

Abstract
Mo-catalyzed endo-cycloisomerizations of alkynes tethered to alcohols have been studied using density functional theory, and comparisons were made with the W-catalyzed reaction. The cycloisomerization is initiated with the formation of metal alkyne π complex and is followed by the rate-determining step, which transforms the π complex to a vinylidene carbene complex, considered to be critical for endo-mode cyclization. Several different alkynols have been selected to investigate five- and six-membered ring endo-cycloisomerizations in the presence of Mo(CO)5 catalyst. The energy barriers calculated for five- and six-membered ring cycloisomerizations are within a range of 25–30 kcal/mol for most cases studied, showing no significant energy difference between the two metals. The stabilization effect of THF and Et3N solvents and the substitution reaction of THF by alkynol substrates in the reaction process with Mo and W complexes are studied as well. The principal difference between Mo- and W-catalyzed cycloisomerization processes appears to be the initial formation of a π complex, which is more stabilizing for formation of the W-alkyne vs Mo-alkyne complexes.
1. Introduction
During the past decade, a new family of endo-cycloisomerizations of terminal alkynes tethered to alcohol,1 nitrogen,2 carbon,3 and sulfur nucleophiles4 has been investigated using metal carbonyl catalysts of group VI metals. A principal factor in the reaction design for endo-cyclization involves intramolecular nucleophilic addition to a transition metal vinylidene intermediate, via in situ rearrangement from a terminal alkyne π complex, thus favoring formation of endocyclic enol ether products from alkynyl alcohols. In contrast, exo-mode cyclizations are generally observed when the metal-alkyne π complex is either unable to rearrange to metal vinylidene (for instance, with internal alkynes) or if the rate of nucleophilic addition to one carbon of the π complex is faster than the rate of rearrangement to the metal vinylidene complex (for instance, with higher valent Lewis acidic reagents or catalysts).5 Experimental evidence for the intermediacy of a metal carbene anion for cycloisomerization reaction in the presence of a Mo- or W-pentacarbonyl complex prompted a reaction mechanism proposed in Scheme 1.6 After initial rearrangement of the metal alkyne π complex to the vinylidene carbene complex, base-induced cyclization of the alcohol nucleophile then affords the cyclic anionic intermediate, which further provides the endocyclic enol ether products by protonation of the metal—carbon bond. These advances in the field of synthetic methodology have paved the way to the efficient synthesis of antiviral nucleosides, polycyclic ethers, and oligosaccharides.7,8
Scheme 1.

Mechanism of the Catalytic, endo-Selective Alkynol Cycloisomerization Reaction, Proposed in Refs 6 and 12, and Modified in This Paper
Molybdenum pentacarbonyl was the first catalyst developed for endo-selective alkynol cycloisomerization to five-membered ring dihydrofurans.1a,b The active catalyst, (Et3N)Mo(CO)5, is best prepared by photolysis of Mo(CO)6 in a mixture of diethyl ether and triethylamine.9 The Mo-catalyzed cycloisomerization proceeds at room temperature by addition of the alkynol substrate after removal of the light source. The reaction is critically dependent on the presence of triethylamine, whereas solvent such as tetrahydrofuran (THF) or acetonitrile inhibits the reaction.
For the analogous endo-cycloisomerization to sixmembered ring endocyclic enol ethers (dihydropyrans), the molybdenum-catalyzed transformation is generally ineffective. A stoichiometric tungsten-promoted alkynol cyclization to the six-membered ring oxacarbene was originally developed,10 which has since been supplanted by a tungsten-catalyzed cyclization process (10–25 mol% W(CO)6 involving photolysis at 350 nm, at or near the reflux point of THF, 50–65 °C, in the presence of the alkynol substrate and triethylamine).1c
The differences between the chemistry of molybdenum and tungsten carbonyl catalysts in alkynol cycloisomerization transformations are a matter of considerable interest to the experimental chemist. According to the available experimental analysis, the Mo-catalyzed reaction is limited to 4-hydroxy-1-alkyne substrates to give five-membered ring products (Scheme 1, n = 1) and is mostly successful for secondary alkynols, whereas primary alkynols and 5-hydroxy-1-alkynes (n = 2) are generally unreactive.1,6 This is contrary to the W-promoted cycloisomerization, where primary, secondary, and tertiary 5-hydroxy-1-alkyne substrates (n = 2) all undergo the reaction proceeding to a six-membered ring product1c-e,10 (Scheme 1). Further experimental data have also revealed that six-membered ring cycloisomerization in the presence of Mo catalyst is generally unsuccessful and the substrate 4-pentyn-1-ol is recovered essentially unchanged.11
Our recent density functional study on one of the possible mechanisms of tungsten-catalyzed endo-selective cycloisomerizations of 4-pentyn-1-ol in the gas phase has shown that pentacarbonyl tungsten catalyst significantly lowers the barrier of the rate-determining step of this specific pathway relative to the metal-free transformation.12 Figure 1 (from ref 12 and modified in this paper) has summarized potential energy profiles of the tungsten-catalyzed process. According to this study, the rate-determining step of this mechanism of tungsten-catalyzed endo-selective cycloisomerization of 4-pentyn-1-ol in the gas phase is the alkyne-alkylidene isomerization. The calculations have shown that this process occurs with 52.5 kcal/mol activation barrier in the absence of catalyst, while the inclusion of the pentacarbonyl tungsten catalyst lowers it by half to 26.4 kcal/mol. This result has been explained by the role of the tungsten center in stabilizing the lone pair electrons at the terminal Cα while alkynyl hydrogen is migrating to the internal Cβ. It has also been shown that further steps to complete cyclization occur with low activation barriers.
Figure 1.

Potential energy profile of endo-cycloisomerization reactions of 4-pentyn-1-ol taken from ref 12 and modified in this paper. Zero-point energy corrections are not included into the reported energies.
An important goal is to discern reasons for the differences of the catalytic activity observed between the Mo and W complexes. As a first step, in the present paper we report the structure and energetics of the key structures on the potential energy surface of molybdenum-catalyzed endo-selective cycloisomerization of 4-pentyn-1-ol in the gas phase. For simplicity, we studied the same mechanism reported previously for the W-catalyzed reaction.12 To understand substrate dependency of this mechanism, we made a systematic comparison between the Mo-catalyzed cycloisomerization of 3-butyn-1-ol (primary alkynol), 1-phenyl-3-butyn-1-ol (secondary alkynol), and acetylene and pent-4-ynyl-carbamic acid (both nonalcohol substrates). We have also carefully analyzed the alkyne–alkylidene rearrangement step, which we previously reported was the rate-determining step of one of the possible mechanisms of gas-phase tungsten-catalyzed endo-selective cycloisomerization of 4-pentyn-1-ol.
2. Methods of Calculation
All calculations were performed with the hybrid density functional method B3LYP using the Gaussian-03 program.13 The B3LYP method uses Becke's 3-parameter hybrid functional,14 which mixes a nonlocal exchange functional with the exact HF exchange functional and Lee–Yang–Parr's nonlocal correlation functional.15 In this calculation we use the Lanl2dz basis set,16 which combines Hay–Wadt's relativistic effective core potential (ECP) for Mo with a valence double-ξ basis set and the 6-31G basis set for main group elements. It was previously shown that the B3LYP/Lanl2dz approximation used in this paper provides reasonable agreement with available experimental data and high-level methods in analogous systems.17 To rationalize the basis set effect, we have repeated all the calculations at the B3LYP level in conjunction with the Stuttgart/Dresden ECP18 and associated triple-ξ SDD basis set for Mo and 6-31G(d) basis set for main group elements. The results presented in the Supporting Information clearly indicate that the improvement of basis sets from Lanl2dz to SDD+6-31G(d) has no significant effect on the calculated geometries and energetics of the reactants, intermediates, transition states, and products. Since previously the mechanism of alkynol cycloisomerization catalyzed by W-carbonyl was reported only at the B3LYP/Lanl2dz level, below, to be able to compare the mechanisms of Mo- and W-carbonyl-catalyzed alkynol cycloisomerization reactions, we report only B3LYP/Lanl2dz-calculated results for Mo systems.
Harmonic vibration frequencies were calculated for each optimized structure to identify the nature of the structure. Zero-point energies were calculated on the basis of harmonic frequencies (at 298.15 K temperature and 1 atm pressure) and were included in the reported energies, unless explicitly stated. Throughout the paper, numbers presented without parentheses are enthalpies, and those in parentheses are Gibbs free energies. Pseudo-IRC calculations19 (10 steps of IRC followed by optimization) were performed for some key transition states to confirm the connectivity of reactants and products.
Previously12 the enthalpy and Gibbs free energies of the reaction of W(CO)6 with 4-pentyn-1-ol were not reported. For the sake of completeness of our comparison of W(CO)6- and Mo(CO)6-catalyzed 4-pentyn-1-ol cycloisomerization, in this paper we have calculated those missing values for some specific steps of the tungsten-catalyzed reaction at the same level of theory used for the Mo systems described above.
The bulk-solvent effects were evaluated at the polarizable continuum model (PCM)20 level by utilizing the gas-phase-optimized geometries and diethyl ether as a solvent media. Zero-point energy, enthalpy, and Gibbs free energy corrections were estimated from the gas-phase studies and included to the PCM-calculated solvation energy.
3. Results and Discussion
A. Generation of Active Catalyst by CO Elimination from the M(CO)6 Complexes. To have a better understanding of catalytic activities of different transition metal carbonyl complexes, the knowledge of metal–carbonyl strength is important. Experimental studies show that photolysis of Mo(CO)6 at 350 nm in the presence of triethylamine leads to the formation of (Et3N)Mo(CO)5,9 which is proposed to be the active catalyst in the cycloisomerization reaction. However, a time-resolved infrared absorption spectroscopy following pulsed excimer laser photolysis of vapor-phase Mo(CO)6 has shown the possibility of a second CO dissociation in the case of a lower wavelength irradiation.21
Therefore, in this paper we investigated the energetics of the first and second CO dissociation from the Mo(CO)6, leading to Mo(CO)5 and Mo(CO)4, respectively. The calculated results are collected in Table 1. A comparison of the energetics of the reactions M(CO)6 → M(CO)5 + CO and M(CO)5 → M(CO)4 + CO for M [unk] Mo and W shows that the first and second Mo—CO bond dissociation energies are 8.2 (8.0) and 8.7 (8.5) kcal/mol lower than the respective W—CO bond energies. The lower CO dissociation energies from the Mo-carbonyl complexes compared to W-analogues can be explained in terms of the fact that the relativistic effects significantly (more than 4d orbitals) destabilize the 5d orbitals, reduce the dπ−πC−O* gap, and thus enhance the back-donation interaction in the case of tungsten.17l,22
Table 1.
CO Dissociation Energy and Metal—C and C—O Bond Lengths for Mo(CO)n and W(CO)n, n = 6, 5, and 4
| compound | sym | ΔE (kcal/mol) |
R(M—C) (Å) |
R(C—O) (Å) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYPa | MP2b | CCSD(T)b | exptl | B3LYP | MP2b | exptlc | B3LYPa | MP2b | exptlc | ||
| Mo(CO)6 | Oh | 39.8 | 46.1 | 40.4 | 40.5 ± 2d | 2.060 | 2.066 | 2.063 | 1.175 | 1.164 | 1.145 |
| (29.3) | 38.2 (PZC) | 35 ± 5e | |||||||||
| Mo(CO)5 | C4v | 39.7 | 27 ± 5e | eq 2.055 | eq 1.177 | ||||||
| (29.4) | |||||||||||
| ax 1.945 | ax 1.186 | ||||||||||
| Mo(CO)4 | C2v | 1.934 | 1.189 | ||||||||
| 2.049 | 1.180 | ||||||||||
| W(CO)6 | Oh | 48.0 | 54.9 | 48.0 | 2.051f | 2.054 | 2.058 | 1.177f | 1.166 | 1.148 | |
| (37.3) | 45.7 (PZC) | ||||||||||
| W(CO)5 | C4v | 77.2f | 46.0 ± 2d | eq 2.042f | eq 1.179f | ||||||
| 48.4 | ax 1.942f | ax 1.188f | |||||||||
| (37.9) | |||||||||||
| W(CO)4 | C2v | 1.928 | 1.192 | ||||||||
| 2.033 | 1.182 | ||||||||||
| D4hf | 2.025f | 1.184f | |||||||||
ΔH and ΔG (in parantheses) calculated in this study. Structures for W-complexes are taken from footnote f.
Ehlers, A. W.; Frenking, G. J. Am. Chem. Soc. 1994, 116, 1514. At MP2-optimized geometries, both geometries and energies with the basis set HW3DZ2P (441/2111/41) for metal and 6-31G(d) for CO.
Jost, A.; Rees, B. Acta Crystallogr. 1975, B31, 2649, Ganske, J. A.; Rosenfeld, R. N. J. Phys. Chem. 1990, 94, 4315.
Lewis, K. E.; Golden D. M.; Smith G. P. J. Am. Chem. Soc. 1984, 106, 3905.
Frenking, G.; Pidun, U. J. Chem. Soc., Dalton Trans. 1997, 1653.
Sheng, Y.; Musaev, D. G.; Reddy, K. S.; McDonald, F. E.; Morokuma, K. J. Am. Chem. Soc. 2002, 124, 4149–4157.
B. Mo(CO)5- and Mo(CO)4-Catalyzed 4-Pentyn-1-ol Cycloisomerization in the Gas Phase: The Alkyne–Alkylidene Rearrangement Mechanism. As mentioned above, in the present paper we report the structure and energetics of the key structures on the potential energy surface of molybdenum-catalyzed endo-selective cycloisomerization of 4-pentyn-1-ol in the gas phase. In our studies we pursue the same mechanism of the reaction reported previously for the W-catalyzed reaction.12 The optimized structures for the key intermediates and transition states of this reaction are shown in Figure 2, and the corresponding potential energy surface is presented in Figure 3. In Figure 4 we compare the initial steps of the Mo- and W-catalyzed endo-selective cycloisomerization of 4-pentyn-1-ol in the gas phase.
Figure 2.
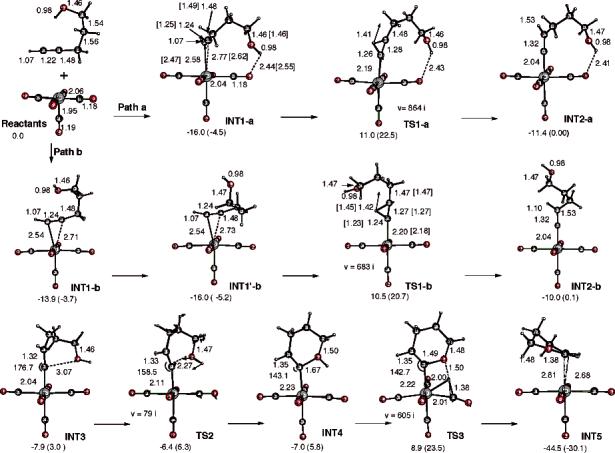
B3LYP/Lanl2dz-optimized geometries (in Å and deg) and relative energies (ΔH and ΔG (in parentheses) in kcal/mol, relative to Mo(CO)5 + 4-pentyn-1-ol dissociation limit) of the reactants, intermediates, and transition states of endo-cycloisomerization of 4-pentyn-1-ol in the presence of the Mo(CO)5 catalyst. The parameters given in brackets are for endo-cycloisomerization of 4-pentyn-1-ol in the presence of the W(CO)5 catalyst. For the transition states, the reaction coordinate vector and imaginary frequency (in cm−1) are also shown.
Figure 3.
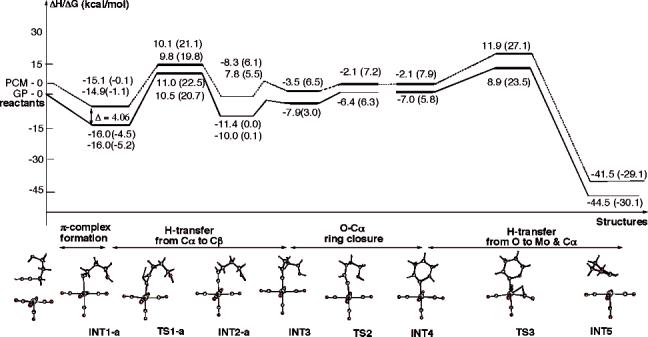
ΔH-scaled potential energy profile of endo-cycloisomerization of 4-pentyn-1-ol catalyzed by Mo(CO)5 in gas phase (solid line) and condensed phase (broken line). ΔG values are reported in parentheses.
Figure 4.

Comparison of the energetics of initial steps of Mo- (solid line) and W-catalyzed (broken line) endo-cycloisomerization of 4-pentyn-1-ol. The given potential energy surface is ΔH scaled, while ΔG values are given in parentheses.
As expected, the first step of the reaction is the coordination of substrate to the transition metal center, leading to the structure INT1. Depending on the position of the hydroxyl group with respect to the acetylenic hydrogen, two optimized conformational structures for the initial η2 complex have been found. In INT1-a, the hydroxyl group forms a weak hydrogen bond with one of the carbonyl oxygen atoms with . In INT1-b, the hydroxyl group is far from the CO groups, and no hydrogen bond is made. As a consequence, two different pathways either through transition state TS1-a or through TS1-b are identified for the hydride migration step from Cα to Cβ, leading to alkylidene complexes INT2-a and INT2-b. There are some major geometrical changes associated with the hydride migration process. The Mo—Cβ is breaking going from INT1 to INT2 structures. The Mo—Cα distance decreases from the single bond of 2.58 Å for INT1-a and 2.54 Å for INT1-b to a double bond of about 2.04 Å for INT2-a and INT2-b. The Cα—Cβ distance increases from 1.24 Å to 1.32 Å, changing from a C[unk]C triple bond in INT1 structures to a C[unk]C double bond in INT2 structures. The bond angle ∠Mo—Cα—Cβ also changes from 85° to 180°. Comparison of η2 complexes of Mo and W indicates that W—CR and W—Cβ distances are about 5% shorter for Mo species.
In contrast to the complexation of 4-pentyn-1-ol with W(CO)5, where the complexation energy is calculated to be 21.2 (9.4) kcal/mol, the corresponding energetics for Mo(CO)5 are only 16.0 (4.5) and 13.9 (3.7) kcal/mol for INT1-a and INT1-b, respectively. Thus, the complexation energy of 4-pentyn-1-ol with W(CO)5 is about 5.2 (4.9) kcal/mol larger than that with Mo(CO)5. This is consistent with the observation that unsuitable substrates for the Mo-catalyzed cycloisomerization reaction are generally recovered unchanged, presumably due to facile reversibility of π complex formation, whereas with the tungsten-catalyzed process, formation of the π complex appears to be irreversible, as the alkyne substrate cannot be recovered once the π complex has been formed.
The next step of the reaction is proposed to be the alkyne–alkylidene rearrangement, which occurs via the transition state TS1. The calculations show that the only imaginary frequency of 864i cm−1 for transition state TS1-a and of 683i cm−1 for TS1-b correspond to nearly pure hydride migration. While the IRC calculation from TS1-a confirmed the connectivity between INT1-a and INT2-a in the reaction coordinate, the IRC from TS1-b, as shown in Figure 2, leads to INT1′-b, which is not the direct π complex from the reactants: the direct π complex from the reactants is INT1-b. Obviously some conformational isomerization has to be considered to complete reaction path b. However, such isomerizations usually occur with a low barrier, and therefore no further study on details of conformational changes has been attempted. The hydride migration barriers calculated from the lowest π complexes are nearly identical for paths a and b: 27.0 (27.0) and 26.5 (25.9) kcal/mol, respectively. Therefore, both pathways would compete in the reaction. These barriers for the experimentally unsuccessful cycloisomerization reaction of 4-pentyn-1-ol in the presence of Mo(CO)5 are about 3.4 (5.1) and 2.9 (4.0) kcal/mol larger than the activation energy for the successful reaction of the same alkynol in the presence of W(CO)5 (see Figure 4).
As seen in Figures 2 and 3, the hydride migration is endothermic by 4.6 (4.5) and 6.0 (5.1) kcal/mol for paths a and b, respectively, which is similar to 4.3 (3.0) kcal/mol for the tungsten-catalyzed reaction, reported earlier.12
The next step of the reaction is the ring closure, which may occur with involvement of a tertiary amine base under experimental conditions. Nevertheless, our gas-phase calculation with no base involvement shows low activation energy at the TS2 of 1.5 (3.3) kcal/mol calculated from INT3. (INT3 can be reached from INT2-a or -b by conformational isomerization. We did not study this process, as a low barrier is expected from our previous study on the W system.) The TS2 has an imaginary frequency of 79i cm−1, which corresponds to the reaction coordinate for ring closure; IRC calculations show that the TS2 connects INT3 and INT4. The calculated Cα—O distance in TS2 is 2.27 Å. Calculations indicate three major geometrical changes in this six-membered ring closure. First, the Cα—O distance decreases from 3.07 Å in INT3 to 1.67 Å in INT4, showing the bond is almost formed. Second, the bond angle ∠Mo—Cα—Cβ changes from 176.7° to 143.1°. Third, the Mo—Cα distance lengthens from double bond 2.04 Å to single bond 2.23 Å, suggesting the involvement of the π* component of the Mo[unk]Cα and the lone pair of the O center to form the Cα—O bond.
The further process to final products proceeds via a transition state structure TS3 bearing one imaginary frequency (605i cm−1) for O—H bond stretching, leading to a structure with stretched O—H and C—H bonds of 1.50 and 1.38 Å and a Mo—H bond of 2.00 Å. As a consequence of this hydride migration, the Cα—O distance shortening from 1.67 Å in INT4 to 1.49 Å in TS3 further stabilizes the formed ring. IRC calculations confirm that TS3 connects INT4 with the final product INT5. Going from INT4 to INT5, hydride migrates from the hydroxyl group to Cα and the η1 coordination of the Cα[unk]CβH fragment rearranges to η2 coordination to Mo. Careful analysis of IRC steps shows at first hydride moves to ca. 1.32 Å from the C with a Mo—H of 1.97 Å. Then, the ring gradually starts rotating up to ca. 90°, elongating the C—H and H—O bonds to ca. 1.45 and 3.3 Å, respectively. At this stage, the hydride begins migration to Cα, shortening the H—Cα bond from 2.25 Å to 1.08 Å, where η1–η2 rearrangement takes place. Overall, hydride migration from the hydroxyl group to Mo and Cα takes place in concert with ring rotation without intermediates. This has also been confirmed by optimization of the slightly disturbed TS3 structure (geometry of the first step of the IRC forward) that converged to INT5. Attempts to locate any intermediate between TS3 and INT5 were also unsuccessful. This is contrary to the W-catalyzed potential surface, where W–H complexes WN11 and WN12 were located as high-energy intermediates. The entire endo-cycloisomerization reaction is calculated to be exothermic by 44.5 (30.1) kcal/mol.
Since the CO dissociation from Mo(CO)5 is calculated to be as facile as from the Mo(CO)6, we have also studied the 4-pentyn-1-ol cycloisomerization reaction catalyzed by Mo(CO)4 (C2v symmetry). As the rate-determining step of the Mo(CO)5-catalyzed 4-pentyn-1-ol cycloisomerization is the alkyne-alkylidene rearrangement step, we carried out calculations only for the formation of the π complex (INT1) and its transformation to the alkylidene complex (INT2). The key intermediates and transition state structures are shown in the Supporting Information (Figure S1), and their energetics are given in Table 2. Calculations show that the Mo—Cα and Mo—Cβ bond distances are shortened in theπ complex (2.17 and 2.22 Å) in comparison with the analogous complex of Mo(CO)5 (2.54 and 2.73 Å), thus indicating a stronger bond between Mo and carbon atoms of the substrate. The bond angle ∠Cα—Mo—Cβ is also about 10° larger than the same one in Mo(CO)5. The activation barrier for the transformation of the π complex to the carbene complex is calculated to be 35.1 (33.9) kcal/mol, about 8–9 kcal/mol higher than that for the reaction of Mo(CO)5, suggesting that Mo(CO)4 is not likely to be the active catalyst for 4-pentyn-1-ol cycloisomerization via the alkyne–alkylidene rearrangement mechanism.
Table 2.
Enthalpy and Gibbs Free Energies (in parentheses) of the π Complexes, Alkylidene Intermediate, and Transition State (TS1) of the Reaction of M(CO)n (where M = Mo and W, and n = 5 and 4) with Alkynes, Calculated Relative to the Reactants
| reactants | enthalpy (Gibbs free energies) |
activation energya | ||
|---|---|---|---|---|
| π complex | TS for H-migration | alkylidene complex | ||
| W(CO)5 + 4-pentyn-1-olb | −21.2 (−9.4) | 2.4 (12.5) | −16.9 (−6.4) | 23.6 (21.9) |
| Mo(CO)5 + 4-pentyn-1-ol (path a) | −16.0 (−4.5) | 11.0 (22.5) | −11.4 (0.0) | 27.0 (27.0) |
| Mo(CO)5 + 4-pentyn-1-ol (path b) | −16.0 (−5.2) | 10.5 (20.7) | −10.0 (0.1) | 26.5 (25.9) |
| Mo(CO)4 + 4-pentyn-1-ol (path b) | −26.2 (−14.6) | 8.9 (19.3) | −14.8 (−4.1) | 35.1 (33.9) |
| Mo(CO)5 +1-phenyl-3-butyn-1-ol | −18.9 (−5.7) | 6.5 (17.3) | −13.2 (−2.6) | 25.4 (23.0) |
| Mo(CO)5 + 3-butyn-1-ol (path b) | −16.2 (−5.4) | 9.1 (19.7) | −12.7 (−2.7) | 25.3 (25.1) |
| W(CO)5 + acetyleneb | −21.5 (−11.3) | 8.2 (17.4) | −27.1 (−16.8) | 29.7 (28.7) |
| Mo(CO)5 + acetylene | −15.8 (−6.1) | 15.9 (24.8) | −16.8 (−6.7) | 31.7 (30.9) |
| Mo(CO)5 + pent-4-ynyl-carbamic acid | −23.8 (−10.4) | 5.5 (17.0) | −16.1 (−4.3) | 29.3 (27.4) |
The presented activation energies are energy differences between the π complexes and transition states. All values are in kcal/mol.
Structures are taken from: Sheng, Y.; Musaev, D. G.; Reddy K. S.; McDonald, F. E.; Morokuma, K. J. Am. Chem. Soc. 2002, 124, 4149–4157.
C. Mo(CO)5-Catalyzed 1-Phenyl-3-butyn-1-ol and 3-Butyn-1-ol Cycloisomerization in the Gas Phase: The Alkyne–Alkylidene Rearrangement Mechanism. To understand substrate dependency of alkynol cycloisomerization catalyzed by Mo-carbonyl complexes, we have studied the same alkyne–alkylidene rearrangement mechanism of the Mo-catalyzed cycloisomerization of 3-butyn-1-ol (primary alkynol), 1-phenyl-3-butyn-1-ol (secondary alkynol), acetylene, and pent-4-ynyl-carbamic acid (both nonalcohol substrates). The obtained structures of the key intermediates and transition states are given in the Supporting Information (Figures S2–S5), while their energies are collected in Table 2.
Let us begin discussions with the 3-butyn-1-ol and 1-phenyl-3-butyn-1-ol substrates. Experimentally, the secondary alcohol, 1-phenyl-3-butyn-1-ol, undergoes catalytic cycloisomerization in the presence of Et3N/Mo(CO)5 with a significant yield (>80%) of 2-phenyl-2, 3-dihydrofuran,23 whereas 3-butyn-1-ol is recovered essentially unchanged under the same conditions.4 Therefore, it is reasonable to expect significant differences in the energetics of their reactions with the Mo-carbonyl complex. The only significant difference between these two substrates is observed in the relative energies for formation of the initial π complex (Table 2), where the complex from 1-phenyl-3-butyn-1-ol is approximately 2.7 (0.3) kcal/mol more stable than the complex from the parent 3-butyn-1-ol. The calculated results show the subsequent rate-determining alkyne–alkylidene rearrangement barriers of 25.4 (23.0) and 25.3 (25.1) kcal/mol for 1-phenyl-3-butyn-1-ol and 3-butyn-1-ol reactions, respectively. We also note that comparing the π complexes for 3-butyn-1-ol and 1-phenyl-3-butyn-1-ol shows that the Mo—Cα and Mo-Cβ bond distances are slightly elongated (ca. 0.03–0.05 Å) in the presence of an electron-withdrawing phenyl group on C4.
For a better comparison of the effect of nucleophiles, we also calculated the energetics of the formation of π complex and a Mo-vinylidene complex for two other simple model substrates, specifically acetylene and pent-4-ynyl-carbamic acid, which bears a NH—COOH group instead of the hydroxyl group as nucleophile. Reaction of acetylene and Mo(CO)5 shows an almost identical energy barrier compared with the analogous step in W-catalyzed reaction. The geometrical parameters are also very similar to those for the W-catalyzed case. For pent-4-ynyl-carbamic acid, the energy barrier for alkyne–alkylidene transformation is calculated to be 29.3 (27.4) kcal/mol, approximately about 2.3 (0.4) kcal/mol higher than for the similar step with 4-pentyn-1-ol. This is attributed to the more stable π complex provided by a hydrogen bond interaction between the hydroxyl group and one CO group.
D. Effect of Solvent. According to the experiments, solvent plays a major role in Mo- and W-catalyzed alkynol cyclizations. Therefore, below we discuss the influence of the solvent effects on the energetics of the Mo-catalyzed 4-pentyn-1-ol cycloisomerization, which takes place in the presence of diethyl ether. The energies of reactants, transition states, intermediates, and products of this reaction are evaluated at the polarizable continuum model (PCM) level by utilizing the gas-phase-optimized geometries and diethyl ether as a solvent media. The PCM-calculated energetics are compared with the corresponding gas-phase values in Figure 3. A comparison of these data shows no significant changes in the relative energies of the species upon going from gas phase to solution. The energy barrier for the rate-determining step at the transition state TS1 is reduced by about 1.0 kcal/mol, which is mostly due to the additional destabilization of the π complex in the presence of solvent.
Furthermore, it has been shown that THF inhibits the Mo-catalyzed reaction, while it is generally used for W-catalyzed cycloisomerizations. Although the elucidation of the role of solvent effects in the Mo- and W-catalyzed alkynol cyclization reactions needs separate and comprehensive investigations, we have studied the possible first step: the coordination/decoordination of solvent molecule on the vacant site of the transition metal center. In our studies we model this process with single tetrahydrofuran (THF) and triethylamine (Et3N) molecules. The optimized structures of the (THF)Mo(CO)5, (THF)W(CO)5, (Et3N)Mo(CO)5, and (Et3N)W(CO)5 complexes are shown in the Supporting Information (Figure S6). The calculated coordination energies of THF to Mo(CO)5 and W(CO)5 are 24.5 (11.7) and 29.6 (16.5) kcal/mol, respectively. These values for Et3N are 17.9 (3.3) and 23.6 (8.6) kcal/mol.
The initial reaction of the solvated Mo(CO)5 or W(CO)5 is expected to be substitution of the solvent molecule by an alkyne substrate molecule, which can occur via either an associative path through a substitution transition state or a dissociative path through departure of a solvent molecule and coordination of the substrate to the M(CO)5 unit. The calculated energetics in both paths with 4-pentyn-1-ol as substrate and THF as solvent molecule, as well as the optimized substitution transition state structures, are shown in Figure 5. The enthalpies of the associative transition states are found to be 11.4 and 14.9 kcal/mol for Mo(CO)5 and W(CO)5, respectively. Although Gibbs free energies increase these barriers by about 8–9 kcal/mol, the activation energies of 20.0 and 23.8 kcal/mol for Mo(CO)5 and W(CO)5 are still lower than the barriers for the alkyne–alkylidene rearrangement process. The calculated barriers for the dissociative path are 24.5 (11.7) and 29.6 (16.5) kcal/mol for Mo(CO)5 and W(CO)5, respectively. These will be further reduced, because the solvent molecule dissociated from the metal center will in reality interact with other solvent molecules and be stabilized. Therefore, the solvent–substrate exchange is not likely to be the rate-determining step for the reported alkyne–alkylidene mechanism of 4-pentyn-1-ol cyclizations catalyzed by Mo- and/or W-carbonyl complexes. The calculated exchange barrier for the Mo complex is lower than that for the W complex. However, this result does not explain the inhibition effect of THF in Mo cycloisomerization.
Figure 5.
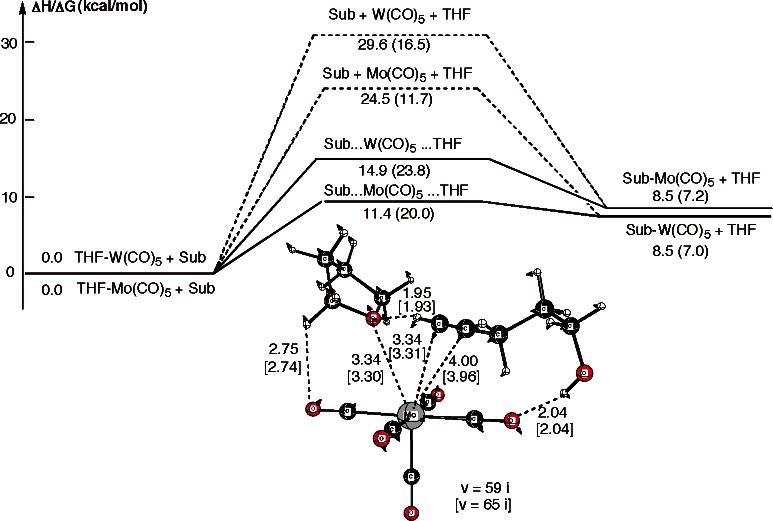
ΔH-scaled potential energy profiles for dissociative (broken line) and associative substitution (solid line) reaction in THF solvent by 4-pentyn-1-ol in the presence of Mo(CO)5 and W(CO)5 complexes. ΔG values are given in parentheses. The optimized structure of the transition state (in Å and deg), its reaction coordinate vector, and associated frequency are also shown. Values given in brackets are (THF)W(CO)5 systems.
4. Conclusions
From the above-presented discussion of the alkynol cyclization via the alkyne–alkylidene rearrangement mechanism catalyzed by Mo- and/or W-carbonyl complexes, we can draw the following conclusions.
(1) The first step of the reaction is CO dissociation from the M(CO)6 species, which occurs with 39.8 and 48.0 kcal/mol enthalpy for M = Mo and W, respectively. Although the CO dissociation from the Mo(CO)5 occurs with almost the same energy as that from the M(CO)6 species, calculations of full potential energy surfaces of Mo(CO)5-, Mo(CO)4-, (THF)M(CO)5-, and (Et3N)M(CO)5- catalyzed cycloisomerization reactions clearly indicate that the catalytically active species is only M(CO)5 or its derivatives such as (THF)M(CO)5 and/or (Et3N)M-(CO)5.
(2) One of the principal factors for catalyst–substrate combinations that undergo successful endo-selective cycloisomerizations appears to be the stability of the initial π complex, which is suitably stabilized for tungsten–alkyne π complexes, but only for a limited number of molybdenum–alkyne π complexes. While the formation of the π complex is not the rate-determining step, this does affect the energy of the transition state for subsequent rearrangement to a vinylidene complex intermediate relative to the uncomplexed alkynol substrate.
(3) The rate-determining alkyne–alkylidene rearrangement barrier for 4-pentyn-1-ol is found to be 27.0 (27.0) (for path a) and 26.5 (25.9) (for path b) kcal/mol for the Mo-catalyzed cycloisomerization reaction, which is 3.4 (5.1) and 2.9 (4.0) kcal/mol larger than the activation energy for the successful reaction of the same alkynol in the presence of W(CO)5.
(4) The alkyne–alkylidene rearrangement on the (THF)M(CO)5 and/or (Et3N)M(CO)5 complexes begins with the substitution of a solvent molecule with the substrate, which is found to proceed via an associative (or substitution) transition state with an 11.4 (20.0) and 14.9 (23.8) kcal/mol activation barrier for Mo(CO)5 and W(CO)5, respectively. Since the barrier of the consequent alkyne–alkylidene rearrangement step is almost twice as large as this solvent–substrate substitution barrier, one may expect that the alkyne–alkylidene rearrangement step will be the rate-determining step of the entire process even in solution.
(5) Calculations show almost similar activation energies for primary alkynol, 3-butyn-1-ol (25.3 (25.1) kcal/mol), and for secondary alkynol, 1-phenyl-3-butyn-1-ol (25.4 (23.0) kcal/mol), indicating almost no significant substrate dependency for the reactions studied.
However, the above presented results and conclusions are related to only one of the possible mechanisms of alkynol cyclization by Mo- and W-carbonyl catalysts, namely, the alkyne–alkylidene rearrangement mechanism, and do not fully include the solvent effects. Our comprehensive studies on the other possible mechanisms and the role of the explicit solvent molecules in this fascinating reaction are in progress and will be reported later.
Supplementary Material
Acknowledgment
The present research is in part supported by an NSF grant (CHE-0209660) to K.M. and D.G.M., and an NIH grant (CA-59703) to F.E.M. Computer resources were provided in part by the Air Force Office of Scientific Research DURIP grant (FA9550-04-1-0321) as well as by the Cherry Emerson Center for Scientific Computation at Emory University.
Footnotes
Supporting Information Available: The optimized stationary point structures (in Å and deg) and relative energies (relative to Mo(CO)4 + 4-pentyn-1-ol) on the reaction pathways of endo-cycloisomerization of 4-pentyn-1-ol in the presence of Mo(CO)4 (Figure S1), of 1-phenyl-3-butyn-1-ol in the presence of the Mo(CO)5 catalyst (Figure S2), of 3-butyn-1-ol in the presence of Mo(CO)5 (Figure S3), of acetylene in the presence of Mo(CO)5 (Figure S4), and of pent-4-ynylcarbamic acid in the presence of Mo(CO)5 (Figure S5). Stabilization energy comparison for Mo(CO)5 or W(CO) in the presence of THF or NEt3 (Figure S6). Geometries and relative energies of the reactants, intermediates, transition states, and products calculated at the B3LYP/SDD+6-31G(d) level (Figure S7). Tables of Cartesian coordinates of all the optimized structures (Tables S1–S7). Complete ref 13.
References
- 1.(a) McDonald FE, Connolly CB, Gleason MM, Towne TB, Treiber KD. J. Org. Chem. 1993;58:6952. [Google Scholar]; (b) McDonald FE, Gleason MM. J. Am. Chem. Soc. 1996;118:6648. [Google Scholar]; (c) McDonald FE, Reddy KS, Díaz Y. J. Am. Chem. Soc. 2000;122:4304. [Google Scholar]; (d) McDonald FE, Reddy KS. Angew. Chem., Int. Ed. 2001;40:3653. doi: 10.1002/1521-3773(20011001)40:19<3653::aid-anie3653>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]; (e) Alcázar E, Pletcher JM, McDonald FE. Org. Lett. 2004;6:3877. doi: 10.1021/ol0483495. [DOI] [PubMed] [Google Scholar]
- 2.McDonald FE, Chatterjee AK. Tetrahedron Lett. 1997;38:7687. [Google Scholar]
- 3.(a) McDonald FE, Olson TC. Tetrahedron Lett. 1997;38:7691. [Google Scholar]; (b) Maeyama K, Iwasawa N. J. Am. Chem. Soc. 1998;120:1928. [Google Scholar]; (c) Miura T, Iwasawa N. J. Am. Chem. Soc. 2002;124:518. doi: 10.1021/ja0113091. [DOI] [PubMed] [Google Scholar]
- 4.McDonald FE, Burova SA, Huffman LG. Synthesis. 2000:970. [Google Scholar]
- 5.(a) Riediker M, Schwartz J. J. Am. Chem. Soc. 1982;104:5842. For representative examples of exo-mode cyclizations of alkynyl alcohols, including discussions of the regioselectivity, see. Hg. [Google Scholar]; (b) Utimoto K. Pure Appl. Chem. 1983;55:1845. Pd. [Google Scholar]; (c) Pale P, Chuche J. Tetrahedron Lett. 1987;28:6447. Ag. [Google Scholar]; (d) Villemin D, Goussu D. Heterocycles. 1989;29:1255. Hg. [Google Scholar]; (e) Luo F-T, Schreuder I, Wang R-T. J. Org. Chem. 1992;57:2213. Pd. [Google Scholar]; (f) Seiller B, Bruneau C, Dixneuf PH. Tetrahedron. 1995;51:13089. Ru, Pd. [Google Scholar]; (g) Gabriele B, Salerno G. J. Chem. Soc., Chem. Commun. 1997:1083. Pd/Cu. [Google Scholar]; (h) Cacchi S, Fabrizi G, Moro L. J. Org. Chem. 1997;62:5327. Pd. [Google Scholar]; (i) Asao N, Noyami T, Takahashi K, Yamamoto Y. J. Am. Chem. Soc. 2002;124:764. doi: 10.1021/ja017366b. Pd. [DOI] [PubMed] [Google Scholar]; (j) Wipf P, Graham TH. J. Org. Chem. 2003;68:8798. doi: 10.1021/jo034813s. W. [DOI] [PubMed] [Google Scholar]
- 6.(a) McDonald FE, Schultz CC. J. Am. Chem. Soc. 1994;116:9363. [Google Scholar]; (b) McDonald FE, Schultz CC, Chatterjee AK. Organometallics. 1995;14:3628. [Google Scholar]
- 7.(a) McDonald FE. Chem. Eur. J. 1999;5:3103. [Google Scholar]; (b) Bruneau C, Dixneuf PH. Chem. Rev. 1999;99:311. [Google Scholar]; (c) Alonzo F, Beletskaya IP, Yus M. Chem. Rev. 2004;104:3079. doi: 10.1021/cr0201068. [DOI] [PubMed] [Google Scholar]
- 8.(a) Trost BM, Rhee YH. J. Am. Chem. Soc. 2003;125:7482. doi: 10.1021/ja0344258. For other catalysts for alkynol cycloisomerization, see. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Rhee YH. Org. Lett. 2004;6:4311. doi: 10.1021/ol048165q. [DOI] [PubMed] [Google Scholar]
- 9.(a) Gray HB, Wrighton M, Hammond GS. J. Am. Chem. Soc. 1971;93:4336. [Google Scholar]; (b) Stiegman AE, Stieglitz M, Tyler DR. J. Am. Chem. Soc. 1983;105:6032. [Google Scholar]
- 10.McDonald FE, Bowman JL. Tetrahedron Lett. 1996;37:4675. [Google Scholar]
- 11.McDonald FE, Zhu HYH. Tetrahedron. 1997;53:11061. For an example of alkynol cycloisomerization where Mo(CO)5 catalysis was modestly successful in six-membered ring formation, see. [Google Scholar]
- 12.Sheng Y, Musaev DG, Reddy KS, McDonald FE, Morokuma K. J. Am. Chem. Soc. 2002;124:4149. doi: 10.1021/ja017668d. [DOI] [PubMed] [Google Scholar]
- 13.Frisch MJ, et al. Gaussian 03. Gaussian, Inc.; Wallingford, CT; 2004. Revision C.02. [Google Scholar]
- 14.(a) Becke AD. J. Chem. Phys. 1993;98:5648. [Google Scholar]; (b) Becke AD. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 15.Lee C, Yang W, Parr RG. Phys. Rev. B. 1998;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 16.(a) Dunning TH., Jr. In: Modern Theoretical Chemistry. Schaefer HF III, editor. Plenum; New York: 1976. pp. 1–28. [Google Scholar]; (b) Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:270. [Google Scholar]; (c) Wadt WR, Hay PJ. J. Chem. Phys. 1985;82:284. [Google Scholar]; (d) Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:299. [Google Scholar]
- 17.(a) Cui Q, Musaev DG, Svensson M, Sieber S, Morokuma K. J. Am. Chem. Soc. 1995;117:12366. [Google Scholar]; (b) Musaev DG, Morokuma K. J. Phys. Chem. 1996;100:6509. [Google Scholar]; (c) Erikson LA, Pettersson LGM, Siegbahn PEM, Wahlgren U. J. Chem. Phys. 1995;102:872. [Google Scholar]; (d) Ricca A, Bauschlicher CW., Jr. J. Phys. Chem. 1994;98:12899. [Google Scholar]; (e) Heinemann C, Hertwig RH, Wesendrup R, Koch W, Schwarz H. J. Am. Chem. Soc. 1995;117:495. [Google Scholar]; (f) Hertwig RH, Hrusak J, Schroder D, Koch W, Schwarz H. Chem. Phys. Lett. 1995;236:194. [Google Scholar]; (g) Schroder D, Hrusak J, Hertwig RH, Koch W, Schwerdtfeger P, Schwarz H. Organometallics. 1995;14:312. [Google Scholar]; (h) Fiedler A, Schroder D, Shaik S, Schwarz H. J. Am. Chem. Soc. 1994;116:10734. [Google Scholar]; (i) Fan L, Ziegler T. J. Chem. Phys. 1991;95:7401. [Google Scholar]; (j) Berces A, Ziegler T, Fan L. J. Phys. Chem. 1994;98:1584. [Google Scholar]; (k) Lyne PD, Mingos DMP, Ziegler T, Downs A. J. Inorg. Chem. 1993;32:4785. [Google Scholar]; (l) Li J, Schreckenbach G, Ziegler T. J. Am. Chem. Soc. 1995;117:486. [Google Scholar]
- 18.(a) Dolg M, Wedig U, Stoll H, Preuss H. J. Chem. Phys. 1987;86:866. [Google Scholar]; (b) Schwerdtfeger P, Dolg M, Schwarz WH, Bowmaker GA, Boyd PDW. J. Chem. Phys. 1989;91:1762. [Google Scholar]; (c) Andrae D, Haubermann U, Dolg M, Stoll H, Preuss H. Theor. Chim. Acta. 1990;77:123. [Google Scholar]; (d) Bergner A, Dolg M, KŸchle W, Stoll H, Preuss H. Mol. Phys. 1993;80:1431. [Google Scholar]
- 19.Fukui K. Acc. Chem. Res. 1981;14:363. [Google Scholar]
- 20.(a) Tomasi J, Persico M. Chem. Rev. 1994;94:2027. [Google Scholar]; (b) Tomasi J, Cammi R. J. Comput. Chem. 1996;16:1449. [Google Scholar]
- 21.Ganske JA, Rosenfeld RN. J. Phys. Chem. 1990;94:4315. [Google Scholar]
- 22.Ehlers AW, Ruiz-Morales Y, Baerends EJ, Ziegler T. Inorg. Chem. 1997;36:5031. [Google Scholar]
- 23.McDonald FE, White BH. Org. Synth. 2002;79:27. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.