

Abstract
Alzheimer’s disease (AD) is characterized by neurofibrillary tangles and by the accumulation of β-amyloid (Aβ) peptides in senile plaques and in the walls of cortical and leptomeningeal arteries as cerebral amyloid angiopathy (CAA). There also is a significant increase of interstitial fluid (ISF) in cerebral white matter (WM), the pathological basis of which is largely unknown. We hypothesized that the accumulation of ISF in dilated periarterial spaces of the WM in AD correlates with the severity of CAA, with the total Aβ load in the cortex and with Apo E genotype. A total of 24 AD brains and 17 nondemented age-matched control brains were examined. CAA was seen in vessels isolated from brain by using EDTA-SDS lysis stained by Thioflavin-S. Total Aβ in gray matter and WM was quantified by immunoassay, ApoE genotyping by PCR, and dilatation of perivascular spaces in the WM was assessed by quantitative histology. The study showed that the frequency and severity of dilatation of perivascular spaces in the WM in AD were significantly greater than in controls (P < 0.001) and correlated with Aβ load in the cortex, with the severity of CAA, and with ApoE ɛ4 genotype. The results of this study suggest that dilation of perivascular spaces and failure of drainage of ISF from the WM in AD may be associated with the deposition of Aβ in the perivascular fluid drainage pathways of cortical and leptomeningeal arteries. This failure of fluid drainage has implications for therapeutic strategies to treat Alzheimer’s disease.
INTRODUCTION
Alzheimer’s disease (AD) is characterized clinically by dementia and pathologically by the accumulation of ubiquitinated tau and other proteins within neurons and by deposition of β-amyloid (Aβ) in the extracellular compartments of gray matter (GM) and in blood vessel walls as cerebral amyloid angiopathy (CAA) (1–3). Tracer studies in experimental animals have shown that in CAA, Aβ accumulates within the perivascular channels around capillaries and arteries that have been identified as interstitial fluid (ISF) drainage pathways (4–11). Biochemical determinations have shown that it is the levels of soluble Aβ in Alzheimer brains, rather than the insoluble Aβ plaque load, that correlate with severity of cognitive decline (12,13). These findings support the hypothesis that the failure to eliminate Aβ along ISF drainage pathways is a major factor in Aβ accumulation in the cortex and in the AD pathogenesis.
In addition to the pathological changes in cerebral gray matter (GM) in AD, there is significant pathology in the underlying cerebral white matter (WM). This is characterized by a relative increase in ISF detectable through imaging and by histological techniques (14). There is some evidence that vascular insufficiency may be involved in the pathogenesis of WM changes in AD (15). However, there also is the distinct possibility that deposition of Aβ in periarterial ISF drainage pathways may interfere with the drainage of ISF from cerebral WM particularly as dilation of peri-arterial spaces (état criblé) is a feature of white matter in AD.
Cerebral cortex and underlying WM both receive their blood supply from branches of leptomeningeal arteries on the surface of the brain. Extensive studies by Duvernoy and others (16) have shown that arteries supplying the cerebral hemispheres penetrate the cortex perpendicular to the surface and exhibit various branching patterns and capillary fields within the cortex. Some arteries supply the WM branch as they pass through the cortex, but others do not. The arteries supplying the WM, therefore, are derived from the same leptomeningeal arteries that supply the overlying cortex and exhibit prominent CAA in AD.
CAA is a feature of a number of familial diseases and is present in the brains of the majority if not all patients with AD (15). The pattern of deposition of Aβ in capillary and artery walls correlates with the ISF drainage pathways for interstitial fluid (17). A major risk factor for CAA is possession of the ApoE ɛ4 genotype (18).
In the present study, we test the hypothesis that accumulation of fluid in dilated periarterial ISF pathways in the WM in AD correlates with severity of CAA, total Aβ load in the overlying cortex, and Apo E genotype. In order to test this hypothesis, we employed a novel strategy to analyze the topology of cerebrovascular amyloid that took advantage of the extreme insolubility and resistance to disruption of amyloid deposits. This technique involves the combined action of EDTA-SDS lysis and allows detailed analysis of entire tufts of blood vessels spanning the GM. Total Aβ peptide in GM and WM was quantified by immunoassay and ApoE genotyping by PCR. The dilation of perivascular spaces in the WM was assessed by quantitative histology.
MATERIALS AND METHODS
Human Subjects—Brain Tissue
Brain tissue was obtained from the Brain Donation Program of the Sun Health Research Institute. The cases were chosen to provide a mixture of Apo E genotypes in both control nondemented (ND) and AD groups. Characteristics of the cases in this study are given in Table 1 and include age, Apo E genotype, density of neocortical neuritic plaques, Braak stage, and the National Institute of Aging (NIA)-Reagan Institute diagnostic status. The terms “high” and “intermediate” reflect the probability that symptoms of dementia were due to AD. The mean age of ND cases was 83.2 y, whereas that of AD cases was 82.2 y. Nondemented cases (n = 17) comprised 8 females and 9 males while the AD cohort (n = 24) contained 11 females and 13 males. Mean postmortem intervals were 2.6 h for ND subjects and 2.8 h for AD subjects (difference not significant on 2-tailed unpaired t-test). Leptomeninges were dissected from the brain surface in the immediate post-mortem period and stored at −80 ºC. Cerebral hemispheres were cut in the coronal plane as 8- to 10-mm thick slices. Right hemisphere slices were frozen between sheets of dry ice and stored at −80 ºC.
Table 1.
Study Subjects
| Case ID | Age/Sex | ApoE | White matter score | Plaque density | Break stage | NIA-Reagan Score |
|---|---|---|---|---|---|---|
| Control cases | ||||||
| 1 | 94/F | 3/3 | 1 | Mod | III | ND |
| 2 | 94/M | 3/3 | 1 | Mod | III | ND |
| 3 | 85/M | 3/3 | 1 | None | II | ND |
| 4 | 86/F | 3/3 | 0 | None | III | ND |
| 5 | 89/F | 3/3 | 1 | Mod | IV | ND |
| 6 | 76/F | 3/3 | 1 | Mod | I | ND |
| 7 | 84/M | 3/3 | 0 | Mod | III | ND |
| 8 | 78/M | 3/3 | 1 | None | I | ND |
| 9 | 92/M | 3/3 | 0 | Sparse | III | ND |
| 10 | 74/M | 3/3 | 0 | Mod | IV | ND |
| 11 | 86/M | 3/3 | 1 | None | II | ND |
| 21 | 80/F | 3/4 | 1 | Mod | III | ND |
| 22 | 83/F | 3/4 | 1 | Mod | II | ND |
| 23 | 86/F | 3/4 | 1 | Mod | III | ND |
| 24 | 85/F | 3/4 | 0 | None | III | ND |
| 25 | 69/M | 3/4 | 1 | Sparse | I | ND |
| 26 | 73/M | 3/4 | 0 | None | III | ND |
| Alzheimer’s disease cases | ||||||
| 51 | 93/F | 3/3 | 0 | Freq | VI | High |
| 52 | 60/F | 3/3 | 1 | Freq | VI | High |
| 53 | 90/F | 3/3 | 2 | Freq | V | High |
| 54 | 96/F | 3/3 | 3 | Mod | V | High |
| 55 | 80/F | 3/3 | 3 | Freq | VI | High |
| 56 | 85/F | 3/3 | 2 | Freq | V | High |
| 61 | 91/M | 3/4 | 2 | Freq | V | High |
| 62 | 87/M | 3/4 | 0 | Freq | V | High |
| 63 | 60/M | 3/4 | 0 | Freq | VI | High |
| 64 | 84/M | 3/4 | 1 | Freq | V | High |
| 65 | 89F | 3/4 | 3 | Freq | V | High |
| 66 | 87/M | 3/4 | 2 | Freq | V | High |
| 71 | 79/M | 4/4 | 1 | Mod | V | High |
| 72 | 81/F | 4/4 | 0 | Freq | VI | High |
| 73 | 79/F | 4/4 | 3 | Freq | IV | Int |
| 74 | 68/M | 4/4 | 1 | Freq | VI | High |
| 75 | 77/M | 4/4 | 0 | Freq | VI | High |
| 76 | 87/M | 4/4 | 2 | Mod | IV | Int |
| 77 | 79/M | 4/4 | 2 | Freq | V | High |
| 78 | 80/M | 4/4 | 3 | Freq | VI | High |
| 79 | 80/F | 4/4 | 1 | Freq | VI | High |
| 80 | 93/F | 4/4 | 2 | Freq | VI | High |
| 81 | 84/M | 4/4 | 1 | Freq | VI | High |
| 82 | 83/M | 4/4 | 1 | Freq | VI | High |
Mod, moderate; Freq, frequent; Int, intermediate; ND, nondemented.
Tissue Processing
Left hemisphere slices were fixed in 4% paraformaldehyde for 48 h and then dissected into small tissue blocks for paraffin embedding or larger blocks for microtome sectioning. Paraffin blocks were sectioned at 5 μm and stained with hematoxylin and eosin (H&E). Frozen sections (40 μm) were cut after cryoprotection in 2% dimethyl sulphoxide and 20% glycerol using a sledge-type microtome, stained with H&E for general morphology and observation of WM. The lesions of AD were visualized using the Campbell-Switzer and Gallyas stains. Brain sections also were stained with 1% aqueous (wt/vol) Thioflavin-S (Sigma, St. Louis, MO, USA) and rinsed 10 times with 70% ethanol to remove unbound fluorochrome. The sections were mounted with Apathy’s media and observed in an epifluorescent microscope.
Neuropathological Tissue and Patient Grading
Rarefaction of the WM in each cerebral lobe was graded on the fraction of the centrum semiovale affected in thick (40 μm) quarter-hemisphere sections stained with H&E (mild < 25%; moderate 25% to 50%; severe > 50%). Individual percentages for each lobe were converted to a numeric code (mild = 1; moderate = 2; severe = 3) from which a mean score for each brain was obtained. Neuritic plaque density in cerebral cortex was graded according to criteria established by the Consortium to Establish a Registry for Alzheimer’s Disease (1). Neurofibrillary tangles were staged according to the method of Braak (2). The diagnosis of AD was assigned whenever a subject was demented and had at least a “moderate” plaque density. In addition, the protocol developed by the NIA and the Reagan Institute (3) was used to assign to each demented case a probability estimate that dementia was due to AD.
Transmission Electron Microscopy
Cerebral cortex was taken fresh postmortem (1.2 h) from 3 AD individuals, cut into small fragments (about 1 mm3), and processed for electron microscopy. The specimens were fixed in 3% buffered glutaraldehyde and placed in 4% unbuffered osmium tetroxide. The blocks were stained with 1% aqueous uranyl acetate for 30 min, embedded in epon araldite resin, thin-sectioned, and treated with lead citrate. The specimens were examined by a Phillips 400 electron microscope at 80 kV.
Apolipoprotein E Genotyping
Genomic DNA was extracted from 50 mg of cerebellar tissue and amplified by PCR using allele-specific oligonucleotide probes spanning positions 112 and 158 of the Apo E gene sequence. The PCR products were HhaI-restricted, and the fragments were separated by polyacrylamide electrophoresis. Genotypes were distinguished on the basis of differential HhaI band fragment mobilities as described by Hixson and Vernier (19).
Quantification of Cortical Aβ by Immunoassay
Cerebral cortex fractions containing Aβ were purified by fast performance liquid chromatography, and Aβ peptide was quantified by immunoassay as described previously (20).
Preparation of Leptomeninges
Leptomeningeal tissue was prepared by partially transecting the larger vessels to facilitate the release of entrapped blood. The specimens then were rinsed 20 times with cold distilled water containing 0.01% sodium azide to promote hemolysis and prevent contamination. Blood vessels larger than 1-mm diameter were removed. The cleaned, colorless membranes of leptomeninges and the vascular network were then spread on disposable 15-cm Petri dishes and dried in an oven at 50 ºC for 18 h. The leptomeninges were stained with 1% aqueous Thioflavin-S, as described above.
Preparation of Cerebral Cortex Vascular Tufts
From a slice of cerebrum (parietal lobe), approximately 8- to 10-mm thick, the GM was dissected from the underlying WM and sectioned into small cubes measuring about 4 to 5 mm. A total of 25 pieces of cerebral cortex from each individual case were stirred for approximately 24 h in 1 liter of lysis buffer (5% sodium dode-cyl sulfate, 50 mM Tris HCl, 3 mM ethylenediaminetetraacetic acid [EDTA], pH 7.4). After 24 h, pieces of the tissue were allowed to settle and the lysis buffer decanted. A 2nd liter of lysis buffer was added, and after complete brain tissue lysis, the white tufts of blood vessels were isolated by filtration through a nylon mesh (0.12 mm) mounted on a 10-cm diameter plastic embroidery hoop. Those vessels retained on the nylon mesh were immediately released by vigorous shaking of the nylon mesh in 1 liter of distilled water containing 0.01% sodium azide. The vessel preparations were filtered twice more using the same technique. After settling at room temperature for 16 h, the water phase was removed by aspiration and the vascular suspension, approximately 30 mL in volume, was poured onto a Petri dish. The vascular tufts were removed from the Petri dish and spread onto a glass microscope slide. The blood vessel preparations were air dried at 60 ºC for 6 h, fixed in absolute ethanol for 30 min, rinsed with distilled water, and Thioflavin-S stained. The stained preparations were observed using an epifluorescence microscope (Zeiss Axiophot) equipped with a 450 to 490 nm excitation filter and a 520 nm barrier filter.
Morphometric Assessment of État Criblé–like Lesions
Brain tissue sections stained with H&E were viewed at 25× magnification with the Leica DMLB microscope system (Leica Microsystems AB, Ernst-Leitz-Strasse, Wezlar, Germany) with a 100 W transmitted light source. The whole area of each slide was scanned for evidence of dilated perivascular spaces in the WM; their outlines were digitized with an Optronics Magnafire SP camera (model s99805) using the Optronics Magnafire SP software program (Optronics, Goleta, CA) and stored in TIF format. All measurements were carried out using Image-Pro Express software v. 4.0 (Media Cybernetics, Silver Springs, MD), which was calibrated to 25× magnification. The diameter of each enlarged perivascular space, that is, larger than 1 mm in perimeter, and the blood vessel within were measured using the straight line tool and computer mouse. The data were transferred to Microsoft Excel®, and the mean ratios of perivascular space diameters to blood vessel diameters for each case were calculated and then grouped by ApoE genotype and diagnosis.
RESULTS
Cerebral Amyloid Angiopathy
A detailed analysis of entire tufts of cerebral vessels spanning the GM of the cerebral cortex was performed on preparations of cortex after complete cell lysis by the combined action of EDTA-SDS. No vascular amyloid was detected by Thioflavin-S–staining in control brains, but amyloid deposits were present in AD brains. All vessels less than 500 μm in diameter had amyloid deposits (Figure 1A and 1B). The deposits of amyloid tend to disappear in larger leptomeningeal arteries. The arteriolar and capillary vessels demonstrated a unique abundance of discreet ‘core-like’ amyloid deposits firmly attached to the vessel wall and, in many cases, the cores appeared to be fused together (see Figure 1C and 1D). All the spherical amyloid cores associated with capillaries displayed a brush-like surface. On average, there was a moderate amount of amyloid associated with the vessels of the cerebral cortex in AD patients with ApoE ɛ3/ɛ3 and ɛ3/ɛ4 genotypes. However, the most severely affected individuals, in terms of amyloid load, were those with ApoE ɛ4/ɛ4 genotype (see Figure 1). Whole mount preparations of all ɛ4/ɛ4 AD subjects revealed an overwhelming amount of vascular amyloid in the cerebral cortex. Electron microscopy of the GM arteries showed a virtual disappearance of smooth muscle cells from artery walls containing large amounts of amyloid and obliteration of the perivascular spaces (Figures 1A and 2B, 2C, and 2D).
Figure 1.

Thioflavin-S–stained whole-mounted tufts of cortical vessels after complete brain parenchyma lysis by EDTA-SDS. A: Ring-like amyloid deposits in leptomeningeal arterial walls. The pattern of deposition follows the orientation of the smooth muscle cells. Magnification 25×. B: Tufts of cortical-penetrating arteries heavily loaded with amyloid from an AD patient with Apo E ɛ4/ɛ4 genotype. The perivascular spaces are probably occluded in the saturated amyloid vessels, and in some cases may be totally destroyed by the encroaching amyloid, thus hindering the elimination of interstitial fluid. Magnification 100×. C: Cortical arterioles and capillaries showing an abundant deposition of amyloid cores, in an individual with Apo E ɛ4/ɛ4 genotype, at different stages of development that are intimately linked to the basal laminae of the vessels. The profuse deposits of amyloid at the arteriolar/capillary junction may block the openings of the perivascular spaces that drain the brain’s interstitial fluid. Magnification 200×. D: At a higher magnification, each of the fluorescent blebs represents a fully developed globular deposit of amyloid evenly spread around and constricting the microvessel, probably creating areas of ischemia and alterations in blood-brain barrier. Magnification 400×.
Figure 2.

Electron micrographs of a normal cortical artery and of arteries destroyed by amyloid deposition in 3 AD cases homozygous for Apo E ɛ4/ɛ4. A: An electron micrograph of a small normal cortical artery. The endothelial cells are joined by tight junctions (small arrows) that participate in the blood-brain barrier. Both their nuclei and thin cytoplasm have a normal morphology. These cells are in contact with the subjacent basal lamina (opposing large arrows). The smooth muscle cells (white arrows) have a normal-looking fibrillar cytoplasm, and a typical nucleus is seen on the left margin of the photograph. The thin periarterial space (black arrowheads) contains the extracellular matter of the adventitia and is on its outer margin limited by a single layer of pial cells (8). Magnification 5500×. B and C: Small cortical arteries surrounded by heavy amyloid deposition. The endothelial cells are apparently swollen. The tight junctions have an abnormal morphology (black arrows). The dense amyloid deposits are fused with the basal lamina of the vascular cells. The myocytes have vanished or are reduced to a collection of cellular debris (white arrows) that is enclosed by concentric layers and peripheral wisps of amyloid surrounded by a large amount of tissue debris (open stars). The tunica adventitia, perivascular spaces, and the glia limitans have degenerated as the result of heavy amyloid deposition. Magnification 4300× and magnification 3500×, respectively. D: An abnormal cortical artery with abundant fine wisps of amyloid perpendicular to the main axis of the vessel. The amyloid bundles, interspersed by large amounts of cellular debris, pushed out the glia limitans (open arrows). On the left margin of the micrograph, the amyloid has destroyed the glia limitans invading the surrounding cortical tissue (black stars). All myocytes and the tunica adventitia have been replaced by amyloid fibrils and debris. Magnification 2000×. One may assume from the extensive vascular pathology that the delivery of oxygen and nutrients as well as arterial contractility are largely compromised.
In AD cases, most of the arteries in the leptomeninges, up to a diameter of 500 μm, demonstrated a heavy load of ring-like deposits similar to those seen in the cerebral cortical arteries (see Figure 1A). However, in those individuals with ApoE ɛ3/ɛ3 and ɛ3/ɛ4 genotypes, the distribution of amyloid in the leptomeningeal vessels was segmental; that is, portions of artery with a heavy load of amyloid were interspersed with lengths of vessel that were free of amyloid. As in the cerebral cortex, those individuals with ApoE ɛ4/ɛ4 genotypes had a more prominent amount of vascular amyloid involving almost all the small (< 500 μm) leptomeningeal arteries. As the leptomeningeal arteries increased in diameter, the amyloid load gradually declined to isolated streaks associated with the more peripheral layers of the tunica media. The large branches of the circle of Willis were entirely devoid of microscopically visible (Thioflavin-S positive) amyloid. Occasionally in the leptomeninges, areas of old microhemorrhages were revealed by the presence of hemosiderin deposits along vessels burdened with amyloid (data not shown).
Dilation of Perivascular Spaces in AD
Macroscopic inspection of brain sections from ND controls (Figure 3A) and from individuals with AD (see Figure 3B) revealed a large number of dilated perivascular spaces in the WM of AD cases. The dilated perivascular spaces were more prominently displayed by light microscopy (Figure 4). Very few dilated perivascular spaces were seen in control material. Areas of rarefaction in the WM were more abundant in AD than in the control cases. As shown in Table 1, the average WM rarefaction score was significantly higher in the AD cases (mean = 1.5, range 0 to 3) than in the control cases (mean = 0.65, range 0 to 1; P = 0.004).
Figure 3.
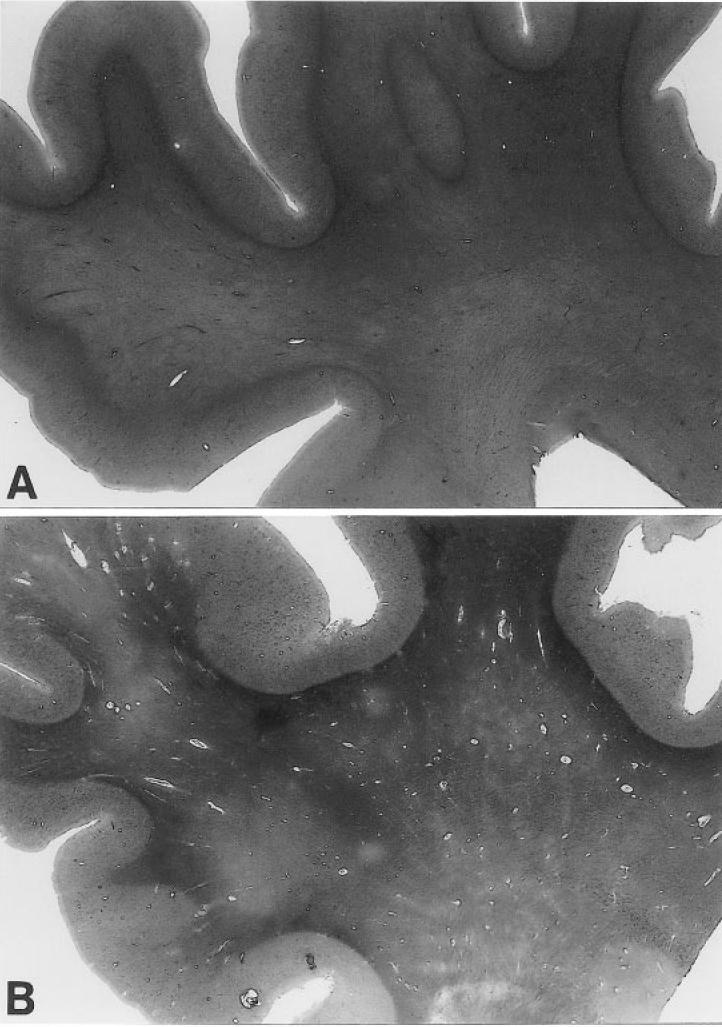
Hematoxylin- and eosin-stained superior frontal gyrus and underlying WM sections from an ND individual and an AD case. Magnification: about 2.5×. A: Cerebral section from a 74-y-old ND individual with ApoE ɛ3/ɛ3 genotype, who died as the result of metastatic prostate cancer. The WM appears homogeneously stained without noticeable dilated periarterial spaces. B: Shows a section from an 80-y-old AD patient with ApoE ɛ4/ɛ4 genotype. The numerous arteries with enlarged perivascular spaces are evident throughout the entire extent of the WM, whereas the GM is comparatively free of these alterations. The paler blotches in the WM represent areas of myelin rarefaction.
Figure 4.

Perivascular spaces in Alzheimer’s disease white matter. Row A: The fine periarterial spaces are not normally visible at light microscopic level. Magnifications: left and center 100×; right 200×. Row B: The periarterial spaces appear to be slightly dilated. Magnifications: left 200×; center and right 100×. Rows C, D, and E demonstrate a series of severely distended perivascular spaces. The centrally located arteries apparently have a normal morphology. All captions were taken at 100×. All histological slides were stained with hematoxylin and eosin.
Quantitation of Aβ and Dilated Perivascular Spaces in the White Matter
Quantitation of total cortical Aβ (Aβ 40 and Aβ 42) by immunoassay from ApoE ɛ4/ɛ4 AD subjects yielded an average of 54 μg/g of tissue with 2 of 3 of the Aβ peptides ending at residue 42; this is supported by our previous observations (21). Total Aβ in the cortex quantified by immunoassay in the ApoE ɛ3/ɛ4 and ɛ3/ɛ3 cohorts showed mean values of 26 and 35 μg/g of tissue, respectively, with a predominance of Aβ 42. In these cases, core-like and ring-like amyloid deposition prevailed, resembling the type 2 deposits described by Thal and others (22). In contrast to the AD subjects, the total Aβ in control individuals carrying the ApoE ɛ3/ɛ3 and ɛ3/ɛ4 genotypes only amounted to 3 and 4 μg/g of tissue, respectively.
Total Aβ in GM of AD patients was 12 times greater than in control cases (Figure 5A). The amount of Aβ was significantly higher in the AD patients with ApoE ɛ4/ɛ4 than in those AD individuals with ApoE ɛ3/ɛ4 and ɛ3/ɛ3 genotypes (see Figure 5A). Likewise, the number of perivascular spaces in WM as well as the ratio of the diameters of perivascular spaces over the diameters of centrally located blood vessels were significantly higher in the AD cohort when compared with ND controls (see Figure 5B and 5C). When the AD cohorts were grouped by ApoE genotype, there was a positive correlation between the average number of enlarged perivascular spaces and the average cortical concentration of Aβ (see Figure 5D). The ratio of the perivascular space diameter over blood vessel diameter also had a significant positive correlation with the cortical Aβ concentration (see Figure 5E) and the number of enlarged perivascular spaces (see Figure 5F). In contrast to the ND population, the AD cohort had an increased load of cortical Aβ, a higher count of distended perivascular spaces (état criblé–like lesions). In addition, the distended perivascular spaces were larger relative to the size of their respective blood vessels. Furthermore, individuals with the ApoE ɛ4/ɛ4 genotype have more of the aforementioned pathology than those with 1 or no ApoE ɛ4 alleles.
Figure 5.

Quantitative and statistical analyses among the 3 variables investigated in this study: cortical Aβ concentrations, number of dilated perivascular spaces (état criblé–like lesions), and ratio of perivascular space diameter/blood vessel diameter. In the 3 left panels, ▪ and □ histograms represent the total numbers of ND and AD individuals and these populations divided by Apo E genotype, respectively. The 3 panels on the right side of the figure demonstrate the linear correlations and P values among the 3 studied variables. A: The total levels of cortical Aβ in ND and AD subjects. P value represents the 2-tailed unpaired Student t-test probability. * and ** represent P < 0.05 and P < 0.01, respectively, in post-hoc analysis (Newman-Keuls Multiple Comparison Test) after 1-way analysis of variance demonstrated that there was a significant difference among the AD subgroups. B: The average number of WM enlarged perivascular spaces (état criblé–like lesions) in the ND and AD groups. P value represents the 2-tailed unpaired Student t-test probability. C: Mean ratios of perivascular space diameter/blood vessel diameter observed in ND and AD cohorts. P value represents the 2-tailed unpaired Student t-test probability. D: Linear correlation (R = 0.97) between the mean number of WM enlarged perivascular spaces and average levels of cortical Aβ when the AD and control subjects were grouped by Apo E genotype. E: Linear correlation between the ratio of perivascular space diameter to blood vessel diameter and the total levels of cortical Aβ (R = 0.55; P < 0.001). F: Linear correlation between the mean number of WM enlarged perivascular spaces and the ratio of perivascular space diameter to blood vessel diameter (R = 0.70; P < 0.001).
Relationship Between Dilated Perivascular Spaces and Cerebral Atrophy
We also explored the possibility that enlargement of perivascular spaces may result from retraction of WM due to overall brain atrophy. Because there is a significant difference in brain size between genders, these cohorts were evaluated separately. No correlation was found between brain weight and the average number of enlarged perivascular spaces in the WM or between brain weight and the average ratio of perivascular space diameter to blood vessel diameter.
DISCUSSION
Our results suggest that there is a correlation between the severity of cortical and leptomeningeal CAA, the amount of Aβ in the cerebral cortex, and the accumulation of ISF in dilated perivascular spaces (état criblé–like lesions) in AD WM. Conventional histological techniques do not allow full evaluation of CAA. By stripping the leptomeningeal arteries from the cortex and then removing all traces of blood and Thioflavin-S staining, a clear picture of the extent of amyloid involvement of cerebral vessels emerges. Leptomeningeal arteries with CAA have a ring-like pattern of Aβ deposition that follows the orientation of smooth muscle cells within the media. A similar pattern is seen in the larger cortical arteries. A ring-like distribution of Aβ in which whole lengths of vessel are virtually replaced by Aβ is seen in smaller arteries and arterioles. Core-like deposits of Aβ form in capillary walls, and they are often arranged in confluent rows. It is difficult to assess the effect of arterial and capillary CAA on the blood supply of the cerebral cortex. We observed that some of the capillaries are occluded by amyloid deposits. In other studies, capillary lumina remained patent even in the presence of heavy deposition of Aβ in the capillary basement membrane (17). Even if occlusion did occur, extensive capillary anastomoses exist within the cortex (16), making any physiological effects difficult to predict.
Accumulation of Aβ in the cerebral cortex and cerebral vessels correlates with dilation of perivascular spaces in the underlying WM. Thus the number of dilated perivascular spaces in Alzheimer brains is significantly greater than in ND individuals, and the number of dilated spaces is proportional to the level of Aβ in the overlying cortex and associated arteries. Furthermore, Aβ concentrations in the cortex were higher for ApoE ɛ4/ɛ4 cases than in other ApoE genotypes. Total Aβ load in the WM was much higher in Alzheimer’s disease than in ND individuals, (20) which suggests that Aβ elimination from the WM is reduced in AD.
In the present and previous investigations, we have observed that the amount of Aβ deposited along the cortical and leptomeningeal vessels directly correlates with Apo E genotype (12). In AD, the relative amount of Aβ in these vessels decreases as follows: ɛ4/ɛ4 >> ɛ3/ɛ3 > ɛ3/ɛ4. It appears that slowly evolving occlusion of the perivascular spaces in the cortical and leptomeningeal arteries results in dilation of perivascular spaces in the WM, that is, état criblé, and subclinical chronic edema of the WM parenchyma. The extent of the edema is related to tissue compliance, which is greater in the WM than in the GM and greater in the deep WM than in the superficial WM (23). In humans, the WM of the centrum semiovale is supplied by long penetrating medullary arteries that are a continuation of cortical perforating vessels derived from the leptomeningeal arteries (16) and by perforating arteries directly derived from the circle of Willis (24).
Little attention has been paid in the past to the effects of CAA on WM in AD despite the evidence from Duvernoy and others (16) that branches of leptomeningeal arteries supply the cerebral WM as well as the cortex. Although CAA is rarely seen in arteries in the WM, impedance of ISF flow from the WM still could occur due to involvement of leptomeningeal and cortical arteries by CAA. It is of interest for the future development of immunotherapy for Alzheimer’s disease that some of the patients treated by Aβ immunization develop severe rarefaction and significant increase of ISF in the WM. In 1 post-mortem case, the WM edema was accompanied by severe CAA (25).
État criblé has been classically described as the consequence of dilation of perivascular spaces due to enlargement, distortion, and formation of loops in cerebral arterioles and small arteries that result from aging, hypertension, or a combination of both factors. In our AD cohort, we rarely observed the characteristic thickening and hyalinosis of artery walls commonly seen in the WM as the état criblé associated with hypertension (14). Rather, in the état criblé–like condition of AD, most of the WM arteries appear to have a normal configuration.
The état criblé quantified in the present study resembles the dilated perivascular spaces occasionally seen in the WM of younger individuals, with or without an accompanying dementia (26). Dilated perivascular spaces also are seen in the GM of the basal ganglia as état lacunaire but rarely if at all in the cortex. Giant lacunes in the basal ganglia or in the mid-brain may obstruct CSF flow and result in hydrocephalus (27). It has been suggested that such lacunes are due to blockage of ISF drainage pathways (28). The anatomy of perivascular spaces of arteries in the WM appears to be similar to that of arteries in the basal ganglia but differs from cortical vessel (26,28). Thus the structure of the perivascular spaces may play a key role in the preferential formation of état lacunaire in the basal ganglia and état criblé in the WM.
We have found that the WM of AD subjects is in general more fragile and softer than the WM of control elderly individuals, which under pressure has a higher resilience (20). From a biochemical viewpoint, the AD WM has significantly less cholesterol and myelin proteins and an elevated amount of Aβ peptides, even though parenchymal amyloid deposits and obvious perivascular amyloid accumulations are lacking (20). Ultrastructural studies have demonstrated that the AD WM, relative to controls, has a decreased number of axons and myelin levels, which are accompanied by the presence of large astrocytes with swollen-like cytoplasm (B Bohrmann and AE Roher, unpublished results). The astrocytes, by virtue of being interconnected by gap junctions, apparently regulate the amount of water and ions present in the extracellular space of relatively large areas of the brain (29). We postulate that obstruction of ISF drainage caused by deposition of amyloid in cortical and leptomeningeal blood vessel walls not only dilates the perivascular spaces in the WM and produces ISF stasis but also forces the sub-ependymal astrocytes to eliminate excessive amounts of water into the ventricles. The production of CSF by the choroid plexi in a normal adult is around 600 mL/d, whereas in AD patients it is substantially reduced to about 300 mL/d (30). In view of these observations, it is possible that the remarkable enlargement of the lateral ventricles in AD results from, among other factors, the sustained drainage of extra fluid from WM into the lateral ventricles. Thus, the ventricles may act as a sink for interstitial WM fluid that could not be cleared through the amyloid-obstructed perivascular spaces.
In conclusion, the results of this study strongly support our hypothesis that compromised drainage of interstitial fluid from the WM in AD is due to blockage of the cortical and leptomeningeal perivascular spaces by amyloid deposition, thereby causing dilatation of perivascular spaces in the WM (Figure 6). Examination of AD vessel topology has clarified the pattern of vascular amyloid deposition and suggests a possible pathophysiological chain of events. Our analyses suggest that dysfunctional Aβ transport and attendant vascular amyloid deposition results in an impairment of brain fluid dynamics and leads to some of the deleterious changes linked to advanced AD. Fundamental structural changes in the integrity of the arterial tree may be the key factor initiating amyloid deposition (31). As the stability of the microvasculature and the blood-brain barrier declines with normal aging or through ischemic and traumatic injury, amyloid nucleation sites become available in the vasculature. Continued and relentless accretion of Aβ along the cortical and leptomeningeal vessels may result in the ultimate impairment of the entire regional circulation, causing cerebral ischemia, stasis of interstitial fluid, and chronic subclinical edema of WM. These findings underscore the complex and multifactorial nature of AD pathology.
Figure 6.

Diagram suggesting the morphological changes that lead to enlargement of WM perivascular spaces. A: Under normal conditions, the brain’s interstitial fluid is collected by the patent periarterial spaces around the blood vessels of the WM, GM, and leptomeninges. B: The leptomeningeal and perforating cortical arteries supplying the brain GM and WM are surrounded by heavy amyloid deposits that destroy the vascular walls and block the perivascular spaces at the level of the cerebral cortex and leptomeninges. This obstruction results in WM congestion and stasis of interstitial fluid with simultaneous dilation of the WM perivascular spaces. The greater density and rigidity and lesser compliance of the GM matter leave this tissue substantially less affected than the WM.
Acknowledgments
This study was supported by National Institutes of Health grants AG 17490, AG 18345, and NF 38674 and by the State of Arizona Alzheimer’s Research Center.
REFERENCES
- 1.Mirra SS, et al. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II: standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–6. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 3.The National Institute on Aging and Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer’s disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 4.Weller RO. Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol. 1998;57:885–94. doi: 10.1097/00005072-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid-β accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998;153:725–33. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weller RO, Kida S, Zhang ET. Pathways of fluid drainage from the brain—morphological aspects and immunological significance in rat and man. Brain Pathol. 1992;2:277–84. doi: 10.1111/j.1750-3639.1992.tb00704.x. [DOI] [PubMed] [Google Scholar]
- 7.Yamada S, DePasquale M, Patlak CS, Cserr HF. Albumin outflow into deep cervical lymph from different regions of rabbit brain. Am J Physiol. 1991;261:H1197–204. doi: 10.1152/ajpheart.1991.261.4.H1197. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ET, Inman CB, Weller RO. Interrelationships of the pia matter and the perivascular (Virchow-Robin) spaces in the human cerebrum. J Anat. 1990;170:111–23. [PMC free article] [PubMed] [Google Scholar]
- 9.Weller RO, Massey A, Kuo YM, Roher AE. Cerebral amyloid angiopathy: accumulation of Aβ in interstitial fluid drainage pathways in Alzheimer’s disease. Ann NY Acad Sci. 2000;903:110–7. doi: 10.1111/j.1749-6632.2000.tb06356.x. [DOI] [PubMed] [Google Scholar]
- 10.Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol. 1992;2:269–76. doi: 10.1111/j.1750-3639.1992.tb00703.x. [DOI] [PubMed] [Google Scholar]
- 11.Van Dorpe J, Smeijers L, Dewachter I. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am J Pathol. 2000;157:1283–98. doi: 10.1016/S0002-9440(10)64644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lue LF, et al. Soluble amyloid-β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLean CA, et al. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.van Swieten JC, et al. Periventricular lesions in the white matter on magnetic resonance imaging in the elderly: a morphometric correlation with arteriolosclerosis and dilated perivascular spaces. Brain. 1991;114:761–74. doi: 10.1093/brain/114.2.761. [DOI] [PubMed] [Google Scholar]
- 15.Kalaria RN. Advances in molecular genetics and pathology of cerebrovascular disorders. Trends Neurosci. 2001;24:392–400. doi: 10.1016/s0166-2236(00)01836-1. [DOI] [PubMed] [Google Scholar]
- 16.Duvernoy HM, Delon S, Vannson JL. Cortical blood vessels of the human brain. Brain Res Bull. 1981;7:519–79. doi: 10.1016/0361-9230(81)90007-1. [DOI] [PubMed] [Google Scholar]
- 17.Preston SD, Steart PV, Wilkinson A, Nicoll JAR, Weller RO. (2003) Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: defining the perivascular route for the elimination of amyloid-β from the human brain. Neuropathol. Appl. Neurobiol Forthcoming. [DOI] [PubMed]
- 18.Cho HS, Hyman BT, Greenberg SM, Rebeck GW. Quantitation of ApoE domains in Alzheimer disease brain suggests a role for ApoE in Aβ aggregation. J Neuropathol Exp Neurol. 2001;60:342–9. doi: 10.1093/jnen/60.4.342. [DOI] [PubMed] [Google Scholar]
- 19.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with Hhal. J Lipid Res. 1990;31:545–8. [PubMed] [Google Scholar]
- 20.Roher AE, et al. Increased Aβ peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–90. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- 21.Roher AE, et al. β-amyloid 1-42 is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Nat Acad Sci USA. 1993;90:10836–40. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Tredici KD, Braak H. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61:282–93. doi: 10.1093/jnen/61.3.282. [DOI] [PubMed] [Google Scholar]
- 23.O’Brien MD. (1995) Ischemic cerebral edema. In: Brain Ischemia Caplan LR (ed.) Springer, New York, pp. 43–50.
- 24.Waddington MM. (1974) Atlas of cerebral angiography with anatomic correlation. Boston: Little, Brown and Co. p 72-5.
- 25.Nicoll JAR, Wilkinson D, Holmes C, Weller RO (2003) Neuropathology of human Alzheimer’s disease following immunization with amyloid-β-peptide. Neuropathol. Appl. Neurobiol. Forthcoming.
- 26.Vital C, Julien J. Widespread dilation of perivascular spaces: a leukoencephalopathy causing dementia. Neurology. 1997;48:1310–3. doi: 10.1212/wnl.48.5.1310. [DOI] [PubMed] [Google Scholar]
- 27.Kanamalla US, Calabro F, Jinkins JR. Cavernous dilation of mesencephalic Virchow-Robin spaces with obstructive hydrocephalus. Neuroradiology. 2000;42:881–4. doi: 10.1007/s002340000440. [DOI] [PubMed] [Google Scholar]
- 28.Pollack H, Hutchings M, Weller RO, Zhang ET. Perivascular spaces in the basal ganglia of the human brain: their relationship to lacunes. J Anat. 1997;191:337–46. doi: 10.1046/j.1469-7580.1997.19130337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsson M, Thorlin T, Blomstrand F, Hansson E. The star shaped cells: astrocytes are involved in the pathogenesis and progress of neurological diseases. Lakartidningen. 2000;97:3604–10. [PubMed] [Google Scholar]
- 30.Silverberg GD, et al. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology. 2001;57:1763–6. doi: 10.1212/wnl.57.10.1763. [DOI] [PubMed] [Google Scholar]
- 31.Yow HY, Weller RO. A role for cerebrovascular disease in determining the pattern of β-amyloid deposition in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2002;28:149. [Google Scholar]