagonizes camptothecin-mediated DNA cleavage. Reactions were as described in Materials and Methods. Lane 1, supercoiled pHOT1 DNA; lane 2, same amount of DNA plus 50 units of topoisomerase I; lane 3, same as lane 2 plus 20 μM camptothecin; lanes 4 to 6, same as lane 3 but in presence of 1, 5, and 10 μM of DHBA added simultaneously; lanes 7 to 9, same as lanes 4 to 6 but enzyme was preincubated with different concentrations of DHBA before addition of camptothecin and DNA. E: DHBA does not induce topoisomerase II–mediated DNA cleavage and antagonizes etoposide-induced cleavage. Reactions were as described in Materials and Methods. Lane 1, control pRYG DNA; lanes 2 and 3, DNA with 25 and 50 units of L. donovani topoisomerase II; lanes 4 and 5, same as lane 3 but in presence of 10 and 50 μM etoposide; lanes 6 to 8, same as lane 3, but in presence of 1, 5, and 10 μM DHBA added simultaneously with DNA and topoisomerase II; lanes 9 to 11, same as lane 5, but enzyme was preincubated with increasing concentration of DHBA before addition of etoposide. F: DHBA did not induce cleavage of kDNA minicircles in L. donovani AG83 promastigotes. Reactions were as described in Materials and Methods. Lane 1, DNA from control cells; lane 2, cells treated with 0.5% DMSO; lanes 3 and 4, cells treated with etoposide at 100 and 200 μM; lanes 5 to 8, from cells treated with increasing concentration of DHBA. I, covalently closed minicircles; II, minicircles containing nicks or gaps; III, linearized minicircles. G: DHBA inhibits etoposide-promoted minicircle cleavage. Lanes 1 and 2, same as lanes 1 and 2 in F; lane 3, DNA from cells treated with 50 μM etoposide; lanes 4 and 5, DNA from cells treated with 50 μM etoposide along with 5 and 20 μM DHBA added simultaneously; lanes 6 and 7, DNA from cells preincubated with 5 and 10 μM DHBA followed by treatment with etoposide.
Since the binary complex formation precedes the enzyme-DNA cleavable complex formation, it was expected that DHBA would not stabilize the cleavable complex. To confirm this, topoisomerase I–mediated DNA cleavage was assayed with different DHBA concentrations, keeping camptothecin as a positive control. Unlike camptothecin, which induced stabilization of the cleavable complex, DHBA had no effect (see Figure 2C). Moreover, when topoisomerase I was preincubated with DHBA prior to the addition of camptothecin, the camptothecin-mediated cleavage was markedly reduced and completely inhibited at higher concentrations of DHBA (see Figure 2D). Similar results were obtained with DHBA and topoisomerase II, where etoposide-mediated cleavage also was inhibited (see Figure 2E). As both camptothecin and etoposide bind to the enzyme-DNA binary complex and no such complex formation is possible in presence of DHBA, this cleavage antagonism is expected.
To understand whether such in vitro observations were comparable in vivo, DHBA treated L. donovani AG83 promastigotes were assayed for drug-mediated linearization of kDNA minicircles. Unlike etoposide, which promoted DNA cleavage and resulted in a large increase in linear minicircle population, DHBA had no effect (see Figure 2F). On the other hand, when promastigotes were incubated with DHBA for 30 min before the addition of etoposide, induction of minicircle linearization was inhibited as expected. It was interestng to note that when etoposide and DHBA together were added simultaneously, there was no appreciable change in linear minicircle population, indicating DHBA-enzyme interaction was dependent on time (see Figure 2G). These experiments confirm that mode of inhibition is same, both in vitro and in vivo.
DHBA Inhibits Growth of L. donovani Promastigotes and Amastigotes
L. donovani AG83 promastigotes (2.5 × 106 cells/mL) were incubated with 3 different concentrations of DHBA for 3 d. Next the numbers of live promastigotes were counted by trypan blue exclusion. On day 1, 65% percent growth was inhibited by 5 μM DHBA, and 98% growth was inhibited on day 3, which was comparable to the inhibition achieved by 50 μM DHBA on day 1. Dihydrolupeol had no effect on the survival of AG83 promastigotes. With 5 μM DHBA, a meager 2% parasite population survived after 3-d incubation (Figure 3A). To ensure that this was not due to the emergence of a resistant subpopulation, 5 μM DHBA-treated cells after incubation for 2 (AG83D5II) and 3 d (AG83D5III) were washed with media and PBS and resuspended in fresh media. Compared with normal AG83 promastigotes, only 66% of AG83D5II cells were recovered after 3 d of incubation and recovery was as low as 26% for AG83D5III cells. With the addition of 5 μM DHBA, both cultures were equally vulnerable with approximately 10% cells surviving after 1 d of incubation (see Figure 3B). Therefore, DHBA is not only very effective against L. donovani promastigotes, but it also does not induce the generation of a resistant subpopulation. The IC50 value, calculated to be 2.6 μM (see Figure 3C), varied inversely with incubation time (see inset, Figure 3C).
Figure 3.

In vitro cytotoxicity of DHBA on cultured L. donovani promastigotes. A: Cells (2.5 × 106 cells/mL) were cultured for 3 d in M199 media supplemented with 10% fetal calf serum in the presence of 5 μM DHBA (♦), 20 μM DHBA (▪), 50 μM DHBA (▴), and 100 μM dihydrolupeol (•). Data represent mean ± SD of 3 independent experiments. B: DHBA-induced cell death does not give rise to resistant subpopulation. AG83 promastigotes treated with 5 μM DHBA for 2 d (AG83D5II) or 3 d (AG83D5III) were washed and cultured in absence of DHBA for another 3 d. AG83D5II, ⋄; AG83D5III, ▵. Similar experiments were repeated but in the presence of 5 μM DHBA. AG83D5II + 5 μM DHBA, □; AG83D5III + 5 μM DHBA, ×. Data are expressed as percentage of live promastigotes and represent mean ±SD from 3 independent experiments. C: Calculation of IC50. The IC50 values were calculated for each type of incubation from a plot of live promastigotes percentage compared with DHBA concentration. 1 d, ○; 2 d, □; and 3 d, ▵. All experiments were performed 3 times and representative data from 1 set of these experiments are expressed as mean ±SD. Inset, variation of IC50 with incubation time. D: Thioglycollate-activated peritoneal macrophages were allowed to adhere on cover slips in RPMI-1640 supplemented with 10% FCS before in vitro infection with L. donovani promastigotes, followed by further incubation for 2 d at 37 °C with graded concentrations of DHBA, ♦; and with dihydrolupeol, •. Experiments were performed 3 times and representative data from 1 set of these experiments are expressed as mean ±SD.
Peritoneal macrophages of BALB/c mice were infected with L. donovani AG83 promastigotes in vitro. Infected macrophages, after subsequent washing were incubated with 3 different concentrations of DHBA for 2 d. Similar to its effect on promastigotes, 5 μM DHBA reduced the parasitic burden by 68%. Only 2% survival was observed upon incubation with 50 μM DHBA (see Figure 3D). IC50 of inhibition was calculated to be 4.1 μM. Dihydrolupeol, however, failed to decrease the parasitic burden.
DHBA Induces DNA Damage but Not Dyskinetoplastidy
DHBA does not bind DNA, as evidenced by filter-binding assay, using labeled DNA and DHBA, and determination of melting temperature of DNA in presence and absence of DHBA (data not shown). Many DNA binding agents like acriflavine and berenil induce dyskinetoplastidy, a phenomenon where parasitic cells, though having mitochondrial membrane, completely lack kDNA (39). Confocal microscopy of L. donovani AG83 promastigotes treated with 5 μM DHBA for 24 h shows that DHBA is incapable of inducing dyskinetoplastidy. However, DHBA causes an extensive degranulation and defragmentation of total DNA. Some of the cells showed initial signs of nuclear compartmentalization and reduction of DNA intensity (Figure 4A). Berenil-treated parasites were mostly dyskinetoplastid (see Figure 4B) whereas cells grown in media supplemented with 0.5% DMSO were completely healthy (see Figure 4C).
Figure 4.
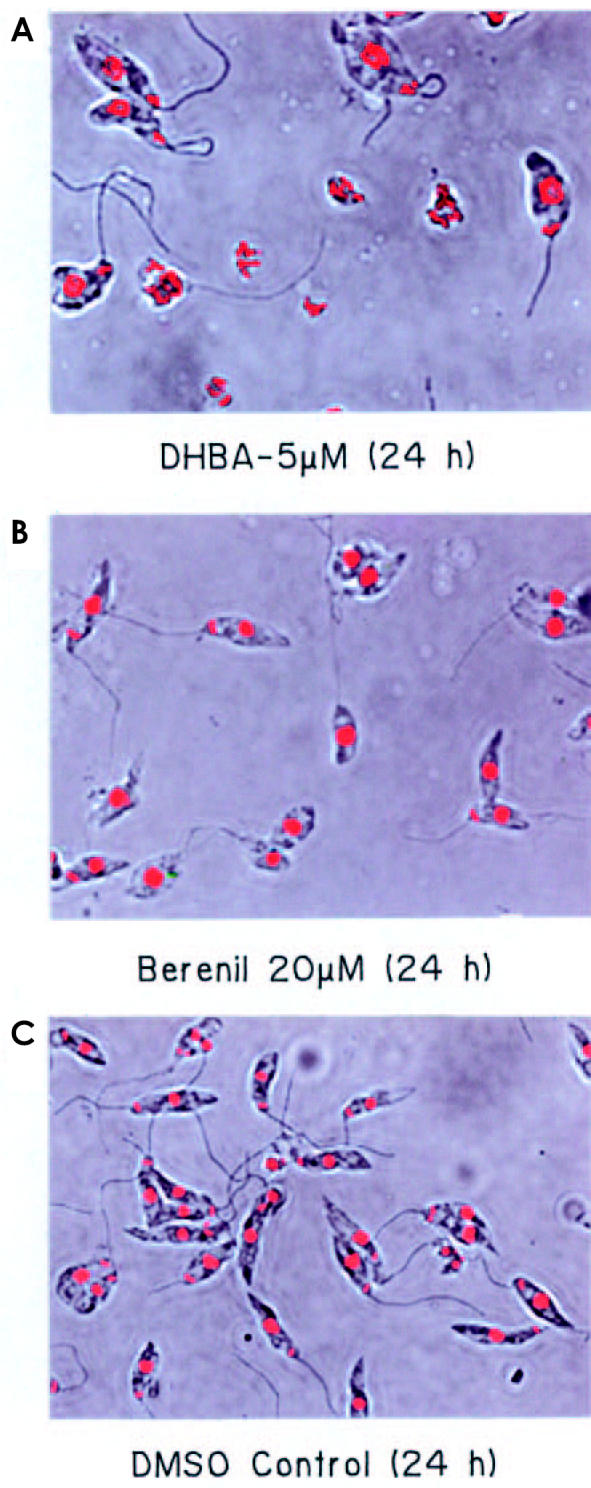
Confocal microscopy of L. donovani AG 83 promastigotes. Parasites incubated with 5 μM DHBA (A), 20 μM berenil (B) and 0.5% DMSO (C) were stained with ethidium (0.1 μg/mL) in 1× phosphate-buffered saline containing 10% glycerol. The cells were viewed under a Leica DM IRB inverted microscope. Phase contrast and fluorescence images for the same fields were merged and are represented here. Magnification 1000×.
In order to have a closer look at the effect of DHBA on L. donovani genomic DNA, transmission electron microscopy (TEM) was performed with both DHBA-treated and untreated promastigotes by embedding in spurr-blocks as described in Materials and Methods. Cells treated for 8 h with 5 μM DHBA had intact plasma membrane (Figure 5). However, the nuclei of such cells showed a particular chromatin margination, followed by its compaction toward the nuclear periphery, to form 1 or several frequent cup-shaped masses, which are typical characteristics of apoptosis (40). Examination of cells treated with 5 μM DHBA for 16 h showed the disruption of outer cell membrane and blebbing formation. Upon examination of parasites incubated with 5 μM DHBA for 24 h, total loss of cellular morphology and the generation of distinct compartments similar to apoptotic bodies were visible. Therefore, TEM data indicate that DNA degradation, as visualized by confocal microscopy, might actually be caused by programmed cell death. In order to substantiate these cellular observations in vitro, a pulsed field gradient gel electrophoresis was performed with DHBA-treated AG83 promastigotes. No genomic DNA was visible upon 48 h incubation with 5 μM DHBA indicating complete fragmentation of DNA (Figure 6).
Figure 5.

Transmission electron microscopy of DHBA-treated promastigotes. L. donovani AG83 cells were cultured in media containing 0.5% DMSO or 5 μM DHBA for different time periods. Spurr blocks were prepared as described in Materials and Methods. Control cells treated with 0.5% DMSO; cells treated with 5 μM DHBA for 8 hr (arrow shows cup-shaped masses); cells treated for 16 h (arrows show surface blebbing); cells treated for 24 h (arrows show apoptotic bodies); nucleus of control cell (arrow shows intactness of nucleus); nucleus of cells treated with 5 μM DHBA for 16 h (arrows indicate chromatin margination). Magnification of 15000 (15K) and 20000 (20K) are indicated in each figure.
Figure 6.
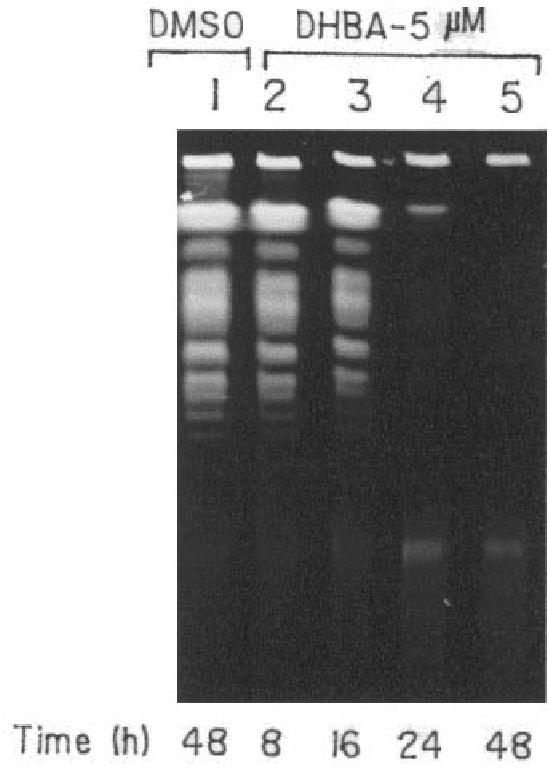
Pulsed field gradient gel electrophoresis of DHBA-treated promastigotes. L. donovani promastigotes were incubated with 5 μM DHBA for different time periods. Agarose gel blocks were prepared for each sample and loaded onto a 1% agarose gel and electrophoresed as described in Materials and Methods.
DHBA Induces Apoptosis and Reduces Parasitic Burden
To further confirm that leishmanial cell death was due to an active process driven by the cell and not just the consequence of a passive degenerative process, cultured AG83 cells were treated with 50 μM camptothecin; 5, 10, and 50 μM DHBA; and 100 μM dihydrolupeol for 24 h. Next their DNAs were isolated and electrophoresed on a 1% agarose gel (Figure 7). Parasites treated with camptothecin showed the formation of DNA ladders indicating apoptosis. Cells treated with DHBA showed similar band patterns with increase in intensity upon increasing DHBA concentration. However, even upon 24 h incubation, 100 μM dihydrolupeol did not induce apoptosis.
Figure 7.
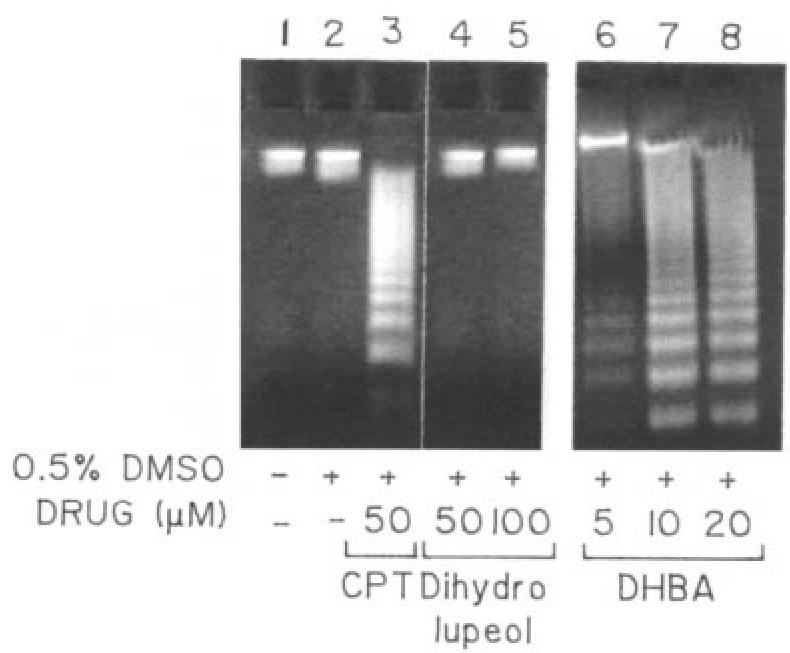
Agarose gel electrophoresis to study apoptosis. Genomic DNAs were isolated from cultured cells, grown in presence of camptothecin, DHBA, or dihydrolupeol for 24 h and electrophoresed.
The inhibition of catalytic activities of DNA topoisomerases, growth of parasites, and induction of apoptosis indicate that DHBA is a highly potent antileishmanial agent. The therapeutic potential of the compound was further substantiated by an in vivo experiment. Golden hamsters were infected with AG83 promastigotes and treated 2-wk after infection with DHBA (10 mg/kg body weight) for 6 wk. Compared with the infected, untreated hamsters, which showed a progressive and fatal visceral leishmaniasis, when DHBA was administered intramuscularly there was a drastic decrease in parasitic burden, 97% and 99% in spleen and liver respectively. Upon oral administration, splenic and liver parasitic burden decreased to 95.5% and 92.5% respectively (Figure 8A). Giemsa-stained micrographs of spleen and liver smears for saline-treated, orally and intramuscularly DHBA-treated hamsters also show this profound effect and nearly complete reduction of parasitic burden (see Figure 8B). More importantly, DHBA was found to be nontoxic as evident from the analysis of serum glutamate oxaloacetate transferase and serum glutamate pyruvate transferase of DHBA-treated animals. Also, the weight of infected, untreated hamsters reduced drastically compared with the weight of normal and DHBA-treated infected hamsters, indicating healthy recovery (data not shown).
Figure 8.
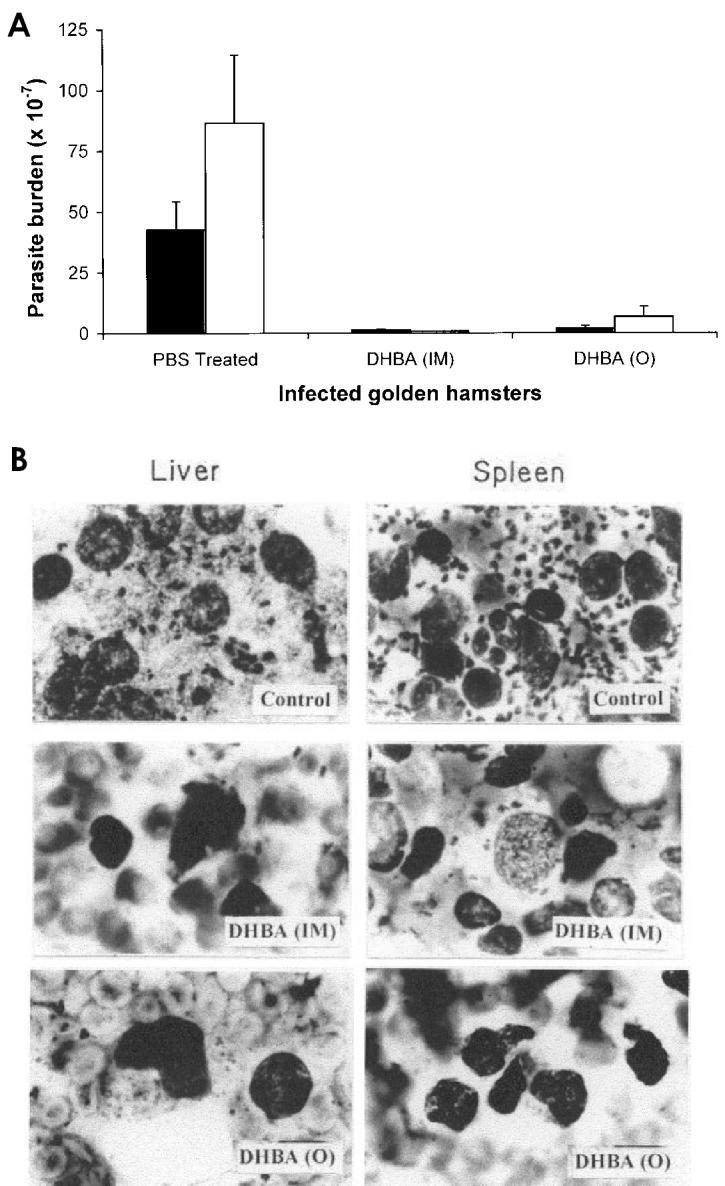
DHBA induces apoptosis and reduces parasitic burden. A: DHBA reduces both splenic and hepatic parasitic burden in infected golden hamsters. Experiments with intramuscular (IM) and oral (○) administration of DHBA were performed in triplicate and representative data from 1 set of these experiments for spleen (▪) and liver (□) are expressed as mean ±SD. B: Micrographs of Giemsa-stained splenic and hepatic smears of L. donovani–infected golden hamsters after treatment with DHBA. Infected hamsters were treated both orally (○) and intramuscularly (IM) with either phosphate-buffered saline or 10 mg/kg body weight of DHBA.
DISCUSSION
We report here that DHBA is a dual inhibitor of both parasite DNA topoisomerases. It prevents interaction between the enzyme and substrate DNA, as evidenced from electrophoretic mobility shift assay. As a result, DHBA is unable to stabilize the cleavable complex and also antagonizes camptothecin or etoposide-mediated DNA cleavage. Therefore, the compound is very different from well-known topoisomerase inhibitors, such as, camptothecin (37), etoposide (38), amsacrine (38), doxorubicin (41), and so on, and is more similar to known class II inhibitors like merbarone (42), aclarubicin (41), chloroquine (43), and so on. However, unlike all these compounds, DHBA does not interact with substrate DNA, and thus belongs to a rare group of dual inhibitors.
DHBA has a profound inhibitory effect on the growth of L. donovani promastigotes. IC50 values of DHBA were calculated to be 4.1 μM, 3.2 μM, and 2.6 μM when cells were incubated with the compound for a period of 24, 48, and 72 h. However, when incubated with dihydrolupeol, a 17-decarboxylated analogue of DHBA having no inhibitory effect on topoisomerases, there was no significant inhibition of promastigote growth. This suggests that inhibition of topoisomerases is a major step in the antileishmanial activity of DHBA. The possibility of emergence of a sub-population of promastigotes resistant to DHBA was addressed by assessing the promastigote survival with 5 μM DHBA on the residual viable population. It was observed that the residual population became highly susceptible to DHBA with repeated exposure to the compound. The IC50 value of 4.1 μM after 2-d treatment of amastigotes was comparable to the IC50 value of 3.2 μM upon inhibition of promastigotes growth after 2 d. However, it was not possible to calculate IC50 of intracellular amastigotes for 1- and 3-d periods. Negligible infection was detected in control macrophages upon 1-d incubation with parasites, whereas 3-d incubation led to lysis of macrophages due to heavy infection.
With the establishment of DHBA as a potent antileishmanial agent that targets DNA topoisomerases, it was important to study the effect of the compound on the parasitic ultra structure. Though DHBA did not induce dyskinetoplastidy in L. donovani promastigotes, it caused chromatin margination in the nucleus and also dechromatinization of the genome as visualized under confocal microscope. TEM showed a strongly rearranged nucleus with progressive changes in cell shape along with surface blebbing, which are characteristics of apoptosis (40). That DHBA induces apoptosis of L. donovani promastigotes has been further confirmed by agarose gel electrophoresis. Dihydrolupeol not only loses its ability to inhibit DNA topoisomerases but also fails to induce apoptosis and ultimate cell death in L. donovani. This observation strongly supports a direct correlation between topoisomerase inhibition and apoptosis in L. donovani. The parent compound, betulinic acid, has been previously reported to induce apoptosis (44,45) without any knowledge of a plausible target. Our study, for the first time, places DNA topoisomerase I and II on the shortlist as the primary cellular targets responsible for DHBA-induced apoptosis.
Various topoisomerase inhibitors have been reported to induce apoptosis, for example, doxorubicin (46) and salvicine (47). Signal transduction pathways leading to cell-cycle arrest and apoptosis induced by DNA topoisomerase poisons also have been reported (48). Our laboratory has identified a topoisomerase poison, luteolin, that acts by inducing topoisomerase II–mediated kinetoplast DNA cleavage leading to apoptosis in L. donovani (19). Characteristic features of programmed cell death, such as nuclear condensation and nicking of DNA in the nucleus, already have been reported to exist in L. donovani (49). We show here that the induction of programmed cell death in L. donovani promastigotes by DHBA is effectively translated into in vivo therapeutic effect. DHBA treatment of golden hamsters, both orally and intramuscularly, with established infection, almost completely reduced the parasitic burden in both spleen and liver. The degree of parasite reduction (103 times) was comparable to the effect of established drugs, such as amphotericin B with a degree of reduction of 103 to 105 times.
This profound antileishmanial effect of the compound, along with the absence of side effects, makes DHBA a highly promising candidate for antileishmanial therapy. Structure–function analysis of DHBA-topoisomerase interaction along with modeling studies can lead to the development of more potent drugs and also can facilitate the study of future drug resistance.
Acknowledgments
The authors wish to thank Prof AN Bhaduri, former director, and Prof S Bhattacharya, present director of the Institute of Chemical Biology, for constructive criticism and interest in this work and the Department of Biotechnology, Government of India, for support through Grant No. BT/PRO 493/MED/09/096/96. The expertise and assistance of Mr SN Dey of the Electron Microscopy Division is gratefully acknowledged. Council of Scientific and Industrial Research, Government of India, supported ARC with a Senior Research Fellowship.
REFERENCES
- 1.Walton BC, Peters W, Killick-Kendrick R. (1987) American cutaneous and mucocutaneous leishmaniasis. In: Peters W, Killick-Kendrick R (eds.) The Leishmaniasis In Biology and Medicine. Academic Press, London, pp. 636–44.
- 2.Iwu MM, Jackson JE, Schuster BG. Medicinal plants in the fight against leishmaniasis. Parasitol Today. 1994;10:65–8. doi: 10.1016/0169-4758(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 3.Amato VS, et al. Use of itraconazole in the treatment of mucocutaneous leishmaniasis: a pilot study. Int J Infect Dis. 2000;4:153–7. doi: 10.1016/s1201-9712(00)90077-8. [DOI] [PubMed] [Google Scholar]
- 4.Da-Silva SA, Costa SS, Rossi-Bergmann B. The anti leishmanial effect of kalanchoe is mediated by nitric oxide intermediate. Parasitology. 1999;118:575–82. doi: 10.1017/s0031182099004357. [DOI] [PubMed] [Google Scholar]
- 5.Murray HW, Hariprashad J. Activity of oral atrovaquone alone and in combination with antimony in experimental visceral leishmaniasis. Antimicrob Agents Chemother. 1996;40:586–7. doi: 10.1128/aac.40.3.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dietze R, et al. Phase 2 trial of WR6026, an orally administered 8-aminoquinoline, in the treatment of visceral leishmaniasis caused by Leishmania chagasi. Am J Trop Med Hyg. 2001;65:685–9. doi: 10.4269/ajtmh.2001.65.685. [DOI] [PubMed] [Google Scholar]
- 7.Chen M, Christensen SB, Theander TG, Kharazmi A. Antileishmanial activity of licochalcone A in mice infected with Leishmania major and in hamsters infected with Leishmania donovani. Antimicrob Agents Chemother. 1994;38:1339–44. doi: 10.1128/aac.38.6.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croft SL, Neal RA, Thronton EA, Herrmann DB. Antileishmanial activity of the ether phospholipid ilmfosine. Trans R Soc Trop Med Hyg. 1993;87:217–9. doi: 10.1016/0035-9203(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 9.Berman JD, Keenan CM, Lamb SR, Hanson WL, Waits VB. Leishmania donovani: Oral efficacy and toxicity of formycin B in the infected hamster. Exp Parasitol. 1983;56:215–21. doi: 10.1016/0014-4894(83)90065-6. [DOI] [PubMed] [Google Scholar]
- 10.Gangneux JP, Dullin M, Sulahian H, Garin YJ, Derouin F. Experimental evaluation of second-line oral treatments of visceral leishmaniasis caused by Lesihmania infantum. Antimicrob Agents Chemother. 1999;43:172–4. doi: 10.1128/aac.43.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teixeira MJ, et al. In vitro and in vivo leishmanicidal activity of 2-hydroxy-3-(3-methyl-2-butenyl)-1,4-napthoquinone (lapachol) Phytother Res. 2001;15:44–8. doi: 10.1002/1099-1573(200102)15:1<44::aid-ptr685>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 12.Sundar S, et al. Trial of oral miltefosine for visceral leishmaniasis. Lancet. 1998;352:1821–3. doi: 10.1016/S0140-6736(98)04367-0. [DOI] [PubMed] [Google Scholar]
- 13.Guerin PJ, et al. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Dis. 2002;8:494–501. doi: 10.1016/s1473-3099(02)00347-x. [DOI] [PubMed] [Google Scholar]
- 14.Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–75. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 15.Wang JC. DNA topoisomerases. Annu Rev Biochem. 1996;65:635–92. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 16.Ray S, et al. Amarogentin, a naturally occurring secoiridoid glycoside and a newly recognized inhibitor of topoisomerase I from Leishmania donovani. J Nat Prod. 1996;59:27–9. doi: 10.1021/np960018g. [DOI] [PubMed] [Google Scholar]
- 17.Ray S, Sadhukhan PK, Mandal NB, Mahhato SB, Majumder HK. Dual inhibition of DNA topoisomerases of Leishmania donovani by novel indolyl quinolines. Biochim Biophys Res Commun. 1997;230:171–5. doi: 10.1006/bbrc.1996.5874. [DOI] [PubMed] [Google Scholar]
- 18.Ray S, Hazra B, Mittra B, Das A, Majumder HK. Diospyrin, a bisnapthaquinone: a novel inhibitor of type I DNA topoisomerase of Leishmania donovani. Mol Pharmacol. 1998;54:994–9. doi: 10.1124/mol.54.6.994. [DOI] [PubMed] [Google Scholar]
- 19.Mittra B, et al. Luteolin, an abundant dietary component is a potent anti leishmanial agent that acts by inducing topoisomerase II-mediated kinteoplast DNA cleavage leading to apoptosis. Mol Med. 2000;6:527–41. [PMC free article] [PubMed] [Google Scholar]
- 20.Bodley AL, Shapiro TA. Molecular and cytotoxic effects of camptothecin, a topoisomerase I inhibitor on trypanosomes and Leishmania. Proc Natl Acad Sci USA. 1995;92:3726–30. doi: 10.1073/pnas.92.9.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Syrovets T, Buchele B, Gedig E, Slupsky JR, Simmet T. Acetyl boswellic acids are novel catalytic inhibitors of human topoisomerase I and II. Mol Pharmacol. 2000;58:71–81. doi: 10.1124/mol.58.1.71. [DOI] [PubMed] [Google Scholar]
- 22.Kashiwada Y, et al. Betulinic acid and dihydro betulinic acid derivatives as potent anti-HIV agents. J Med Chem. 1996;39:1016–7. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto F, et al. Anti-AIDS agents-XXVII. Synthesis and anti-HIV activity of betulinic acid and dihydro betulinic acid derivatives. Bioorg Med Chem. 1997;5:2133–43. doi: 10.1016/s0968-0896(97)00158-2. [DOI] [PubMed] [Google Scholar]
- 24.Chowdhury AR, et al. Betulinic acid, a potent inhibitor of eukaryotic topoisomerase I: Identification of the inhibitory step, the major functional group responsible and development of more potent derivatives. Med Sci Monit. 2002;8:BR254–60. [PubMed] [Google Scholar]
- 25.Chatterji N, Rastogi RP, Dhar ML. Chemical examination of Bacopa monniera Wettst: Part I-isolation of chemical constituents. Indian J Chem. 1963;1:212–7. [Google Scholar]
- 26.Simonsen I, Ross WC. The Terpenes. Cambridge University Press. 1957;4:287–367. [Google Scholar]
- 27.Dasgupta S, Adhya S, Majumder HK. A simple procedure for the preparation of pure kinetoplast DNA network free of nuclear DNA from the kinetoplast hemoflagellate Leishmania donovani. Anal Biochem. 1986;158:189–94. doi: 10.1016/0003-2697(86)90608-1. [DOI] [PubMed] [Google Scholar]
- 28.Chakraborty AK, Gupta A, Majumder HK. A type I DNA topoisomerase from the kinteoplast hemoflagellate Leishmania donovani. Biochim Biophys Res Commun. 1993;180:279–85. doi: 10.1016/s0006-291x(05)81289-6. [DOI] [PubMed] [Google Scholar]
- 29.Chakraborty AK, Majumder HK. Decatenation of kinteoplast DNA by an ATP-dependant DNA topoisomerase from the kinteoplast hemoflagellate Leishmania donovani. Mol Biochem Parasitol. 1987;26:215–24. doi: 10.1016/0166-6851(87)90074-0. [DOI] [PubMed] [Google Scholar]
- 30.Ray JC. Cultivation of various Leishmania parasites on solid medium. Indian J Med Res. 1932;20:355–7. [Google Scholar]
- 31.Bonven BJ, Goecke E, Wetsergaard O. A high affinity topoisomerase I binding sequence is clustered at DNase I hypersensitive sites in tetrahymena R-chromatin. Cell. 1985;41:541–51. doi: 10.1016/s0092-8674(85)80027-1. [DOI] [PubMed] [Google Scholar]
- 32.Spitzner JR, Muller MT. A consensus sequence for cleavage by vertebrate topoisomerase II. Nucleic Acids Res. 1988;16:5533–56. doi: 10.1093/nar/16.12.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majumder S, Dey SN, Chowdhury R, Dutta C, Das J. Intracellular development of cholerphage φ149 under permissible and non permissible conditions: an electron microscopic study. Intervirol. 1988;29:27–38. doi: 10.1159/000150026. [DOI] [PubMed] [Google Scholar]
- 34.Benjamin MM, Datta AR. Modified pulsed field gel electropheresis technique using Pefabloc® SC for analyzing Listeria monocytogenes DNA. Biochemica. 1995;2:30–1. [Google Scholar]
- 35.Ward C, et al. NF-κ Buffer activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem. 1999;274:4309–18. doi: 10.1074/jbc.274.7.4309. [DOI] [PubMed] [Google Scholar]
- 36.Strauber LA, Franchino EM, Grun J. An eight day method for screening compound against Leishmania donovani in the golden hamster. J Protozool. 1989;5:269–73. [Google Scholar]
- 37.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. . J Biol Chem. 1985;260:14873–8. [PubMed] [Google Scholar]
- 38.Shapiro TA, Klein VA, Englund PT. Drug promoted cleavage of kinteoplast DNA minicircles, the evidence for type II topoisomerase activity in the trypanosome mitochondria. J Biol Chem. 1989;264:4173–8. [PubMed] [Google Scholar]
- 39.Shapiro TA, Englund PT. Selective cleavage of kinetoplast DNA minicircles promoted by anti trypanosomal drugs. Proc Natl Acad Sci USA. 1990;87:950–4. doi: 10.1073/pnas.87.3.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vitale M, Zauli G, Falcieri E. (1997) Apoptosis vs. necrosis. In: Proceedings of the USPHS Workshop on Laboratory and Epidemiologic Approaches to Determining the Role of Environmental Exposures as Risk Factors for B-Cell Chronic Lymphocytic Leukemia and Other B-Cell Lymphoproliferative Disorders. Marti GE, Vogt RF, Zenger VE (eds.) Purdue University Cytometry Laboratories [CDROM]
- 41.Bridewell DJ, Finlay GJ, Baguley BC. Differential actions of aclarubicin and doxorubicin: the role of topoisomerase I. Oncol Res. 1997;9:535–42. [PubMed] [Google Scholar]
- 42.Fortune JM, Osheroff N. Merbarone inhibits the catalytic activity of human topoisomerase IIα by blocking DNA cleavage. J Biol Chem. 1998;273:17643–50. doi: 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]
- 43.O’Brien RL, Allison JL, Hahn FE. Evidence for intercalation of choloroquine into DNA. Biochim Biophys Acta. 1966;129:622–4. doi: 10.1016/0005-2787(66)90078-5. [DOI] [PubMed] [Google Scholar]
- 44.Fulda S, et al. Betulinic acid triggers CD95(APO-1/Fas)-and-p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997;57:4956–64. [PubMed] [Google Scholar]
- 45.Pisha E, et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat Med. 1995;1:1046–51. doi: 10.1038/nm1095-1046. [DOI] [PubMed] [Google Scholar]
- 46.Bottero V, et al. Activation of nuclear factor κB through the IKK complex buy the topoisomerase poisons SN38 and doxorubicin: a brake to apoptosis in HeLa human carcinoma cells. Cancer Res. 2001;61:7785–91. [PubMed] [Google Scholar]
- 47.Meng L, Ding J. Induction of bulk and e-myc P2 promoter specific DNA damage by an anti-topoisomerase II agent salvicine is an early event leading to apoptosis in HL-60 cells. FEBS Lett. 2001;501:59–64. doi: 10.1016/s0014-5793(01)02633-3. [DOI] [PubMed] [Google Scholar]
- 48.Andoh T. Signal transduction pathways leading to cell cycle arrest and apoptosis induced by DNA topoisomerase poisons. Cell Biochem Biophys. 2000;33:181–8. doi: 10.1385/CBB:33:2:181. [DOI] [PubMed] [Google Scholar]
- 49.Lee N, et al. Programmed cell death in the unicellular protozoan parasite Leishmania. Cell Death Differ. 2002;9:53–64. doi: 10.1038/sj.cdd.4400952. [DOI] [PubMed] [Google Scholar]