

Abstract
To assess whether retinoblastoma formation is associated with the expression of high mobility group (HMG) A2 protein, a transcription factor that is highly expressed during embryogenesis and completely repressed in normal adult tissues, we performed Northern and Western blots and RT-PCR analyses, and immunohistochemistry to test for HMGA2 expression. We used established retinoblastoma cell lines in tumors grown in nude mice and clinical retinoblastoma specimens, and contrasted these tumors with normal embryonic and adult retina. Adenoviral-mediated antisense experiments were conducted on the retinoblastoma cell lines to suppress HMGA2 expression and determine if cell proliferation is HMGA2-dependent. We also transfected a retinoblastoma cell line to identify cis-regulatory elements and transcription initiation sites on the HMGA2 gene promoter. HMGA2 gene expression was silenced in terminally differentiated retina of 6-wk-old mice, but it was detected in retina of a 13.5-d postcoitum embryo. Reactivation of HMGA2 gene expression was observed in the retinoblastoma cell lines Y79, WERI-Rb1, and TOTL-1, in tumors derived from some of these cells propagated in nude mice, and in a high frequency of retinoblastomas excised from human patients. This suggests that expression of HMGA2 gene in retinoblastoma cells involves a derepression process. By using an antisense approach to block HMGA2 expression, we observed a decrease in the number of proliferating retinoblastoma cells. As a 1st step toward understanding HMGA2 gene reactivation in retinoblastoma, we mapped the 2 transcription initiation sites and associated positive regulatory elements within the WERI-Rb1 cells. Our discovery of derepression of HMGA2 gene expression in retinoblastoma provides the 1st evidence that this protein might contribute to neoplastic transformation of retina cells.
INTRODUCTION
Retinoblastoma is a malignant neoplasm composed of embryonic tumor cells arising from retinoblasts of neuroepithelial origin (1). Retinoblastoma development is associated with Rb gene inactivation. The molecular cloning of the Rb gene was accomplished by 3 independent groups—a collaboration between laboratories of Thaddeus Dryja and Robert Weinberg, the lab of Wen-Hwa Lee, and the lab of Yuen-Kai Fung and William Benedict (2–4). Despite this landmark discovery, retinoblastoma formation is not understood completely. Additional “pathogenic” factors, especially those associated with dedifferentiated and/or undifferentiated retinal cells, are thought to exist. The high mobility group (HMG) A2 protein is one of the candidates we have examined, because it appears to coordinate cell proliferation and differentiation during mammalian development (5–9).
HMGA2 is a nuclear protein of 109 amino acid residues. Both HMGA family members, HMGA1 and HMGA2, contain 3 basic DNA-binding domains termed “AT-hooks.” The interaction of HMGA transcription factors with AT-rich DNA sequences results in conformational changes in the DNA helix, which facilitate loading of other transcription factors onto their cis-elements. Because of this feature, HMGA proteins are known as architectural transcription factors (10–12).
The HMGA2 gene is strongly expressed during early stages of fetal development in all cells. In later stages, expression is restricted to particular cell types and ceases before birth (13–17). Aberrant expression of the HMGA2 protein is observed in thyroid cells, transformed by selected oncogenic retroviruses (18–20), and in other kinds of tumor (21–25). The general correlation of HMGA2 expression with malignancy suggests a general role for this factor in the genesis or maintenance of the transformed phenotypes (26). Indeed, overexpression of HMGA2 results in neoplastic transformation of Rat-1 fibroblast cells (27); in addition, pituitary adenomas develop in HMGA2 transgenic mice (28). Reactivation of HMGA2 gene expression also is observed in mesenchymal tumors, where there are frequent rearrangements of HMGA2 gene (29–34). Interestingly, chimeric transcripts (either as an HMGA2 fusion or truncation) were observed in several such tumors. Aberrant expression of truncated forms of HMGA2 protein is thought to contribute to neoplastic transformation through their roles in inducible gene expression. Experimentally, ectopic expression of the truncated HMGA2 protein, deprived of its acidic tail and carrying only the 3 DNA-binding domains, could transform NIH3T3 cells (35). In transgenic mice, ubiquitous expression of the same truncation leads to an abnormally high prevalence of lipomas (36,37). Additional evidence that the HMGA2 product contributes to cell growth stems from the discovery that somatic inactivation of the HMGA2 gene results in growth retardation and a pygmy phenotype in mice (13). This is characterized by a marked inhibition of adipose tissue development. In vitro culturing of embryonic fibroblasts from these mice also exhibits a reduced proliferative capacity.
We report here that HMGA2 expression is derepressed in a high frequency of retinoblastomas (6 of 7 retinoblastoma cell and tumor samples examined). Our finding represents the 1st example of derepression of HMGA2 gene expression in an ocular neoplasm. We provide evidence that derepression of HMGA2 expression in retinoblastoma likely is relevant to retinoblast transformation, and that reduction of HMGA2 expression significantly affects cell proliferation. Finally, we have found 2 regions on the HMGA2 gene promoter responsible for positive transcription activity in a representative retinoblastoma cell line.
MATERIALS AND METHODS
Cell Culture
Human retinoblastoma cells Y79, WERI-Rb1, and TOTL-1 were maintained as described (38,39). Human hepatoma cells PLC/PRF/5 (22), mammary epithelial cells HBL-100 (40), and osteosarcoma cells OSH50T (4) were cultured in Dulbecco’s Modification of Eagle’s Medium (Gibco BRL, Gaitherburg, MD, USA) supplemented with 10% heat inactivated fetal bovine serum (Gibco BRL), 50 μg per mL penicillin/50 units per mL streptomycin (Sigma, St. Louis, MO, USA), and 2 mM glutaMaxI (Gibco BRL).
Northern Blot Analysis
Total RNA was prepared from cells and tumors as described (38). RNA samples were electrophoresed in 1.5% denaturing gels and transferred to Hybond-N membrane (Amersham Pharmacia, Arlington Heights, IL, USA). The blots were hybridized separately by radiolabeled human HMGA2 (22) and HMGA1 (41) coding regions in a stringent manner as described (38); then autoradiography was performed. Comparable amount of RNA loaded between samples was monitored by the probing of 28S rRNA. For reprobing, the membranes were boiled in 0.1% SDS for 30 min to remove the previous probe.
Western Blot Analysis
Perchloric acid extract (5%) of the cells and tumor samples were resolved and transferred to Hybond-P membrane (Amersham Pharmacia) as described (22). The intensity of the histone H1 bands after Coomassie staining was used as a control for uniform loading of the protein level in each extract. HMGA proteins were detected using Western-Light Chemiluminescent Detection System (Tropix, Bedford, MA, USA), with the anti-HMGA (22) and A1-specific polyclonal antibodies (in 1:1000 dilution) (42).
Nude Mouse Injection and Immunohistochemistry
All animal procedures used in this study were conducted in accordance with The Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Six-week-old Swiss nu/nu (nude) mice (Taconic Farms, Germantown, NY, USA) were injected subcutaneously with 10 million retinoblastoma cells or intramuscularly with 1 million osteosarcoma cells in a volume of 0.1 mL. Mice were sacrificed when tumors reached 1 cm (1 to 2 mo after inoculation), and the tumors were excised and embedded in Tissue-Tek OCT medium (Sakura, Torrance, CA, USA). Adult nude mouse thigh muscle, 6-wk-old Balb/c eyeballs, and 129/sv embryos 13.5 d postcoitum were embedded in the same manner. Frozen sections (6 microns) of OCT-embedded samples were incubated in a solution of methanol and 1% hydrogen peroxide for 30 min before immunoperoxidase staining. For HMGA protein detection, the slides were blocked overnight at 4 °C with 2% normal goat serum (NGS; Vector, Burlingame, CA, USA), then incubated with the polyclonal antibodies specifically recognizing HMGA1 (42) and HMGA2 protein (raised against a peptide specific to HMGA2: RGEGAGQPSTSA), diluted 1:100 in phosphate-buffered saline (PBS) and 2% NGS, for 1 h at room temperature. Afterward, the slides were washed in PBS 3 times for 10 min and subsequently incubated with biotinylated goat anti-rabbit IgG for 1 h (Vectastain Elite ABC kits; Vector). The slides were washed for 10 min in PBS containing 0.05% Tween20 followed by 2 washes for 10 min in PBS and then incubated with the secondary antibodies as above. Staining was performed using DAB substrate (Vector).
RT-PCR Analysis
Total RNA was prepared from each surgically removed retinoblastoma samples (RBT952, RBT962, RBT965, and RBT942). First-strand cDNAs were synthesized separately from 2 μg each of the RNA samples using an oligonucleotide with sequence specific to the human HMGA2 exon 4 (5′-CTGAGCAGGCTTCTTCTGAA CAAC-3′) and Random Primers (Gibco BRL), by SuperScript II RNase H– Reverse Transcriptase (Gibco BRL) according to the instructions of the manufacturer, with RNasin ribonuclease inhibitor (Promega, Madison, WI, USA) added. PCR was then performed from the cDNA product using 2.5 units of Promega’s Taq DNA Polymerase, 2.5 units of Taq Extender PCR Additive (Stratagene), 0.2 mM dNTPs, and 1 μM of each primer in a final volume of 50 mL. Amplification involved denaturation at 94 °C for 1 min, primer annealing at 60 °C for 1 min, and extension at 72 °C for 1 min, in the Programmable Thermal Blok II thermal cycler (Labline Instruments, Melrose Park, IL, USA). For the HMGA2 gene amplification, the oligonucleotides used have the sequence 5′-ATGAGCGCACGCGGTGAGGG-3′and 5′-CCATTTCCTAG GTCTGCCTCTTGG-3′, which annealed to exons 1 and 3 of the HMGA2 coding region respectively. To specifically amplify the GAPDH gene, a pair of oligonucleotides with sequence 5′-TGAT GACATCAAGAAGGTGGT-3′ and 5′-CAGTGAGGGTCTCTCTC TTCC-3′ was used. The PCR products were resolved onto 1.5% agarose gels. To detect amplified products of the HMGA2 gene in a specific and sensitive manner, DNA was blotted onto nylon membrane from the agarose gel and the membrane was hybridized with the radiolabeled HMGA2 coding region, and then subjected to autoradiography. GAPDH amplification products were visualized on agarose gels stained with ethidium bromide. Oligonucleotides used were obtained from GenoSys (Woodlands, TX, USA).
Adenoviral-mediated Antisense Expression Analysis
Replication-defective recombinant adenoviruses carrying the HMGA2 coding sequence in an antisense orientation (Ad-A2as), and its parental vector (Ad-GFP), were prepared by Alfredo Fusco using the described method (43). Retinoblastoma cells WERI-Rb1 were exposed to Ad-A2as or Ad-GFP, in the same multiplicity of infection (MOI) of 1000 pfu per cells for 12 h, and then grown in fresh medium for 7 d. Uninfected cells were included as control. The number of viable cells was determined by Trypan blue exclusion and hemacytometry. Proliferation of cells was determined using the ViaLight HS kit (Lumitech, Nottingham, UK), in which measurements for comparison were based on the same number of cells used. Briefly, cells were lysed by the Nucleotide Releasing Reagent, ATP Monitoring Reagent was added to the lysate, and luciferase activity was measured using the Monolight 2010 luminometer (Analytical Luminescence Laboratory, San Diego, CA, USA). Light output was integrated over a 15-s period and displayed as relative light units. Background activity was found to be < 0.1% of all readings. The effectiveness of antisense HMGA2 RNA expression in suppressing the HMGA2 protein level in transduced retinoblastoma cells was confirmed by Western blot analysis as described above.
Transient Transfection Analysis
In each transfection (as described in 44), 3 × 106 WERI-Rb1 cells were electroporated with 10 μg of pGL-2-Basic construct (Promega) harboring the human HMGA2 gene 5′ UTR fragment and 10 μg of pCAT-Control Vector (Promega; served as an internal control to determine transfection efficiency by monitoring CAT expression) that were prepared by Qiagen Plasmid kit (Chatsworth, CA, USA). Transfected cells were maintained in complete RPMI medium for 44 h, before luciferase (Promega) and CAT (Roche, Indianapolis, IN, USA) assays were conducted as suggested by the manufacturers. The pGL2-Promoter Vector (Promega) containing the luciferase gene driven by the SV40 promoter was used as a positive control for comparison of promoter activities. Mock transfection and transfection with the empty reporter were included to determine the background noise. Transfections were carried out in duplicate and repeated 3 times. For OSH50T cell transfection, 2 × 105 cells were seeded onto a 35-mm plate and exposed to LipofectAMINE (Gibco BRL) mixed with 3 μg pGL-2-HMGA2 promoter constructs and 7 μg pCAT-Control Vector.
Primer Extension Analysis
Oligonucleotides of sequence 5′-GGAGATGAGGTGATAGGGC TGGGG-3′ and 5′-TGTGGTGTGTGTGCGTGCACGCGCTTG-3′ (Genosys) were end-labeled with 32P and hybridized with total RNA, as described (45). Extension products obtained by reserve-transcription were resolved through 8% sequencing gels, along with sequencing ladders that were generated using the same oligonucleotides as markers.
RESULTS
High Level of HMGA2 and HMGA1 Gene Expression in Retinoblastoma Cell Lines
Figure 1A shows a Northern blot assessing HMGA2 and HMGA1 transcript levels in the Y79 and WERI-Rb1 retinoblastoma cells. The levels are compared with those of the hepatoma cell line PLC/PRF/5 (previously reported to express high levels of both transcripts) (22), a nonmalignant human mammary epithelial cell line HBL-100 (40), and an osteosarcoma cell line, OSH50T, lacking the Rb protein (4) as also occurs in retinoblastoma cells. The top panel shows that the level of HMGA2 transcripts exceeds that expressed in the human hepatoma cell line PLC/PRF/5. Interestingly, the size of HMGA2 transcripts (that is, approximately 3.9/4.6 kb) appears different from that of PLC/PRF/5 cells, suggesting different transcription initiation/termination site usage. HBL-100 and OSH50T cells do not express HMGA2. As expected, the 2-kb HMGA1 mRNAs are seen in the middle panel of the 5 cell lines examined (as well as many other cultured cell lines), and are abundant in the 2 retinoblastoma cell lines (several fold higher than that in PLC/PRF/5). The same blot was reprobed with 28S rRNA to control for equal RNA loading, as shown in the bottom panel. The experiment was repeated and the same results were obtained.
Figure 1.

High-level expression of HMGA2 and HMGA1 mRNA and protein in cultured retinoblastoma cells. A: Northern blot of total RNA isolated from the Y79 (lane 4) and WERI-Rb1 (lane 5) retinoblastoma cells, PLC/PRF/5 hepatoma cells (lane 1), HBL-100 mammary epithelial cells (lane 2), and OSH50T osteosarcoma cells (lane 3), probed, respectively, with the radiolabeled HMGA2 and HMGA1 coding region DNA (upper and middle panels). 28S and 18S rRNA provide size markers of 4.8 and 1.8 kb. Uniform RNA loading in individual sample was shown by hybridizing the same Northern blot with the 28S rRNA probe (lower panel). B: Western blot of perchloric acid extracts from cells of PLC/PRF/5, HBL-100, OSH50T, Y79, and WERI-Rb1 (lanes 1 to 5), and an additional retinoblastoma cell line TOTL-1 (lane 6); cells were prepared and equal protein loading between lanes confirmed by the histone H1 band stained by Coomassie (lower panel). Anti-HMGA2 and A1 polyclonal antibodies were used to detect HMGA2 (upper panel) and HMGA1a/b (middle panel) proteins.
In Figure 1B, Western blot shows the level of HMGA2 (upper panel) and HMGA1 (middle panel) protein expression in the above 5 cell lines, and an additional retinoblastoma cell line TOTL-1 (39). HMGA2 protein (12 kD) is expressed in all 3 lines of retinoblastoma cells Y79, WERI-Rb1, and TOTL-1, and in PLC/PRF/5 hepatoma cells as previously shown (22), but not in HBL-100 or OSH50T. Results in the middle panel indicate that HMGA1a and HMGA1b proteins (isoforms of each other, 11.8 and 10.6 kD respectively) are expressed at similar levels between the 6 cell lines. In short, there is a good correlation of HMGA2 and HMGA1 transcript levels and protein expression. The polyclonal antibodies recognize predominantly the HMGA2 protein (22), whereas HMGA1 protein was specifically detected with the polyclonal antibodies raised against a N-terminal region of HMGA1 (42). Equal total protein loading of cell extracts was assessed, by monitoring levels of histone H1 on a separate gel stained with Coomassie blue (lower panel). Similar results were obtained in the repeated experiment.
Overexpression of HMGA2 Protein Found in Only Embryonic Retinoblastoma Cells in Tumors Grown in Nude Mice
To determine if HMGA2 and HMGA1 expression in cultured retinoblastoma cells is more than an in vitro phenomenon in tissue culture, 3 cell lines (Y79, WERI-Rb1, and OSH50T) were subsequently injected into and grown in nude mice (Each cell line was implanted in 5 mice). All of the mice developed tumors, and these tumors were excised and analyzed for HMGA2 and HMGA1 expression. Immunohistochemical analysis was performed on cryosections of tumors derived from Y79, WERI-Rb1, and OSH50T in adult nude mice thigh muscle, a retina of 129/sv mouse 13.5 d postcoitum, and a 6-wk-old Balb/c retina. HMGA2 was detected with new and specific polyclonal antibodies; although they were moderate in affinity. HMGA1 was specifically recognized by the polyclonal antibodies described above. In Figure 2A, nuclear staining with HMGA2 and HMGA1 antibodies was observed in the 2 retinoblastoma-induced tumors, derived from Y79 and WERI-Rb1 cells. Not every nucleus was immuno-stained for HMGA2, indicating that some cells of the tumor mass, such as the stromal cells, do not express the protein. HMGA2 and HMGA1 proteins were distinctively stained in the nuclei of embryonic cells, including the retina at 13.5 d postcoitum. This is consistent with previous reports (13,42). It is known that HMGA2 is not expressed in normal adult tissues (13–17). The lack of HMGA2 immunostaining observed in nude mouse thigh muscle and adult mouse retina confirmed this and served as background controls for our staining. Tumors derived from the osteosarcoma cell line OSH50T expressed HMGA1 but not HMGA2 protein. As noted earlier, HMGA2 has a highly restricted expression pattern and the protein has been detected in only a subset of tumors (18–25). The top row of Figure 2A shows the results of the hematoxylin & eosin staining. The bottom row depicts the results of immunostaining without primary antibodies, indicating no endogenous peroxidase enzyme activity was detected. Furthermore, no immunostaining was observed when the tissues were analyzed after the antibodies had been preincubated with the corresponding immunogens (data not shown), demonstrating the specificity of the antibodies in recognizing HMGA2 and HMGA1 proteins. Overall, the immunohistochemical data indicates that HMGA2 and HMGA1 are expressed in experimental retinoblastoma and embryonic retina, and HMGA2 is absent in adult nude mouse thigh muscle and retina. These results were highly reproducible. A high level of HMGA1 expression in adult mouse retina was reported previously by us (38).
Figure 2.

HMGA2 expression is in established lines of retinoblastoma cells propagated in nude mice, surgically removed retinoblastoma samples, and mouse embryonic retina, but absent in adult mouse tissues. A: Immunohistochemistry of cryosections of nude mouse tumors derived from the retinoblastoma cells Y79 and WERI-Rb1, and that from osteosarcoma cells OSH50T were hematoxylin & eosin stained, immunoperoxidase stained with the polyclonal antibodies specifically recognizing only HMGA2 protein (anti-HMGA2 panel), immunostained with the polyclonal antibodies which recognize only HMGA1 protein (anti-HMGA1 panel), and endogenous peroxidase controlled (endog control panel). HMGA proteins were detected in the primary retinoblastoma sample RBT952 (see Figure 3) as well. Retina of 129/sv mouse embryo 13.5-d postcoitum showed positive HMGA2 and HMGA1 immunostaining, whereas 6-wk-old Balb/c retina demonstrated undetectable HMGA2 protein but abundant HMGA1 protein. Thigh muscle from adult nude mouse did not stain with HMGA2 or HMGA1. Photomicrographs are magnified 400×. B: Northern blot of total RNA prepared from PLC/PRF/5 hepatoma cells (lane 1), adult mouse thigh muscle (lane 2), and nude mouse tumors derived respectively from OSH50T (lane 3), Y79 (lane 4), and WERI-Rb1 (lane 5), probed separately with radiolabeled HMGA2 (upper panel) and HMGA1 (middle panel) coding region DNA, and 28S rRNA (lower panel).
Because the HMGA2 antibodies, although specific, do not provide strong immunostaining signals, Northern blot analysis for HMGA2 (and HMGA1) expression of these experimental tumors was performed. The results shown in Figure 2B demonstrate the high level HMGA2 and HMGA1 expression in nude mouse tumors derived from 2 retinoblastoma cell lines, Y79 and WERI-Rb1.
HMGA2 and HMGA1 Gene Expression in Surgically Removed Retinoblastoma Samples
To extend the analysis of HMGA2 and HMGA1 gene expression to clinical retinoblastoma, Northern blot and RT-PCR analysis was performed on intact RNA that was prepared from surgically removed primary retinoblastoma specimens. RNA extracted from WERI-Rb1 and Y79 cells served as positive controls and that from OSH50T as a negative control for the detection. Four specimens (RBT952, RBT962, RBT965, RBT942) were examined. Northern blot analysis detected abundant HMGA2 transcripts in the 2 retinoblastoma cell lines and RBT942, and significant expression in RBT952 and RBT962 (Figure 3A, top panel). HMGA1 mRNA, as expected, was present in all RNA samples (see Figure 3A, middle panel). HMGA2 expression was evaluated also by RT-PCR in a semi-quantitative fashion (that is, linear amplification conditions) followed by blotting the amplification products for radioactive detection. (This more sensitive approach of detection also confirmed specific amplification and has been employed elsewhere [34].) RBT942 gave a strong signal of HMGA2 expression that was comparable to that of cultured retinoblastoma cells (see Figure 3B, top panel). A weaker signal was detected in RBT952 and an even weaker one in RBT962. No reaction products were observed after PCR amplification and hybridization detection of HMGA2 from the RNA of OH50T cells and RBT965. Thus, 3 of 4 samples of retinoblastoma primary tumor were reproducibly shown to express the HMGA2 gene. Taken together, of 7 retinoblastoma cell and tumor samples examined, 6 expressed the HMGA2 gene. These results indicate that HMGA2 and HMGA1 gene expression is a frequent event in retinoblastoma.
Figure 3.

High prevalence of HMGA2 gene expression in surgically removed retinoblastoma samples. Intact RNA was prepared from the cells: OSH50T (lane 1), Y79 (lane 2) and WERI-Rb1 (lane 3), and the surgically removed retinoblastoma specimens: RBT952, RBT962, RBT965, and RBT942 (lanes 4 to 7). A: Northern blot of the HMGA2 (upper panel) and HMGA1 (middle panel) mRNA, in which equal RNA loading was controlled by 28S probing (lower panel). Autoradiograph of the upper panel was overexposed to reveal weak signals. B: RT-PCR analysis of the same RNA samples, in which reverse transcription was performed separately using a gene-specific primer for HMGA2 and random primers. cDNA synthesized were used in PCR amplification with pairs of HMGA2 and GAPDH-specific primers. To ensure amplification is specific, PCR products from HMGA2 gene amplification were size fractionated onto an agarose gel, blotted onto a nylon membrane and hybridized with HMGA2 probe (upper panel). PCR products from GAPDH amplification, electrophoresed through an agarose gel and stained with ethidium bromide (lower panel), showed the equal quantities of cDNA used.
Cryosections of specimen RBT952 were available for HMGA2 and HMGA1 immunohistochemistry. Their positive staining results, shown in Figure 2A, confirm the reactivation of HMGA2 and HMGA1 expression in clinical retinoblastoma.
Adenoviral-mediated Expression of Antisense HMGA2 RNA Blocks A2 Protein Expression and Results in Growth Inhibition of Retinoblastoma Cells
Having established that HMGA2 and HMGA1 proteins are expressed in retinoblastoma, we investigated whether retinoblastoma cells grown in vitro are HMGA2-dependent. We chose to ectopically express antisense HMGA2 RNA in WERI-Rb1 as a means to inhibit HMGA2 expression. This approach has been used previously to successfully block HMGA2 expression (46). Adenoviral-mediated expression was used, in which an MOI of 300 is optimal for Y79 cells transduction (47); in our study, an MOI of 1000 appeared to be the most efficient to infect WERI-Rb1 by recombinant adenovirus. This resulted in some cytotoxicity, but a specific antisense effect was achieved (with respect to both HMGA2 expression and cell numbers). WERI-Rb1 cells were infected separately with the replication-deficient recombinant adenovirus expressing green fluorescent protein (GFP) as control, and the same recombinant carrying the HMGA2 gene in an anti-sense orientation (created by Fusco’s group). A comparable MOI of infections was confirmed by fluorescent microscopy on both infected cells 3 to 4 d after adenovirus exposure (Figure 4C). The WERI-Rb1 cells were harvested 7 d after exposure to the recombinant viruses; cell counts were determined and expressed as a percentage of the number of the uninfected cells (see Figure 4A, upper panel). Proliferation of the same number of infected cells were assayed by measuring the ATP content of lysed cells (that are metabolically active) (see Figure 4A, lower panel). We note that ATP levels drop to 44%, indicative of a drop in cell proliferation. A significant reduction in the mean cell number and proliferation of WERI-Rb1 cells transduced with GFP/antisense HMGA2 adenovirus was observed when compared with uninfected cells or cells transduced with adenovirus-expressing GFP. The fact that a high MOI results in cell death could explain the baseline decrease in cell number when WERI-Rb1 was infected by adenovirus-expressing GFP. Our results suggest that change of cell number is not linearly related to change of cell proliferation, which could be explained by the ATP measurement rather than tritiated thymidine uptake as readouts for proliferation assays.
Figure 4.

Antisense reduction of HMGA2 protein level affects proliferation of retinoblastoma cells. Retinoblastoma cells WERI-Rb1 were harvested after 7 d of infection (at 1000 pfu/cell) with the replication-defective recombinant adenovirus expressing GFP only (sample 1), or GFP and the HMGA2 gene in an antisense orientation (sample 2). Same number of cells was used initially in samples 1 and 2 and in the uninfected control sample 3. A: Cell number was determined in individual infection, and the results of 3 experiments are summarized in the upper panel as mean percentage of uninfected cells, with standard error of mean values (error bars) shown. Cell proliferation was measured from the same number of cells in samples 1 to 3, and the result (mean percentage of uninfected cells and standard error) is shown in the lower panel. B: Western blot showing the level of HMGA2 and HMGA1 protein in samples 1 to 3, in which perchloric acid extracts of the samples were prepared and the amount of HMGA proteins equalized by histone H1. Anti-HMGA2 and A1 polyclonal antibodies were used to detect HMGA2 (upper panel) and A1 protein (lower panel). WERI-Rb1 cells infected with the recombinant adenovirus carrying an antisense of HMGA2 demonstrate a reduced HMGA2 protein but HMGA1 level remained unchanged. C: Fluorescent micrographs reveal the comparable green fluorescence between cells infected with recombinant GFP (panel 1) and GFP/antisense HMGA2 (panel 2) adenovirus, for 3 to 4 d, indicating similar degree of infection. No GFP fluorescence is observed in uninfected cells (panel 3). Identical sensitivity and exposure time were used.
To confirm that expression of antisense HMGA2 RNA resulted in the suppression of HMGA2 protein in WERI-Rb1 cells, perchloric acid extracts were prepared from infected and uninfected cells; the protein content of the individual cell lysate was normalized by histone H1 on a separate gel stained with Coomassie blue (data not shown), for Western blot analysis of level of HMGA2 and HMGA1 proteins. The results of a representative blot, shown in the upper panel of Figure 4B, indicate a strong suppression of HMGA2 protein expression in WERI-Rb1 cells infected with the GFP/antisense HMGA2 adenovirus, and the HMGA2 protein level appears unchanged by infecting with adenovirus expression GFP only. Unaltered HMGA1 protein level was seen in the lower panel, suggesting the event leading to a reduction in cell number is independent to HMGA1.
Similar results were obtained from using Y79 cells for this experiment (data not shown).
Expression of HMGA2 Gene Driven by 2 Promoters Coupled with Distinct Transcription Start Sites
After determining the expression pattern of HMGA proteins, in particular HMGA2, in retinoblastoma, the next goal was to understand the mechanism of derepression of HMGA2 gene expression. The 1st step we took was to identify regulatory regions necessary for HMGA2 gene expression in retinoblastoma. To accomplish this, luciferase reporter constructs harboring various regions of the HMGA2 gene 5′ UTR (48) in the forward orientation were generated and then transiently transfected into WERI-Rb1 and OSH50T cells. The results are summarized in Figure 5. The nucleotide immediately upstream from the methionine containing ATG was numbered as −1. Sequences flanking nt −27 to −476 and nt −871 to −1128 each possess strong promoter activity in WERI-Rb1 cells (82% and 86% compared with the SV40 promoter), whereas such activity was substantially less in OSH50T cells (26% and 37%, respectively). Therefore, these 2 regions function as proximal and distal promoters of the HMGA2 gene in retinoblastoma cells. Their similar positive promoter strength matched the comparative level of the 2 HMGA2 transcripts detected in these cells. Regions covering nt −476 to −2939 and nt −1128 to −2939 contribute minimal promoter activity in WERI-Rb1 cells (23% and 18% of that of the SV40 promoter), suggesting the presence of negative regulatory activity between region nt −1128 to −2939 and/or region nt −476 to −871. These results indicate that expression of the HMGA2 gene involves utilization of alternative transcription start sites. We are investigating whether the 2 transcript species utilize identical polyadenylation signals.
Figure 5.

Identification of 2 HMGA2 gene promoter regions active in retinoblastoma cells. Analysis was conducted in HMGA2-positive WERI-Rb1 and HMGA2-negative OSH50T cells. The schematic diagrams on the left show the organization of various plasmids constructed by subcloning different segments of the human HMGA2 5′ UTR genomic fragments in forward orientation into the upstream region of the firefly luciferase gene (in pGL2-Basic vector) used in the transient transfection experiments. + 1 corresponds to the A of the initiator methionine codon, and residues preceding it are represented by negative numbers. The top bold line shows the restriction sites of the 5′ UTR (A, ApaLI; B, BamHI; E, EcoRI; P, PstI) and the location of the 2 transcription start sites are represented by bent arrows (at nt −154 and −979; Figure 6 and 7). On the right is the relative luciferase activity (expressed as percent of pGL2-SV40 Promoter expression) upon cotransfection of different luciferase reporter plasmids and pCAT-Control Vector (used as internal control). Luciferase activity measured was normalized to the corresponding CAT activity for each sample. Values represent the average of 3 double transfections, and bars of standard error of mean are shown, of which black and white bars show that of WERI-Rb1 and OSH50T cells accordingly.
Subsequently, the transcription start sites associated with the 2 promoters of HMGA2 gene were mapped, by performing primer extension on the RNA prepared from WERI-Rb1 and Y79 cells. Because HMGA2 transcripts were not detected in OSH50T cells, RNA from this cell type served as HMGA2 negative control in the experiment. Several oligonucleotides were made within the promoter regions to test for the presence of extended products only from retinoblastoma RNA. As shown in Figures 6A and 7A, 1 primer each from the extension reactions produced retinoblastoma-specific bands; their size predicts the proximal and distal HMGA2 transcription start sites to be located at nt −154 and nt −979 5′ to the translational start, respectively. The results also support our promoter mapping data generated via transfections.
Figure 6.

Analysis of the proximal HMGA2 gene promoter region (flanking nt −27 to −476 of the genomic sequence with respect to the translational start). A: Primer extension of the HMGA2 RNAs prepared from WERI-Rb1 (lane 1) and Y79 (lane 2) cells using an oligonucleotide complementary to nt −75 to −98 in the proximal promoter region of HMGA2 gene. RNA extracted from OSH50T cells (lane 3) was used as negative control template. Extension products were analyzed in a 7M urea/8% polyacrylamide gel. Sequencing ladder generated from the same primer was electrophoresed simultaneously as a marker. The Davidic star to the left of the sequence indicates the transcription start site at nt −154. B: Nucleotide sequence of the proximal HMGA2 gene promoter region. Consensus transcription factor recognition sites are shown. The arrow depicts the location of primer annealed. The bent arrow indicates the mapped transcriptional start site. Restriction sites are underlined. The uppercase ATG is the methionine start codon.
Figure 7.
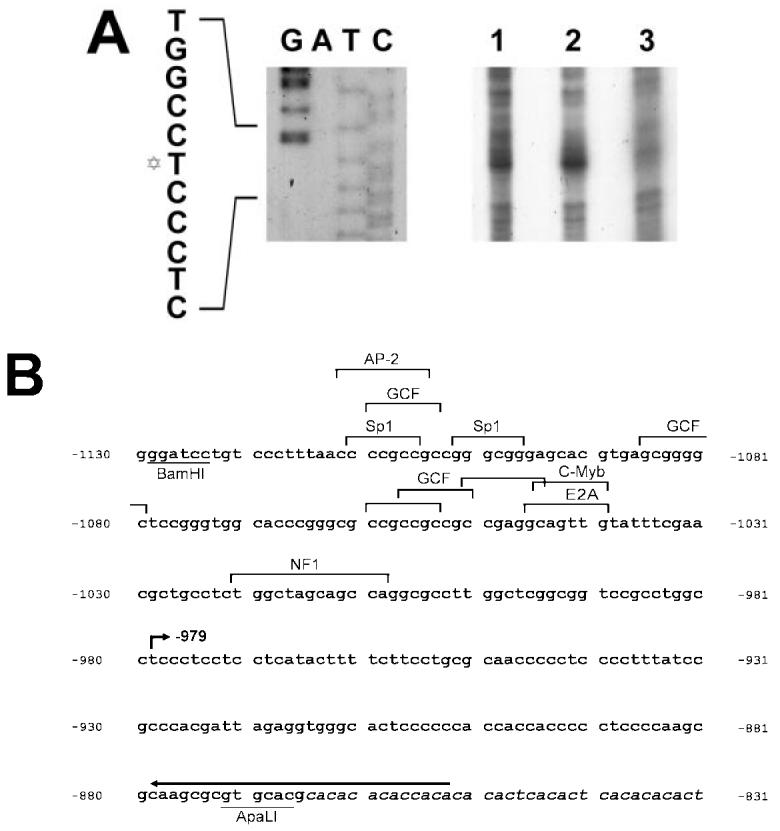
Analysis of the distal HMGA2 gene promoter region (flanking nt −871 to −1128 of the genomic sequence with respect to the translational start). A: Primer extension of the HMGA2 RNA prepared from WERI-Rb1 (lane 1) and Y79 (lane 2) cells using an oligonucleotide complementary to nt −853 to −879 in the distal promoter region of HMGA2 gene. RNA extracted from OSH50T cells (lane 3) was used as negative control template. Extension products were analyzed in a 7M urea/8% polyacrylamide gel. Sequencing ladder generated from the same primer was electrophoresed simultaneously as a marker. The Davidic star to the left of the sequence indicates the transcription start site at nt −979. B: Nucleotide sequence of the distal HMGA2 gene promoter region. Consensus transcription factor recognition sites are shown. The arrow depicts the location of primer annealed. The bent arrow indicates the mapped transcriptional start site. Restriction sites are underlined. Sequence of the CA repeat is shown in italic.
The genomic DNA sequences of the proximal and distal promoter regions of the human HMGA2 are shown in Figures 6B and 7B accordingly. Comparative analysis of the sequences with a database containing known binding sites for transcription factors and regulatory regions of other genes revealed several homologies (49). They are the Sp1 (CCCGCC or GGGCGG), AP-2 (CCMNSSS or YCSCCMNSSS), GCF (SCGSSSC) and S1_HS (TCCTCCTCC) sites. GCF and S1_HS both represent regulatory motifs of the epidermal growth factor receptor gene (50). Neither promoter harbors a TATA box, as also occurs in the human HMGA1 gene 5′ UTR (51).
DISCUSSION
The results of this study suggest that expression of the HMGA2 gene is derepressed in human retinoblastoma, and that this contributes to the proliferation of retinoblastoma cells. HMGA2 is abundantly expressed in embryonic tissue, including the retina, but its expression is repressed in virtually all adult tissues, also including the retina (13–17). Our evidence of derepression of HMGA2 expression stems from the analysis of 3 established retinoblastoma cell lines, namely Y79, WERI-Rb1, and TOTL-1 (grown in culture or in nude mice), and in 3 of a panel of 4 primary tumor specimens. In contrast to HMGA1, whose expression is generally induced in neoplastic cells as well as in many activated and proliferating cells (5–8), HMGA2 gene expression is more tightly regulated. HMGA2 expression is observed in cells of ectodermal origin such as in thyroid, breast, and lung cancers (18–25). Chimeric transcripts are also detected in several tumors of mesenchymal origin (29–34). Full-length HMGA2 proteins were observed in the 3 retinoblastoma cell lines examined, and in 2 of them, genetic translocation of the HMGA2 locus was not detected (data not shown), suggesting genetic alteration is not the cause of inducing HMGA2 expression. Our characterization on HMGA2 (and HMGA1) expression pattern provides the 1st evidence that aberrant HMGA2 expression correlates with and perhaps contributes to the pathogenesis of retinal tumor. In addition, specific blockade of HMGA2 (in which HMGA1 remained unchanged) gene expression, with an antisense adenoviral-mediated approach, resulted in a decrease in cell number/proliferation of retinoblastoma cells. Finally, we isolated 2 regions on the HMGA2 promoter in retinoblastoma cells responsible for such gene reactivation and specifically mapped their corresponding transcription initiation sites. This is the 1st step to assess the mechanisms controlling HMGA2 gene derepression.
The 1st evidence implicating HMGA2 in the pathogenesis of thyroid tumors came from antisense experiments (46). However, HMGA2 protein did not appear to behave as a classical oncogene because ectopic expression did not by itself transform thyroid cells and NIH3T3 fibroblasts (46,35). Instead, it behaves as an oncogene in a subset of tumors, in particular those composed of undifferentiated or dedifferentiated cells and some other highly transformed cells (5–8). Therefore it appears that HMGA2 is necessary but not sufficient for cell transformation in selected cell types.
Although the results of our antisense experiments support a role of HMGA2 in the growth of cultured retinoblastoma cells, we have yet to demonstrate a general requirement in vivo of HMGA2 in retinoblastoma development. The 3 classical retinoblastoma cell lines used in this study, namely WERI-Rb1, Y79, and TOTL-1, are spontaneously transformed cells lacking an Rb signal. Rb inactivation, however, does not seem to directly activate HMGA2 gene expression. The OSH50T cell line established from a patient with bilateral retinoblastoma and a homozygous deletion in the Rb gene (4) did not express HMGA2. Rather, the undifferentiated/dedifferentiated character of retinoblastoma cells appears to permit HMGA2 gene expression. The possibility that HMGA2 expression may be required to maintain the undifferentiated/dedifferentiated nature of this “embryonic” tumor is ruled out by the general observation that HMGA2 is expressed in “differentiated” transformed cells as well, such as malignant PLC/PRF/5 hepatoma cells (22). Indeed, retinoblastoma cells transduced with the recombinant adenovirus to suppress HMGA2 expression did not become differentiated, indicating that HMGA2 level does not control such differentiation.
To investigate the mechanism of HMGA2 gene expression in retinoblastoma, which may allow us in the future to inhibit the derepression process of HMGA2 gene expression, we demonstrated the presence of 2 distinct promoter regions in WERI-Rb1 cells used for transcription, each with its own start sites. This was shown by transfection studies using luciferase reporter constructs carrying various regions of the 5′ flanking sequence of the HMGA2 gene, and by primer extension analysis performed on RNA prepared from the 2 retinoblastoma cells compared with that from the osteosarcoma cells OSH50T that do not express HMGA2. Together, our results demonstrate that the 2 transcriptional start sites are located at nt 154 and 979 5′ upstream from the translational start codon, and they are associated with putative transcriptional elements like AP-2, Sp1, and selected regulatory elements in the epidermal growth factor receptor gene promoter (50). Because 2 different sized transcripts were identified from Northern blot analysis (3.9 and 4.6 kb), these results suggest that expression of HMGA2 gene in retinoblastoma utilizes alternative transcription starts and may have a common terminus.
Understanding the role that HMGA2 plays in pathobiology and molecular mechanisms of retinoblastoma occurrence is biologically and clinically significant, and HMGA2 may become a therapeutic target. One approach would be to block HMGA2 protein expression in retinoblastoma cells, hopefully leading to inhibition of cell proliferation. Another approach is to inhibit HMGA2 gene expression by targeting its promoters.
Our future experiments will determine (1) whether HMGA2 expression is required for, and collaborates with other factors in, retinoblastoma formation by modifying expression of HMGA2 protein and relevant factors (such as p107, Rb) in retinal cells by mouse modeling; and (2) to further identify and characterize functional cis- and trans-factors associated with the HMGA2 promoter, leading to the derepression of HMGA2 gene expression in retinoblastoma, as was performed in PLC/PRF/5 cells (45).
Acknowledgments
We thank Drs Gilbert Jay, C Kathleen Dorey, Patricia Pearson, and other members of our laboratory, in particular: Drs Andrea Keane-Myers, Dai Miyazaki, Paola Arlotta, Takao Nakamura, and Jason Kreisberg who provided advice and help with the manuscript. We also thank Dr Robert Weinberg, Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, for the gifts of Y79, WERI-Rb1, and OSH50T cells; and the Tissue Culture Core Facility of the Lombardi Cancer Center for HBL-100 cells. This work was supported by National Institute of General Medical Science grant RO1 GM49661, the Lucille P Markey Charitable Trust, the Research to Prevent Blindness Foundation, and the “Associazione Italiana per la Ricerca sul Cancro.” GM was supported by “Fondazione Italiana per la Ricerca sul Cancro” while he was a Visiting Scientist of the Ono Lab. SJO was a JSPS Visiting Professor at Kyoto University during the preparation of this manuscript. K-YC was a Fellow of the Research to Prevent Blindness, America Fund (PD97054). SM was supported by the Massachusetts Lions Eye Research Fund and Gifts to the Retina Genetics Fund, Massachusetts Eye & Ear Infirmary.
REFERENCES
- 1.Albert DA, Jakobiec FA (eds). (1994) Principles and practice of ophthalmology Sauders, Philadelphia.
- 2.Friend SH, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–6. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 3.Lee WH, et al. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235:1394–9. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- 4.Fung YK, et al. Structural evidence for the authenticity of the human retinoblastoma gene. Science. 1987;236:1657–61. doi: 10.1126/science.2885916. [DOI] [PubMed] [Google Scholar]
- 5.Goodwin G. Molecules in focus: the high mobility group protein, HMGI-C. Int J Biochem Cell Biol. 1998;30:761–6. doi: 10.1016/s1357-2725(98)00016-8. [DOI] [PubMed] [Google Scholar]
- 6.Zhou X, Chada K. HMGI family proteins: architectural transcription factors in mammalian development and cancer. Keio J Med. 1998;47:73–7. doi: 10.2302/kjm.47.73. [DOI] [PubMed] [Google Scholar]
- 7.Liu F, Chau KY, Arlotta P, Ono SJ. The HMG I proteins: dynamic roles in gene activation, development, and tumorigenesis. Immunol Res. 2001;24:13–29. doi: 10.1385/IR:24:1:13. [DOI] [PubMed] [Google Scholar]
- 8.Reeves R. Molecular biology of HMGA proteins: hubs of nuclear function. Gene. 2001;277:63–81. doi: 10.1016/s0378-1119(01)00689-8. [DOI] [PubMed] [Google Scholar]
- 9.Fedele M, et al. Role of the high mobility group A proteins in human lipomas. Carcinogenesis. 2001;22:1583–91. doi: 10.1093/carcin/22.10.1583. [DOI] [PubMed] [Google Scholar]
- 10.Wolffe A. Architectural transcription factors. Science. 1994;264:1100–1. doi: 10.1126/science.8178167. [DOI] [PubMed] [Google Scholar]
- 11.Mantovani F, et al. NF-κB mediated transcriptional activation is enhanced by the architectural factor HMGI-C. Nucleic Acids Res. 1998;26:1433–9. doi: 10.1093/nar/26.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noro B, et al. Molecular dissection of the architectural transcription factor HMGA2. Biochemistry. 2003;42:4569–77. doi: 10.1021/bi026605k. [DOI] [PubMed] [Google Scholar]
- 13.Zhou X, et al. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature. 1995;376:771–4. doi: 10.1038/376771a0. [DOI] [PubMed] [Google Scholar]
- 14.Zhou X, et al. Genomic structure and expression of the murine HMGI-C gene. Nucleic Acids Res. 1996;24:4071–7. doi: 10.1093/nar/24.20.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogalla P, et al. HMGI-C expression patterns in human tissues. implications for the genesis of frequent mesenchymal tumors. Am J Pathol. 1996;149:775–9. [PMC free article] [PubMed] [Google Scholar]
- 16.Hirning-Folz U, et al. The expression pattern of the HMGIC gene during development. Gene Chrom Cancer. 1998;23:771–4. [PubMed] [Google Scholar]
- 17.Chieffi P, et al. HMGA1 and HMGA2 protein expression in mouse spermatogenesis. Oncogene. 2002;21:3644–50. doi: 10.1038/sj.onc.1205501. [DOI] [PubMed] [Google Scholar]
- 18.Giancotti V, et al. Changes in nuclear proteins following transformation of rat thyroid epithelial cells by a murine sarcoma retrovirus. Cancer Res. 1985;45:6051–7. [PubMed] [Google Scholar]
- 19.Giancotti V, et al. Elevated levels of a specific class of nuclear phosphoproteins in cells transformed with v-ras and v-mos oncogenes and by co-transfection with c-myc and polyma middle T genes. EMBO J. 1987;6:1981–7. doi: 10.1002/j.1460-2075.1987.tb02461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giancotti V, et al. Comparison of multiple forms of the high mobility group I proteins in rodent and human cells. Identification of the human high mobility group I-C protein. Eur J Biochem. 1991;198:211–6. doi: 10.1111/j.1432-1033.1991.tb16003.x. [DOI] [PubMed] [Google Scholar]
- 21.Manfioletti G, et al. cDNA cloning of the HMGI-C phosphoprotein, a nuclear protein associated with neoplastic and undifferentiated phenotypes. Nucleic Acids Res. 1991;19:6793–7. doi: 10.1093/nar/19.24.6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel UA, et al. Expression and cDNA cloning of human HMGI-C phosphoprotein. Biochem Biophys Res Commun. 1994;201:63–70. doi: 10.1006/bbrc.1994.1669. [DOI] [PubMed] [Google Scholar]
- 23.Rogalla P, et al. Expression of HMGI-C, a member of the high mobility group protein family, in a subset of breast cancers: relationship to histologic grade. Mol Carcinog. 1997;19:153–6. doi: 10.1002/(sici)1098-2744(199707)19:3<153::aid-mc2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 24.Rogalla P, et al. HMGIC expression patterns in non-small lung cancer and surrounding tissue. Anticancer Res. 1998;18:3327–30. [PubMed] [Google Scholar]
- 25.Finelli P, et al. The High Mobility Group A2 gene is amplified and overexpressed in human prolactinomas. Cancer Res. 2002;62:2398–405. [PubMed] [Google Scholar]
- 26.Wunderlich V, Bottger M. High-mobility-group proteins and cancer: an emerging link. J Cancer Res Clin Oncol. 1997;123:133–40. doi: 10.1007/BF01214665. [DOI] [PubMed] [Google Scholar]
- 27.Wood LJ, Maher JF, Bunton TE, Resar LM. The oncogenic properties of the HMG-I gene family. Cancer Res. 2000;60:4256–61. [PubMed] [Google Scholar]
- 28.Fedele M, et al. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene. 2002;21:3190–8. doi: 10.1038/sj.onc.1205428. [DOI] [PubMed] [Google Scholar]
- 29.Schoenmakers EFPM, et al. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nature Genet. 1995;10:436–44. doi: 10.1038/ng0895-436. [DOI] [PubMed] [Google Scholar]
- 30.Ashar HR, et al. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell. 1995;82:57–65. doi: 10.1016/0092-8674(95)90052-7. [DOI] [PubMed] [Google Scholar]
- 31.Kazmierczak B, et al. HMG I-C rearrangements as the molecular basis for the majority of pulmonary chondroid hamartomas: a survey of 30 tumors. Oncogene. 1996;12:515–21. [PubMed] [Google Scholar]
- 32.Berner JM, et al. HMGIC, the gene for an architectural transcription factor, is amplified and rearranged in a subset of human sarcomas. Oncogene. 1997;14:2935–41. doi: 10.1038/sj.onc.1201135. [DOI] [PubMed] [Google Scholar]
- 33.Pedeutour F, et al. Dysregulation of HMGIC in a uterine lipoleiomyoma with a complex rearrangement including chromosomes 7, 12, and 14. Genes Chrom Cancer. 2000;27:209–15. [PubMed] [Google Scholar]
- 34.Finelli P, et al. The High Mobility Group A2 gene is amplified and overexpressed in human prolactinomas. Cancer Res. 2002;62:2398–405. [PubMed] [Google Scholar]
- 35.Fedele M, et al. Truncated and chimeric HMGI-C genes induce neoplastic transformation of NIH3T3 murine fibroblasts. Oncogene. 1998;17:413–8. doi: 10.1038/sj.onc.1201952. [DOI] [PubMed] [Google Scholar]
- 36.Battista S, et al. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999;59:4793–7. [PubMed] [Google Scholar]
- 37.Arlotta P, et al. Transgenic mice expressing a truncated form of the high mobility group i-c protein develop adiposity and an abnormally high prevalence of lipomas. J Biol Chem. 2000;275:14394–400. doi: 10.1074/jbc.m000564200. [DOI] [PubMed] [Google Scholar]
- 38.Chau KY, et al. The architectural transcription factor high mobility group I(Y) participates in photoreceptor-specific gene expression. J Neurosci. 2000;20:7317–24. doi: 10.1523/JNEUROSCI.20-19-07317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasabe T, et al. Cyclic nucleotides and differentiation of retinoblastoma. Folia Ophthalmol. 1991;42:1200–6. [Google Scholar]
- 40.Vanhamme L, Szpirer C. Transforming activity of the human mammary line HBL100 DNA is associated with SV40 T antigen genetic information integrated in its genome. Carcinogenesis. 1988;9:653–5. doi: 10.1093/carcin/9.4.653. [DOI] [PubMed] [Google Scholar]
- 41.Johnson KR, et al. Complete murine cDNA sequence, genomic structure, and tissue expression of the high mobility group protein HMG-I(Y) J Biol Chem. 1988;263:18338–42. [PubMed] [Google Scholar]
- 42.Chiappetta G, et al. High level expression of the HMGI(Y) gene during embryonic development. Oncogene. 1996;13:2439–46. [PubMed] [Google Scholar]
- 43.Scala S, et al. Adenovirus-mediated suppression of HMGI(Y) protein synthesis as potential therapy of human malignant neoplasias. Proc Natl Acad Sci USA. 2000;97:4256–61. doi: 10.1073/pnas.070029997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chau KY, Ono SJ. Gene transfer into retinoblastoma cells. Biotechniques. 1999;26:444–6. doi: 10.2144/99263bm17. [DOI] [PubMed] [Google Scholar]
- 45.Chau K, et al. A novel downstream positive regulatory element mediating transcription of the human high mobility group (HMG) I-C gene. FEBS Lett. 1999;457:429–36. doi: 10.1016/s0014-5793(99)01100-x. [DOI] [PubMed] [Google Scholar]
- 46.Berlingieri MT, et al. Inhibition of HMGI-C protein synthesis suppresses retrovirally induced neoplastic transformation of rat thyroid cells. Mol Cell Biol. 1995;15:1545–53. doi: 10.1128/mcb.15.3.1545. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.White JB, Taylor RE, Pittler SJ. Reproducible high efficiency gene transfer into Y79 retinoblastoma cells using adenofection. J Neurosci Methods. 2001;106:1–7. doi: 10.1016/s0165-0270(00)00368-x. [DOI] [PubMed] [Google Scholar]
- 48.Chau K-Y, et al. The gene for the human architectural transcription factor HMGI-C consists of five exons each coding for a distinct functional element. Nucleic Acids Res. 1995;23:4262–6. doi: 10.1093/nar/23.21.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prestridge DS. Signal scan: a computer program that scan DNA sequences for eukaryotic transcriptional elements. CABIOS. 1991;7:203–6. doi: 10.1093/bioinformatics/7.2.203. [DOI] [PubMed] [Google Scholar]
- 50.Ishii S, et al. Characterization and sequence of the promoter region of the human epidermal growth factor receptor gene. Proc Natl Acad Sci USA. 1985;82:4920–4. doi: 10.1073/pnas.82.15.4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedmann M, et al. Organization, inducible-expression and chromosome localization of the human HMG-I(Y) nonhistone protein gene. Nucleic Acids Res. 1993;21:4259–67. doi: 10.1093/nar/21.18.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]