

Abstract
This review outlines the mechanisms underlying the interaction between the nervous and immune systems of the host in response to an immune challenge. The main focus is the cholinergic anti-inflammatory pathway, which we recently described as a novel function of the efferent vagus nerve. This pathway plays a critical role in controlling the inflammatory response through interaction with peripheral α7 subunit–containing nicotinic acetylcholine receptors expressed on macrophages. We describe the modulation of systemic and local inflammation by the cholinergic anti-inflammatory pathway and its function as an interface between the brain and the immune system. The clinical implications of this novel mechanism also are discussed.
INTRODUCTION
Inflammation is a normal response to disturbed homeostasis caused by infection, injury, and trauma. The host responds with a complex series of immune reactions to neutralize invading pathogens, repair injured tissues, and promote wound healing (1,2). The onset of inflammation is characterized by release of pro-inflammatory mediators including tumor necrosis factor (TNF), interleukin (IL)-1, adhesion molecules, vasoactive mediators, and reactive oxygen species (1–3). The early release of pro-inflammatory cytokines by activated macrophages has a pivotal role in triggering the local inflammatory response (2). Excessive production of cytokines, such as TNF, IL-1β, and high mobility group B1 (HMGB1), however, can be more injurious than the inciting event, initiating diffuse coagulation, tissue injury, hypotension, and death (2,4–6). The inflammatory response is balanced by anti-inflammatory factors including the cytokines IL-10 and IL-4, soluble TNF receptors, IL-1 receptor antagonists, and transforming growth factor (TGF)β. Although simplistic (7,8), the pro-/anti- terminology is widely used in the discussion of the complex cytokine network. Apart from their involvement in local inflammation, TNF and IL-1β are signal molecules for activation of brain-derived neuroendocrine immunomodulatory responses. Neuroendocrine pathways, such as the hypothalamo-pituitary-adrenal (HPA) axis and the sympathetic division of the autonomic nervous system (SNS) (9–15), control inflammation as an anti-inflammatory balancing mechanism. The host thereby mobilizes the immunomodulatory resources of the nervous and endocrine systems to regulate inflammation.
Restoration of homeostasis as a logical resolution of inflammation does not always occur. Insufficient inflammatory responses may result in increased susceptibility to infections and cancer. On the other hand, excessive responses are associated with autoimmune diseases, diabetes, sepsis, and other debilitating conditions. When control of local inflammatory responses is lost, pro-inflammatory mediators can spill into the circulation, resulting in systemic inflammation that may progress to shock, multiple organ failure, and death. Effective therapies for diseases of excessive inflammation are needed.
We recently discovered the anti-inflammatory role of the vagus nerve (16,17) in an animal model of endotoxemia and shock. This previously unrecognized immunomodulatory circuit termed the “cholinergic anti-inflammatory pathway” is a mechanism for neural inhibition of inflammation (18,19), and interfaces the brain with the immune system. Can it be a “missing link” in neuroimmunomodulation that will validate the notion of a mind-body connection?
This review outlines brain-derived control mechanisms of immune function and specifically the role of cholinergic anti-inflammatory pathway in the regulation of inflammation.
INTERACTION BETWEEN THE IMMUNE SYSTEM AND BRAIN IN RESPONSE TO IMMUNE CHALLENGE
Communication between the immune, nervous, and endocrine systems is essential for host defense and involves a variety of mediators including cytokines, neurotransmitters, hormones, and humoral factors. The influence of the brain on immune function and the mechanisms involved in these interactions have been elucidated over the past 3 decades (17–19). Two important questions arise when describing the brain-derived immunomodulation: 1) How is the brain initially signaled by cytokines to trigger corresponding neural and neuroendocrine responses; and 2) how is immunomodulation achieved through these mechanisms?
Immune-To-Brain Communication
The brain can monitor immune status and sense peripheral inflammation through 2 main pathways: neural and humoral (Figure 1; for a review, see 20, 21).
Figure 1.
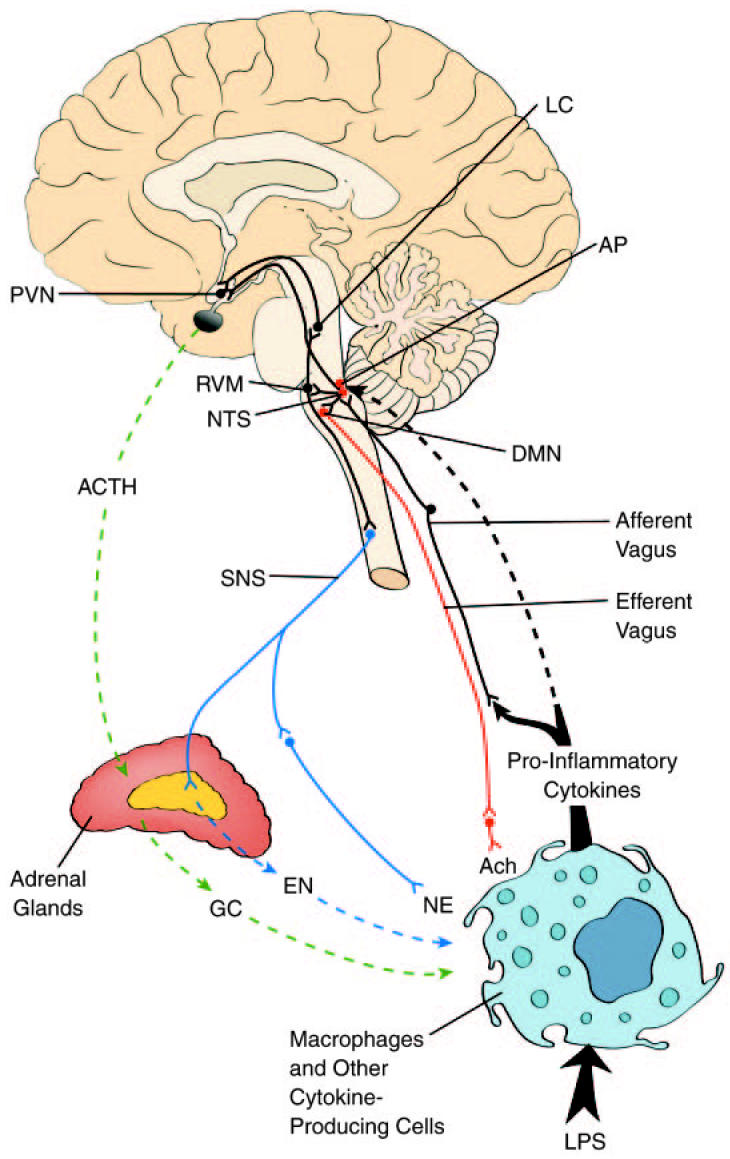
Neural and humoral pathways in immunomodulation. During immune challenge activated macrophages and other immune and nonimmune cells release cytokines that signal the brain for activation of immunomodulatory mechanisms. Central immunomodulation is achieved by the cholinergic anti-inflammatory pathway, HPA axis, and SNS (see text for details). AP, area postrema; NTS, nucleus tractus solitarius; DMN, dorsal motor nucleus of the vagus; PVN, paraventricular nucleus; RVM, rostral ventrolateral medulla; LC, locus coeruleus; SNS, sympathetic nervous system; ACTH, adrenocorticotropin hormone; GC, glucocorticoids; EN, epinephrine; NE, norepinephrine; ACh, acetylcholine; LPS, lipopolysaccharide (endotoxin).
Neural pathway
The neural mechanism relies upon activation of vagus nerve afferent sensory fibers that signal the brain that inflammation is occurring. Immunogenic stimuli activate vagal afferents either directly by cytokines released from dendritic cells, macrophages, and other vagal-associated immune cells, or indirectly through the chemoreceptive cells located in vagal paraganglia (22). For instance, intraperitoneal administration of endotoxin can induce IL-1β immunoreactivity in dendritic cells and macrophages within connective tissues associated with the abdominal vagus nerve and subsequently in vagal paraganglia and afferent fibers (23). Visceral vagus afferent fibers, residing in the nodose ganglion, terminate primarily within the dorsal vagal complex (DVC) of the medulla oblongata. The DVC consists of the nucleus tractus solitarius (NTS), the dorsal motor nucleus of the vagus (DMN), and the area postrema (AP) (24). The DMN is the major site of origin of preganglionic vagus efferent fibers; cardiovascular vagal efferents also originate within the medullar nucleus ambiguous. The AP, which lacks a blood-brain barrier, is an important circumventricular organ and site for humoral immune-to-brain communication, as described below. The main portion of vagal sensory input is received by neurons in the NTS that coordinate autonomic function and interaction with the endocrine system (25). Ascending projections from the NTS reach forebrain sites including hypothalamic nuclei, amygdala, and insular cortex. One of the hypothalamic nuclei receiving input from the NTS is the paraventricular nucleus (PVN). The PVN is associated with the synthesis and release of corticotropin releasing hormone (CRH), an important substance in the HPA axis. This ascending link between the NTS and PVN provides a pathway that can modulate neurohormonal anti-inflammatory responses. Synaptic contacts also exist between the neurons in the NTS and C1 neurons in the rostral ventrolateral medulla (RVM), which occupies an important role in control of cardiovascular homeostasis. The RVM neurons in turn project to the locus coeruleus (LC), which is the main source of noradrenergic innervations of higher brain sites, including the hypothalamus and PVN. Projections emanate from the RVM and LC to sympathetic preganglionic neurons in the spinal cord. There are also descending pathways from the PVN to the RVM and NTS. These ascending and descending connections provide a neuronal substrate for interaction between HPA axis and SNS as an immunomodulatory mechanism.
The transmission of cytokine signals to the brain through the vagal sensory neurons depends upon the magnitude of the immune challenge. Subdiaphragmatic vagotomy inhibits the stimulation of the HPA axis (26) and norepinephrine (NE) release in hypothalamic nuclei (27) in response to intraperitoneal administration of endotoxin or IL-1β. Intravenous endotoxin administration induces expression of the neural activation marker c-Fos in the brainstem medulla, regardless of the integrity of the vagus nerve (28). Vagotomy fails to suppress high dose endotoxin-induced IL-1β immunoreactivity in the brain (29) and increases blood corticosterone levels (30). It is likely that the vagal afferent neural pathway plays a dominant role in mild to moderate peripheral inflammatory responses, whereas acute, robust inflammatory responses signal the brain primarily via humoral mechanisms.
Humoral pathway
A large body of evidence supports the involvement of humoral mechanisms in the immune-to-brain communication, especially in cases of systemic immune challenge (21, 31–33). The question remains, however, of how the circulating cytokines interact with brain structures involved in the anti-inflammatory response, and how circulating cytokines induce central cytokine production associated with fever and sickness. Blood-borne IL-1β and TNF can cross the blood-brain barrier and enter cerebrospinal fluid and the interstitial fluid spaces of the brain and spinal cord by a saturable carrier-mediated mechanism (34) that may function only at very high plasma cytokine concentrations. Cytokines also can bind to receptors at the surface of the endothelium of the brain capillaries and can enhance the synthesis and release of soluble mediators such as prostaglandins and nitric oxide, which diffuse into the brain parenchyma and modulate the activity of specific groups of neurons (21,35,36). It has been suggested that prostaglandins mediate fever and HPA axis activation (13).
Cytokine-to-brain communication also may occur via circum-ventricular organs that lack normal blood-brain barrier function. Among the circumventricular organs, the AP appears to represent the best candidate for such a transduction site (for a review, see 37). The AP is located in the floor of the caudal 4th ventricle (38) and dendrites of neurons in the NTS and DMN penetrate both the AP and floor of the 4th ventricle (39,40). The close proximity of AP to NTS and RVM and the existing neural connections provide a way of signaling the SNS and HPA axis. Cytokine-induced production of prostaglandins within the AP, NTS, and RVM may activate the catecholamine projections to the PVN, resulting in subsequent HPA axis activation (37). This is one possible interaction between the neural and humoral mechanisms of immune-to-brain communication through which the brain mediates anti-inflammatory responses.
Apart from their function in signaling the brain for immunomodulatory responses, cytokines play a multifunctional role in brain injury and neurodegenerative diseases (for review, see 41–43).
Brain-To-Immune Communication
The brain exerts strong modulatory effects on immune function by activation of the HPA axis and the SNS, which results in increased synthesis and release of glucocorticoids and catecholamines (see Figure 1). The immunomodulating properties of α-melanocyte stimulating hormone (α-MSH) and estrogens also are known. The HPA axis is a neurohormonal pathway; its role in the regulation of the immune function has been widely studied (for reviews, see 14,44–46). The components of the HPA axis are the hypothalamic PVN, the anterior pituitary gland, and the adrenal cortex. Specialized neurons in PVN synthesize CRH, which is released into the pituitary portal blood system and stimulates the synthesis of adrenocorticotropin hormone (ACTH) from the anterior pituitary. ACTH is the main inducer of the synthesis and release of immunosuppressive glucocorticoids (cortisol in humans and corticosterone in rats) from the adrenal cortex. Pro-inflammatory cytokines trigger the HPA axis via the neural or humoral mechanisms described above. At both the hypothalamic and pituitary level, the HPA axis is subject to a classical negative feedback loop by the final product: glucocorticoids. ACTH also inhibits the synthesis of CRH from the PVN. The hypothalamo-pituitary circuit of the HPA axis is regulated by neural mechanisms including acetylcholine (ACh)-, catecholamine-, GABA-, serotonin-, and histamine-mediated modulation.
Glucocorticoids exert their effects by binding to intracellular receptors and subsequently triggering up-regulation or down-regulation of gene expression (47). Apart from triggering the activation of the HPA axis, cytokines such as IL-1 and IL-6 also can alter peripheral glucocorticoid effects by directly influencing the function of corresponding glucocorticoid receptors (45). Immunosuppressive glucocorticoid influence is mainly linked to suppression of nuclear factor-κB activity (14,48,49), which plays an important role in regulating cytokine synthesis (50). As summarized by Webster and others (14), glucocorticoids inhibit the synthesis of pro-inflammatory cytokines, such as TNF, IL-1, IL-8, IL-11, IL-12, and interferon-γ; and they activate the synthesis of the anti-inflammatory cytokines IL-4 and IL-10. Inhibition of neutrophil, eosinophil, monocyte, and macrophage infiltration, and adhesion molecule expression are attributed to glucocorticoid suppression of local inflammation (14,45). Glucocorticoids are also potent clinical anti-inflammatory agents (45).
The SNS plays a dual role in the regulation of inflammation, because it mediates both pro- and anti-inflammatory activities; it is thus an integral component of the host defense system against injury and infection (15,51). The locus coeruleus (LC) and RVM, brain functional components of the SNS, project to sympathetic preganglionic cholinergic neurons in the spinal cord. Sympathetic innervation of primary (thymus and bone marrow) and secondary (spleen, lymph nodes, and tissues) lymphoid organs is the anatomic basis for modulation of immune function by the SNS (15,51,52). Sympathetic postganglionic norepinephrine (NE)-ergic and neuropeptide Y-ergic neurons also innervate blood vessels, the heart, the liver, and the gastrointestinal tract (15,52). NE, released from sympathetic postganglionic nerve endings, exerts anti-inflammatory effects by interacting with adrenoceptors expressed on lymphocytes and macrophages. Adrenoceptors belong to the G-protein coupled receptor superfamily and are divided into α and β subtypes, which can be further subdivided. Sympathetic control on cytokine production is achieved by 2 mechanisms: (1) synaptic-like junctions with corresponding adrenoceptors (as in the spleen) and (2) nonsynaptic, distant mechanisms, such as NE diffusion through the parenchyma before interaction with the receptor. The nonsynaptic mechanism plays a dominant physiological role (15). The release of NE is subject to complex presynaptic regulation, involving the effects of neuropeptide Y, acetylcholine, dopamine, prostaglandins, and other micro-environmental factors. Sympathetic immunomodulation also is mediated via epinephrine, and to a lesser extent, by NE released from the chromaffin cells of the adrenal medulla. The chromaffin cells represent homologs of the sympathetic ganglia. Activation of the preganglionic sympathetic neurons innervating these cells leads to an increase in the release of catecholamines in the bloodstream, which can act systematically as hormones. Thus, sympathetic neural regulation is converted into “hormonal regulation” within the adrenal glands. The adrenals are therefore an important peripheral component of the CNS-controlled immunoregulation responsible for the synthesis of glucocorticoids (from the cortical cells) and catecholamines (from the medullar chromaffin cells).
SNS activation protects the organism from the detrimental effects of pro-inflammatory cytokines. Activation of β-adrenoceptors leads to marked inhibition of endotoxin induced serum TNF, IL-1, IL-12, interferon-γ, and nitric oxide production, and elevation of IL-6 and IL-10. Stimulation of β-adrenoceptors also is accompanied by suppression of the TNF and IL-1 expression caused by hemorrhagic shock. NE and epinephrine inhibit production of pro-inflammatory cytokines through stimulation of β2-adrenoceptor-cAMP-protein kinase A pathway, and they stimulate the synthesis of anti-inflammatory cytokines. Healthy volunteers receiving epinephrine and subsequent endotoxin treatment developed significantly decreased TNF levels and elevated IL-10 levels, as compared with controls exposed to endotoxin alone (53), raising the possibility that pharmacological control of cytokine production in septic patients could be achieved by selective adrenoceptor agonists and antagonists. A catecholamine-based strategy for treating sepsis and its complications is difficult, however, because of the broad effects of these agents and the need to balance the cardiovascular and immunomodulatory drug effects (15).
During the early stages of some cases of inflammation, stimulation of the SNS can be associated with activation of the local inflammatory responses and neutrophil accumulation (1,54). NE stimulation of the α2–subtype adrenoceptor has been linked to an increase in endotoxin-induced production of TNF and other cytokines (15,55). This dual role of the SNS in immunomodulation agrees with the general mode of SNS function, depending on the peripheral receptors involved. A classic example of dual effects is the “fight-or-flight” response, leading to increased perfusion of the heart, skeletal muscles, and brain, and release of glucose from the liver. Gastrointestinal motility and the blood supply to the skin are simultaneously depressed.
Anti-inflammatory properties of α-MSH have been shown in animal models of inflammation including sepsis and rheumatoid arthritis (for a review, see 56, 57). In rodents α-MSH is mainly produced from the intermediate lobe of the pituitary gland. The cells of the pituitary pars distalis and various extrapituitary cells, such as monocytes, astrocytes, and keratinocytes, are the source of this hormone production in humans. The activity of α-MSH is mediated through G-protein coupled melanocortin receptors widely distributed in peripheral tissues and in the brain. Immunomodulatory effects of α-MSH are associated with stimulation of its receptors on peripheral immune target cells and on glial cells in brain; α-MSH may also exert indirect effects via a brain-spinal cord pathway and sympathetic neurons (58). Although the exact molecular mechanisms of α-MSH immunosuppression and pro-inflammatory cytokine downregulation are not well understood, hormonal inhibition of nuclear factor κB may play an important role. The administration of endotoxin to normal human subjects is accompanied by an increase in α-MSH blood concentrations (in addition to ACTH) and decreased TNF plasma levels, demonstrating the role of α-MSH in the inflammatory response (59). It is not clear how pro-inflammatory cytokines cause stimulation of pituitary α-MSH secretion, but the ascending pathways from NTS to the hypothalamus underlying the HPA axis activation may play a role.
Female sex hormones and estrogens, in particular, also exert immunomodulatory and anti-inflammatory effects. Because their synthesis and blood concentrations are under control of hypothalamo-pituitary hormones, estrogen-affected immune functions can be imputed to the brain. Female sex hormones play immunoregulatory roles during pregnancy and in diseases like rheumatoid arthritis and osteoporosis (60). The classic mechanism of steroid hormone action (as described above for glucocorticoids) may contribute to estrogen immunomodulating activity. Estrogens are neuroprotectors (61); they prevent cartilage degradation during inflammation associated with increased production of IL-1 (62); and they inhibit the production of pro-inflammatory cytokines at different stages of their synthesis (63,64).
CHOLINERGIC ANTI-INFLAMMATORY PATHWAY
The sympathetic and parasympathetic parts of the autonomic nervous system rarely operate alone; autonomic responses represent the interplay of both parts. A link between the parasympathetic part of the autonomic nervous system and immunoregulatory processes was suggested 30 years ago, when alleviation of T-lymphocyte cytotoxicity by muscarinic cholinergic stimulation was noted (65). Despite this observation, the role of the parasympathetic/vagal efferents in immunomodulation is not completely understood (66–68).
We recently demonstrated the existence of a parasympathetic pathway of modulation of systemic and local inflammatory responses (16,69), which focuses attention on neural immunomodulatory mechanisms via the vagus nerve.
Evidence for Parasympathetic (Vagus Nerve) Control of Systemic And Local Inflammation
Acetylcholine is an important neurotransmitter and neuromodulator in the brain. It mediates neural transmission in the ganglion synapses of both sympathetic and parasympathetic neurons, and is the principle neurotransmitter in the postganglionic parasympathetic/vagal efferent neurons. Acetylcholine acts through 2 types of receptors: muscarinic (metabotropic) (70) and nicotinic (ionotropic) (71). In addition to the brain and “wire-innervated” peripheral structures, the RNA for these receptor subtypes (muscarinic) and subunits (nicotinic) has been detected on mixed populations of lymphocytes and other immune and non-immune cytokine-producing cells (72–77). Most of these cells can also produce acetylcholine (78).
We recently discovered that the α7 subunit of the nicotinic acetylcholine receptor is expressed on macrophages (16). Acetylcholine significantly and concentration-dependently decreases TNF production by endotoxin-stimulated human macrophage cultures via a post-transcriptional mechanism. Using specific muscarinic and nicotinic agonists and antagonists, we demonstrated the importance of an α-bungarotoxin-sensitive nicotinic receptor in the inhibition of TNF synthesis in vitro by acetylcholine. Acetylcholine also is effective in suppressing other endotoxin-inducible pro-inflammatory cytokines, such as IL-1β, IL-6, and IL-18, by a post-transcriptional mechanism; release of the anti-inflammatory cytokine IL-10 from endotoxin-stimulated macrophages is not affected by acetylcholine (16).
Because of the immunosuppressive effects of acetylcholine in vitro, we studied the possible immunonodulatory role of the parasympathetic division of the autonomic nervous system in vivo. In a rat model, vagotomy without electrical stimulation significantly increases serum and liver TNF levels in response to intravenously administered endotoxin (Figure 2A and 2B), suggesting a direct role of efferent vagus neurons in the regulation of TNF production in vivo. Augmentation of efferent vagus nerve by direct electrical stimulation significantly attenuates endotoxin-induced serum and hepatic TNF (see Figure 2A and 2B). TNF amplifies inflammation by activating the release of pro-inflammatory mediators such as IL-1, HMGB1, nitric oxide, and reactive oxygen species (3,6). TNF also plays an essential role in endotoxin-induced shock by inhibiting cardiac output, activating microvascular thrombosis, and modulating capillary leakage syndrome (4,79). These activities of TNF are consistent with the finding that attenuation of serum TNF via cervical vagus nerve stimulation prevents hypotension and shock in animals exposed to lethal doses of endotoxin (see Figure 2C) (16). Animals subjected to vagotomy without vagus nerve stimulation develop profound shock more quickly than sham-operated animals (see Figure 2C), demonstrating a role for vagus nerve efferent signaling in maintaining immunological homeostasis. Importantly, the immunomodulatory effects of the efferent vagus nerve also play a role in localized peripheral inflammation, because electrical stimulation of the distal vagus nerve also inhibits the local inflammatory response in a standard rodent model of carrageenan-induced paw edema (69). Pretreatment with acetylcholine, muscarine, or nicotine localized within the site of inflammation also prevents the development of hind paw swelling (69). Vagal efferents are distributed throughout the reticuloendothelial system and other peripheral organs, and the brain-derived motor output through vagus efferent neurons is rapid. The cholinergic anti-inflammatory pathway is therefore uniquely positioned to modulate inflammation in real time.
Figure 2.
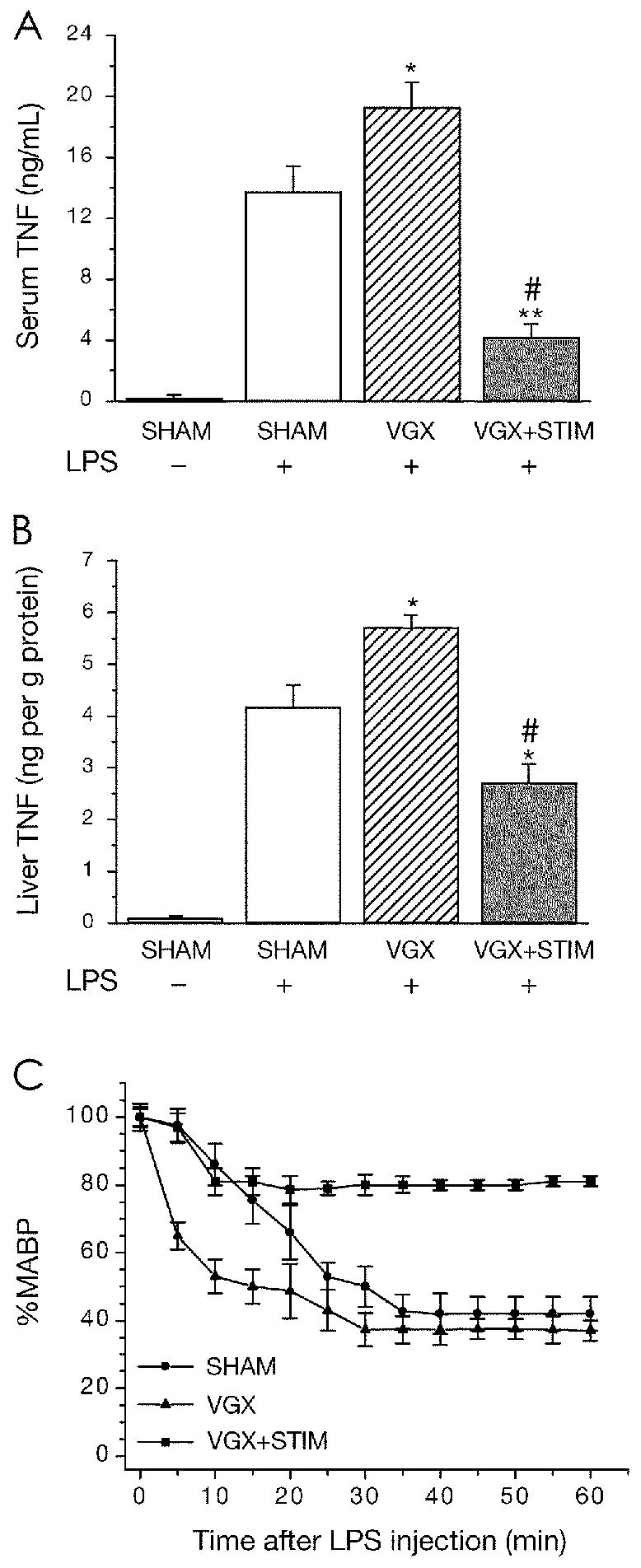
Vagus nerve stimulation attenuates the endotoxin-induced serum TNF response, hepatic TNF response, and development of endotoxemic shock. Rats were subjected to sham surgery (A and B: Sham, □; C:•; n = 7), bilateral cervical vagotomy (A and B: VGX, ▧, C:▴; n = 7), or vagotomy and electrical stimulation (A and B: VGX + STIM, ▩ C: ▪; n = 7). A: Serum TNF response. B: Hepatic TNF response. C: Development of endotoxemic shock. Mean arterial blood pressure data are normalized to MABP at time = 0. Sham-surgery, vagotomy, and electrical stimulation with vagotomy did not significantly affect MABP in vehicle-treated controls (not shown). Data are means +/− SEM. *P < 0.05; **P < 0.005 compared with SHAM + endotoxin; #P < 0.05 compared with VGX + endotoxin. Taken from Borovikova and others (16) with Nature’s copyright permission <http://www.nature.com>.
Pharmacological Activation of the Cholinergic Anti-inflammatory Pathway
It may be possible to activate the cholinergic anti-inflammatory pathway with centrally acting pharmacological agents, because the tetravalent guanylhydrazone CNI-1493 induces vagus nerve firing (69) and confers anti-inflammatory effects through activation of the cholinergic anti-inflammatory pathway in both local and systemic models of inflammation (69,80). Extensive research on the anti-inflammatory activities of CNI-1493 reveals that it confers protection in experimental models of cancer, pancreatitis, rheumatoid arthritis, endotoxin shock, and sepsis (81–83). These studies led to current testing of CNI-1493 in Phase II clinical trials for Crohn’s disease. CNI-1493 was originally developed as an inhibitor of macrophage activation that prevented phosphorylation of p38 mitogen-activated protein kinase (84–86). In models of cerebral ischemia, CNI-1493 delivered via the intracerebroventricular (ICV) route suppresses cerebral TNF synthesis and reduces infarct volume (87). Surprisingly, CNI-1493 administered via the same route suppresses systemic TNF production in response to peripheral endotoxin challenge. Further studies revealed that CNI-1493 stimulates efferent vagus nerve firing (69), suggesting that the cholinergic anti-inflammatory pathway may mediate the peripheral anti-inflammatory activity of this compound.
Intravenous (IV) pretreatment of endotoxemic rats with CNI-1493 leads to a significant and dose-dependent inhibition of serum TNF-release and prevents the development of endotoxin-induced hypotension (80). The lowest IV dose tested in these experiments (100 μg/kg body weight) failed to cause down-regulation of TNF release or prevent hypotension. When 1/100 of this ineffective IV dose was injected ICV, it significantly attenuated serum TNF release and protected against hypotension. Much lower ICV drug doses (in the range 10 to 0.1 ng/kg) also inhibited TNF release and protected against the development of endotoxin-induced shock, suggesting that brain-dependent mechanisms contribute to the systemic anti-inflammatory action of CNI-1493. Similarly, CNI-1493 delivered into the cerebral ventricles at doses significantly lower than peripherally active doses also suppressed local inflammation in a standardized model of carrageenan-induced paw edema (69).
The anti-inflammatory effects of CNI-1493 require the vagus nerve, regardless of the route (ICV or IV) of drug administration, because bilateral cervical vagotomy eliminates its effects. These results indicate that CNI-1493 mediates protection against local and systemic inflammation and shock via the cholinergic anti-inflammatory pathway. Activation of the cholinergic anti-inflammatory pathway by centrallyacting compounds such as CNI-1493, therefore, may hold significant clinical potential for treatment of sepsis and other cytokine-mediated diseases.
It is plausible to consider two potential mechanisms through which CNI-1493 activates vagus nerve efferent signaling: this compound may either gain access to the DMN and activate vagus efferent signals directly, or it may activate other neurons within the DVC or higher brain structures and indirectly activate the efferent vagus nerve. A variety of receptors have been identified within the DVC, the primary termination site of afferent vagus fibers. Within the DMN, for example, studies have revealed the presence of muscarinic receptors, but they have not been associated with vagal efferent neurons (88,89). Muscarinic (especially the M2 subtype) and nicotinic binding sites have been detected on the caudal and medial parts of the NTS (90). The cholinergic system in the NTS has been identified on the basis of the presence of choline acetyltransferase, acetylcholine-esterase, and acetylcholine (88,91). The NTS cholinergic system participates in the regulation of cardiovascular output and modulation of the baroreceptor reflex, which is centrally mediated by glutamate. The presence of glutamate receptors is also well documented within the DVC (92–94). Vagal afferents (cardiovascular and abdominal visceral) terminating in the NTS are predominantly glutamatergic; they synapse on NTS neurons through both NMDA and non-NMDA receptors (94,95). Exposure to either IL-1 or endotoxin can activate the vagal glutamatergic system in the NTS. Both types of glutamate receptors transmit AP neuron excitatory inputs to the NTS (96). NMDA glutamate receptors also are present on AP and DMN neurons. Vagus preganglionic motor neurons located in the DMN extend dendritic fields to the NTS. Nicotinic (especially the α7 subtype), neuropeptide Y, GABA (A and B), neurokine-1, and neurokine-3 receptors have each been localized to vagus efferent neurons in the DMN (97–100). Ionotropic P2X purinoceptors of ATP (acting as a neurotransmitter) are present on the DMN and AP neurons (101). It is possible that some of these receptor mechanisms represent critical components of the pathways leading to pharmacological activation of the cholinergic anti-inflammatory pathway. CNI-1493 may also activate other receptors in higher brain regions, and intermediate neurons may relay CNI-1493 signaling to the efferent vagus nerve.
Nicotinic Acetylcholine Receptor α7 Subunit Is an Essential Component of the Cholinergic Anti-inflammatory Pathway
Inhibition of peripheral TNF synthesis by the efferent vagus nerve implicates a mechanism for signaling from the vagus nerve to TNF-producing cells, such as macrophages. In a series of experiments using specific cholinergic agonists and antagonists, as well as antisense and gene knockout approaches, we have shown that the α7 subunit of the nicotinic acetylcholine receptor is expressed on macrophages and mediates the anti-inflammatory activity of the efferent vagus nerve.
Nicotinic acetylcholine receptors are a family of ligandgated, pentameric ion channels. The main function of this receptor family is to transmit acetylcholine signals at neuromuscular junctions and in the central and peripheral nervous systems (71,102–104). In humans, 16 different nicotinic acetylcholine receptor subunits (α1–7, α9–10, β1–4, δ, ɛ,γ) have been identified (71,104). These subunits have the potential to form a large number of homo- and heteropentameric receptors with distinct properties and functions. Among the 16 subunits, only the α1, α7, and α9 subunits bind an antagonist derived from snake venom, α-bungarotoxin; the α-bungarotoxin-binding activity of the α10 subunit is unknown.
Macrophages specifically bind FITC-labeled α-bungarotoxin, an antagonist of a subset of nicotinic receptors. This surface binding can be competed with nicotine, suggesting that macrophages express functional α1, α7, and/or α9 subunits of the nicotinic acetylcholine receptor. RT-PCR analyses revealed mRNA expression of the α1, α7, and α10 subunits, but not α9. Further characterization by western blotting analyses and pull-down methods showed that fully differentiated primary human macrophages specifically express the α7 subunit (105).
The functional relevance of the macrophage nicotinic receptor α7 subunit in the cholinergic anti-inflammatory pathway was tested using antisense oligonucleotides to the α7 subunit. Inhibition of α7 subunit expression restores the endotoxin-stimulated TNF responses in the presence of nicotine, whereas antisense oligonucleotides to the α1 and α10 subunits, under similar conditions, fail to significantly change TNF release in the presence of nicotine (105). The essential role of the α7 subunit in mediating the activity of the cholinergic anti-inflammatory pathway in vivo has been defined in α7 subunit-deficient mice. These gene knockout mice develop normally and show no gross anatomical defects (106); however, these animals are more sensitive to inflammatory stimuli, because α7 subunit-deficient mice release significantly more TNF, IL-1, and IL-6 into the serum during endotoxemia as compared with wild type mice (105). Electrical stimulation of the vagus nerve inhibits serum and tissue levels of pro-inflammatory cytokines in endotoxemic wild type mice, but is ineffective in α7-deficient mice (Figure 3) (105). Peritoneal macrophages isolated from mice lacking the α7 subunit do not respond to acetylcholine and nicotine, and continue to produce TNF in the presence of these cholinergic agonists. α7 Nicotinic receptor subunits have been found in the superior cervical and pancreatic ganglia, but no functional role for this subunit in ganglionic transmission has been demonstrated in vivo. α3 Nicotinic receptor subunits mediate fast synaptic transmission in the autonomic ganglia. These observations suggest that the higher sensitivity of α7 subunit-deficient mice to inflammatory stimuli cannot be attributed to impaired parasympathetic and/or sympathetic ganglionic transmission. Taken together these data indicate that the nicotinic acetylcholine receptor α7 subunit is a necessary component of the cholinergic anti-inflammatory pathway. Moreover, the cholinergic anti-inflammatory pathway represents a highly specific function of the efferent vagus nerve, because it can signal through nicotinic α7 receptors on macrophages, rather than “classical” muscarinic acetylcholine receptors.
Figure 3.
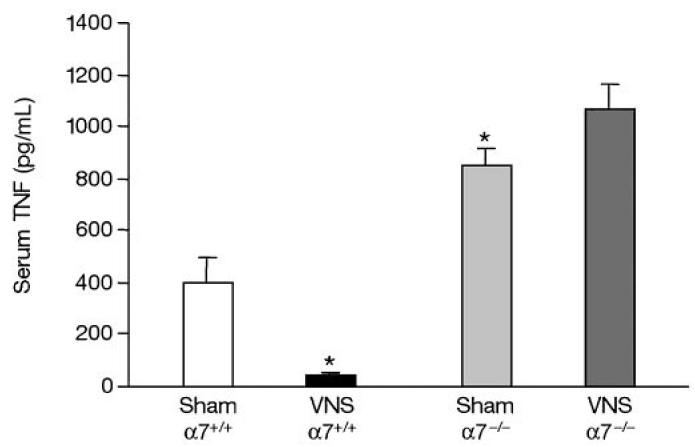
Vagus nerve stimulation does not inhibit TNF in nicotinic acetylcholine receptor α7 subunit–deficient mice. α7 Subunit–deficient mice (−/−) or age- and sex-matched wild-type littermates (+/+) were subjected to either sham operation (Sham) or vagus nerve stimulation (VNS, left vagus, 1 volt, 2 ms, 1 Hz); blood was collected 2 h after endotoxin administration. Serum TNF levels were determined by ELISA. Sham α7+/+, n = 10; VNS α7+/+, Sham α7−/−, VNS α7−/−, n = 11. *P < 0.05 compared with Sham α7+/+. Taken from Wang and others (105) with Nature’s copyright permission: <http://www.nature.com>.
Integration of the Cholinergic Anti-inflammatory Pathway in Brain-Derived Immunomodulation
Involvement of vagus efferent neurons in neuroimmunomodulation is supported by the protective role of the cholinergic anti-inflammatory pathway in systemic and local inflammation. This previously unrecognized neural-immune circuit sheds new insight on brain regulation of immune function (19), and several aspects of this “missing link” in neuroimmunomodulation are under investigation in our laboratory.
The role of the efferent vagus nerve in minute-to-minute modulation of immune activation remains uncertain. Bilateral cervical vagotomy renders animals more sensitive to endotoxemic shock, and results in increased serum and organ TNF levels (see Figure 2A and 2B), suggesting that efferent vagus activity exerts a tonic effect on peripheral inflammatory responses. Electrical stimulation appears to potentiate the tonic immunosuppression conferred by the vagus nerve. These findings might be related to the intriguing question of whether the parasympathetic part of the autonomic nervous system is depressed during sepsis. In the systemic inflammatory response syndrome, the initial sympathocardiac activation decreases during the progression to sepsis (107,108). Spectral analysis of heart rate variability, or the length of time between heartbeats, can provide important information about the status of the autonomic nervous system. Analysis of instantaneous heart rate measurements identifies low frequency and high frequency oscillations, which are indicative of sympathetic and parasympathetic tone, respectively. In comparison to survivors of critical illness, nonsurvivors have significantly less heart rate variability, reduced sympathovagal balance, and reduced parasympathetic tone; reduced sympathovagal balance is associated with an increased risk for death (107,108). Interestingly, low sympathovagal balance (as indicated by a low frequency/high frequency heart rate oscillation ratio of <1) may be an early marker for sepsis in the critically ill, since patients who went on to develop sepsis were admitted to the intensive care unit with a lower low frequency/high frequency ratio than those patients who remained sepsis-free (107). In total, these findings suggest that autonomic dysfunction is an important aspect of lethal critical illness and sepsis. The precise influence of the parasympathetic division of the autonomic nervous system, particularly in terms of afferent or efferent signaling, is unclear from these studies; however, the sensitivity of animals devoid of the nicotinic acetylcholine receptor α7 subunit to endotoxin challenge (105) implies a critical role of the cholinergic anti-inflammatory pathway in regulation of peripheral inflammation.
How is the cholinergic anti-inflammatory pathway activated in response to inflammatory stimuli? It appears that the general pathways of signaling the brain for triggering the HPA axis and the SNS described above may play a role. Proinflammatory cytokines released upon immune challenge can activate vagal afferent signaling and subsequent direct or indirect (through NTS neurons) activation of vagal efferents in the DMN. Thus, the sensory vagal afferents, together with the regulatory vagus efferents, form an inflammatory reflex that continually monitors and modulates the inflammatory status in the periphery (18). The cholinergic anti-inflammatory pathway also can be activated by cytokine signals via AP. Thus, in addition to the cholinergic efferent fibers, the cholinergic anti-inflammatory pathway may be comprised of at least two brainstem medullar structures (such as NTS and DMN) that may be signaled by pro-inflammatory cytokines through either neural (vagus afferents) or humoral (AP) mechanisms. Intravenous administration of IL-1β induces activation of the efferent vagus fibers, innervating the thymus (67). Indeed, administration of endotoxin induces neuronal activation in the DMN as well as the NTS and AP (28). Absence of this neuronal surveillance results in higher levels of inflammation as seen in vagotomized endotoxemic mice (16). This centrally integrated vagal anti-inflammatory reflex is similar for example to the vago-vagal reflex mechanism controlling the gastrointestinal tract. These observations suggest that the cholinergic anti-inflammatory pathway is activated during the inflammatory response.
The NTS may integrate the cholinergic anti-inflammatory pathway with other central immunomodulatory responses, because the NTS can transmit afferent vagus nerve signals to two areas of the brain involved in neuroimmunomodulation. For example, bidirectional neuronal circuits between the NTS and the hypothalamic PVN can activate the HPA axis, leading to glucocorticoid-mediated immunosuppression. NTS neurons also project to brainstem nuclei, such as the RVM and the LC, which may activate the SNS and modulate immune responses (see Figure 1). The key structures in neuroimunomodulation are (1) the NTS, which is associated with the reception and further transmission of the cytokine signal to the cholinergic anti-inflammatory pathway, the SNS, and the HPA axis; (2) the hypothalamic PVN, which is responsible for conversion of the neural signal into a hormonal; and (3) the adrenal glands, which release epinephrine from the chromaffin cells under activation of the SNS and complete the main neurohormonal route via release of glucocorticoids.
Both divisions of the autonomic nervous system are activated by immunogenic stimuli and both contribute to modulation of inflammation. The SNS down-regulates inflammation via β-adrenoceptors. In some cases, however, norepinephrine may increase TNF-α release, most likely through α -adrenoceptors (55). Activation of the cholinergic anti-inflammatory pathway may counteract instances of excessive TNF release. The cholinergic anti-inflammatory pathway and the SNS also act synergistically to control inflammation. While the SNS can cause immunosupression through β-adrenoceptors, vagus nerve downregulation of cytokine production is mediated, at least in part, through nicotinic acetylcholine receptors containing the α7 subunit. In contrast to endocrine-mediated mechanisms, neural regulation of immune responses is rapid and more precisely localized, and thus may be an important early response to peripheral inflammation.
Sympathetic and vagus innervation of the thymus, liver, heart, lungs, gastrointestinal tract, pancreas, and kidneys may provide the anatomical basis for coregulation of tissue macrophages, dendritic cells, mast cells, Kupffer cells (in the liver), and other immune and nonimmune cytokine-producing cells in the tissues. The liver is an important organ in the acute phase of the inflammatory response, supplying the necessary components for the host defense at the site of inflammation and coordinating the activation of acute phase plasma proteins (2). Hepatic sinusoidal macrophages (Kupffer cells) are thought to be the major source of cytokines in endotoxemia (109); therefore, stimulation of efferent vagus neurons may modulate the hepatic inflammatory response. The heart is well-innervated by the 2 divisions of the autonomic nervous system. The autonomic dysfunction (high sympathetic, low parasympathetic tone) that occurs after myocardial infarction is a powerful predictor of early mortality (110). In the heart, resident macrophages and cardiac myocytes are the main sources of TNF, and TNF receptors have been found in cardiac myocytes (111,112). TNF released from both myocardial macrophages and cardiac myocytes contribute to myocardial dysfunction and cardiomyocyte death in sepsis, chronic heart failure, ischemia-reperfusion injury, viral myocarditis, and cardiac allograft rejection (111). The effectiveness of vagus nerve stimulation in inhibiting cardiac TNF (80) warrants further exploration of the immunomodulatory role of the cholinergic anti-inflammatory pathway in the heart and other organs.
Therapeutic Implications of the Cholinergic Anti-inflammatory Pathway
Many current approaches for treatment of unrestrained inflammation are based on direct suppression of pro-inflammatory cytokines or cytokine activity. The identification of the cholinergic anti-inflammatory pathway now suggests several new approaches to modulate cytokines and inflammatory responses to therapeutic advantage. For example, stimulation of the vagus nerve may represent a novel approach to inhibit TNF production and protect against pathological inflammation. Permanently implanted vagus nerve stimulators are clinically approved devices for treatment of epilepsy and depression (113–115). Vagus nerve stimulation prevents seizures by stimulating sensory vagal afferents associated with limbic and cortical function. Although vagal efferents also may be activated as a result of vagus nerve stimulation, no cardiac, gastric, or pulmonary complications have been observed (116). In animals, vagus nerve stimulation causes neural activation (assessed by c-Fos technique) in NTS, LC, DMN, and hypothalamic nuclei, including PVN (117,118). Vagus nerve stimulation enhances the activity of key components of the brain-derived anti-inflammatory response. In addition to the vagal efferents, the HPA axis and the SNS may also be activated as a “side effect” of vagus nerve stimulation. The occurrence of such activation remains to be evaluated by testing the fluctuations in serum glucocorticoid levels in patients with vagus nerve stimulators. If so, it may be possible to use currently existing, approved devices to control inflammatory responses.
The discovery of the cholinergic anti-inflammatory pathway identifies at least 1 receptor type that may be pharmacologically targeted to modulate cytokine activity. The nicotinic acetylcholine receptor α7 subunit is essential for regulation of peripheral inflammatory responses; activation of this receptor via vagus nerve stimulation (105) or cholinergic agonists (119,120) suppresses cytokine release and protects against lethal murine endotoxemia and sepsis. CNI-1493, the tetravalent guanylhydrazone that was instrumental in the discovery of the cholinergic anti-inflammatory pathway, exerts its anti-inflammatory effects in vivo by a central mechanism involving activation of the vagus nerve (69,80). It is possible that other experimental and clinically approved therapeutics function through the unanticipated mechanism of activating neural pathways. For example, low doses of centrally administered α-MSH or salicylates elicit specific peripheral anti-inflammatory responses. Likewise, the cardiac anti-arrhythmic drug amiodarone, as well as aspirin, indomethacin, and ibuprofen substantially increase vagus nerve activity (reviewed in 18). The precise identification of the brain receptor(s) that mediate these effects will facilitate the development of effective, specific receptor agonists to pharmacologically activate the cholinergic anti-inflammatory pathway.
It also is interesting to reconsider alternative therapeutic approaches in light of the cholinergic anti-inflammatory pathway. For instance, hypnosis, meditation, prayer, biofeedback, acupuncture, and even Pavlovian conditioning of immunological responses are believed to involve central mechanisms that modulate experimental systemic or peripheral inflammatory responses. Moreover, autonomic dysfunction occurs not only in association with lethal critical illness and sepsis but also is considered a complication of diabetes, rheumatoid arthritis, and other autoimmune diseases (reviewed in 18). Whether physiological augmentation of vagus nerve activity through any of these methods can modulate disease activity remains an open question.
SUMMARY
We have described the cholinergic anti-inflammatory pathway, a previously unknown neural circuit that provides a new physiological mechanism for immunomodulation. This specific function of the motor branch of the vagus nerve provides new insight into CNS control of peripheral inflammatory responses. Better understanding of the receptor mechanisms and neuronal circuits involved in the cholinergic anti-inflammatory pathway, and evaluation of its stimulation for treatment of inflammatory disorders, may improve the lives of patients with diseases characterized by cytokine excess.
Acknowledgments
Grant support came from the Dept. of Defense (DARPA) and NIH (NIGMS).
REFERENCES
- 1.Sell S. (2001) Immunology, immunopathology, and immunity (6th ed). ASM Press, Washington, D.C.
- 2.Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 3.Koj A. Initiation of acute phase response and synthesis of cytokines. Biochim Biophys Acta. 1997;1317:84–94. doi: 10.1016/s0925-4439(96)00048-8. [DOI] [PubMed] [Google Scholar]
- 4.Tracey KJ, et al. Shock and tissues injury induced by recombinant human cachectin. Science. 1986;234:470–4. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164:1768–73. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 7.Sporn MB. The importance of context in cytokine action. Kidney Int. 1997;51:1352–4. doi: 10.1038/ki.1997.184. [DOI] [PubMed] [Google Scholar]
- 8.Kushner I. Semantics, inflammation, cytokines and common sense. Cytokine Growth Factor Rev. 1998;9:191–6. doi: 10.1016/s1359-6101(98)00016-1. [DOI] [PubMed] [Google Scholar]
- 9.Reichlin S. Neuroendocrine-immune interactions. New Engl J Med. 1993;329:1246–53. doi: 10.1056/NEJM199310213291708. [DOI] [PubMed] [Google Scholar]
- 10.Peristein RS, Whitnall MH, Abrams JS, Mougey EH, Neta R. Synergistic roles of interleukin-6, interleukin-1 and tumor necrosis factor in adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology. 1993;132:946–52. doi: 10.1210/endo.132.3.8382602. [DOI] [PubMed] [Google Scholar]
- 11.Gaillard RC. Neuroendocrine-immune system interactions. Trends Endocrinol Metab. 1995;7:303–9. doi: 10.1016/1043-2760(94)p3206-m. [DOI] [PubMed] [Google Scholar]
- 12.Mulla A, Buchingham JC. Regulation of the hypothalamo-pituitary-adrenal axis by cytokines. Baillieres Best Prac Res Clin Endocriol Metab. 1999;13:503–21. doi: 10.1053/beem.1999.0041. [DOI] [PubMed] [Google Scholar]
- 13.Rivest S. How circulating cytokines trigger the neural circuits that control the hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology. 2001;26:761–88. doi: 10.1016/s0306-4530(01)00064-6. [DOI] [PubMed] [Google Scholar]
- 14.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125–63. doi: 10.1146/annurev.immunol.20.082401.104914. [DOI] [PubMed] [Google Scholar]
- 15.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- 16.Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–61. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 17.Tracey KJ, Czura CJ, Ivanova S. Mind over immunity. FASEB J. 2001;15:1575–6. doi: 10.1096/fj.01-0148hyp. [DOI] [PubMed] [Google Scholar]
- 18.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–9. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 19.Blalock JE. Harnessing a neural-immune circuit to control inflammation and shock. J Exp Med. 2002;195:F25–8. doi: 10.1084/jem.20020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: a review and analysis of alternative mechanisms. Life Sci. 1995;57:1011–26. doi: 10.1016/0024-3205(95)02047-m. [DOI] [PubMed] [Google Scholar]
- 21.Elmquist JK, Scammell TE, Saper CB. Mechanisms of CNS response to systemic immune challenge: the febrile response. Trends Neurosci. 1997;20:565–9. doi: 10.1016/s0166-2236(97)01138-7. [DOI] [PubMed] [Google Scholar]
- 22.Goehler LE, Gaykema RPA, Hansen MK, Anderson K, Maier SF, Watkins L. Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci. 2000;85:49–59. doi: 10.1016/S1566-0702(00)00219-8. [DOI] [PubMed] [Google Scholar]
- 23.Goehler LE, et al. Interleukin-1 in immune cells of the abdominal vagus nerve: a link between the immune and nervous system? J Neurosci. 1999;19:2799–806. doi: 10.1523/JNEUROSCI.19-07-02799.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berthhoud HR, Neuhuber WL. Functional anatomy of afferent vagal system. Auton Neurosci. 2000;85:1–17. doi: 10.1016/S1566-0702(00)00215-0. [DOI] [PubMed] [Google Scholar]
- 25.Iversen S, Iversen L, Saper CB. (2000) The autonomic nervous system and the hypothalamus. In: Principles in Neural Science. 4th ed. Kendel ER, Schwartz JH, Jessel TM (eds.) McGraw Hill, New York. pp. 960–81.
- 26.Gaykema RPA, Dijkstra I, Tilders FJH. Subdiaphragmic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormones neurons and ACTH secretion. Endocrinology. 1995;136:4717–20. doi: 10.1210/endo.136.10.7664696. [DOI] [PubMed] [Google Scholar]
- 27.Ishizuka Y, et al. Effects of area postrema lesion and abdominal vagotomy on interleukin-1β–induced norepinephrine release in the hypothalamic paraventricular nucleus region in the rat. Neurosci Lett. 1997;223:57–60. doi: 10.1016/s0304-3940(97)13388-2. [DOI] [PubMed] [Google Scholar]
- 28.Hermann GE, Emch GS, Tovar CA, Rogers RC. C-Fos generation in the dorsal vagal complex after systemic endotoxin is not dependent on the vagus nerve. Am J Physiol Regulatory Integrative Comp Physiol. 2001;280:R289–99. doi: 10.1152/ajpregu.2001.280.1.R289. [DOI] [PubMed] [Google Scholar]
- 29.Van Dam AM, et al. Vagotomy does not inhibit high dose LPS-indiced interleukin-1β immunoreactivity in the rat brain and pituitary gland. Neurosci Lett. 2000;285:169–72. doi: 10.1016/s0304-3940(00)01031-4. [DOI] [PubMed] [Google Scholar]
- 30.Hansen MK, et al. Effects of vagotomy on serum endotoxin, cytokines and corticosterone after intraperitoneal lipopolysaccharide. Am J Physiol. 2000;278:R331–6. doi: 10.1152/ajpregu.2000.278.2.R331. [DOI] [PubMed] [Google Scholar]
- 31.Hopkins SJ, Rothwell N. Cytokines and nervous system I: expression and recognition. Trends Neurosci. 1995;18:83–8. [PubMed] [Google Scholar]
- 32.Szelényi J. Cytokines and the central nervous system. Brain Res Bull. 2001;54:329–459. doi: 10.1016/s0361-9230(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 33.Rivest S, et al. How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammatory and infectious stimuli. Proc Soc Exp Biol Med. 2000;223:22–38. doi: 10.1046/j.1525-1373.2000.22304.x. [DOI] [PubMed] [Google Scholar]
- 34.Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation. 1995;2:241–8. doi: 10.1159/000097202. [DOI] [PubMed] [Google Scholar]
- 35.Nadeau S, Rivest S. Effects of circulating tumor necrosing factor on the neuronal activity and expression on the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–64. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- 36.Ek M, Engblom D, Saha S, Blomqvist A, Jacobsson P-J, Ericsson-Dahlstrand A. Inflammatory response: pathway across the blood-brain barrier. Nature. 2001;410:430–1. doi: 10.1038/35068632. [DOI] [PubMed] [Google Scholar]
- 37.Buller KM. Circumventricular organs: gateways to the brain. Role of circumventricular organs in pro-inflammatory cytokine-induced activation of the hypothalamic-pituitary-adrenal axis. Clin Experiment Pharmacol Physiol. 2001;28:581–9. doi: 10.1046/j.1440-1681.2001.03490.x. [DOI] [PubMed] [Google Scholar]
- 38.Afifi AK, Bergmann RA. (1998) Medulla oblongata. In: Functional neuroanatomy McGraw-Hill, New York, p. 117.
- 39.Rogers RC, McCann MJ. Intramedullary connections of the gastric region in the solitary nucleus: a biocytin histochemical tracing study in the rat. J Auton Nerv Syst. 1993;42:119–30. doi: 10.1016/0165-1838(93)90043-t. [DOI] [PubMed] [Google Scholar]
- 40.Rogers RC, Hermann GE, Travagli RA. Brainstem pathways responsible for the oesophageal control of gastric motility and tone in the rat. J Physiol. 1999;514:369–83. doi: 10.1111/j.1469-7793.1999.369ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothwell NJ, Hopkins SL. Cytokines and the nervous system II: actions and mechanism of action. Trends Neurosci. 1995;18:130–6. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- 42.Benveniste EN. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998;9:259–75. doi: 10.1016/s1359-6101(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 43.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nature Med. 2002;8:1363–8. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 44.Lilly MP, Gann DS. The hypothalamic-pituitary-adrenal-immune axis: a critical assessment. Arch Surg. 1992;127:1463–74. doi: 10.1001/archsurg.1992.01420120097017. [DOI] [PubMed] [Google Scholar]
- 45.McEwen BS, et al. The role of adrenocorticoides as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- 46.Karalis K, Muglia LJ, Bae D, Hildebrand H, Majzoub JA. CRH and the immune system. J Immunology. 1997;72:131–6. doi: 10.1016/s0165-5728(96)00178-6. [DOI] [PubMed] [Google Scholar]
- 47.Adcock IM. Molecular mechanisms of glucocorticoid action. Pulmon Pharmacol Ther. 2000;13:115–26. doi: 10.1006/pupt.2000.0243. [DOI] [PubMed] [Google Scholar]
- 48.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS. Role of transcriptional activation of I κB alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–6. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 49.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor κB and steroid receptor–signaling pathways. Endocrine Rev. 1999;20:435–59. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionary conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 51.Hasko G, Szabo C. Regulation of cytokine and chemokine production by transmitters and co-transmitters of the autonomic nervous system. Biochem Pharmacol. 1998;56:1079–87. doi: 10.1016/s0006-2952(98)00153-1. [DOI] [PubMed] [Google Scholar]
- 52.Bellinger DL, Lorton CL, Felton DL. (2001) Innervation of lymphoid organs: association of nerves with cells of the immune system and their implications in disease. In: Psychoneuroimmunology Ader R, Felten DI, Cohen N (eds.). 3rd ed. Academic Press, New York, pp. 55.
- 53.van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-α and potentiates interleukin-10 production during human endotoxemia. J Clin Invest. 1996;97:713–9. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madden KS, Sanders VM, Felten DL. Catecholamine influences and sympathetic neuronal modulation of immune responsiveness. Annu Rev Pharmacol Toxicol. 1995;35:417–48. doi: 10.1146/annurev.pa.35.040195.002221. [DOI] [PubMed] [Google Scholar]
- 55.Zhou M, Yang S, Koo DJ, Ornan DA, Chaudry IH, Wang P. The role of Kupffer cell α2-adrenoceptors in norepinephrine-induced TNF-α production. Biochim Biophys Acta. 2001;1537:49–57. doi: 10.1016/s0925-4439(01)00055-2. [DOI] [PubMed] [Google Scholar]
- 56.Lipton JM, Catania A. Anti-inflammatory actions of the neuroimmunomodulator α-MSH. Immunol Today. 1997;140:140–5. doi: 10.1016/s0167-5699(97)01009-8. [DOI] [PubMed] [Google Scholar]
- 57.Catania A, Airaghi L, Colombo G, Lipton JM. α-Melanocyte-stimulating hormone in normal human physiology and disease states. Trends Exp Med. 2000;11:304–8. doi: 10.1016/s1043-2760(00)00296-4. [DOI] [PubMed] [Google Scholar]
- 58.Macaluso A, et al. Anti-inflammatory influences of α-MSH molecules: central neurogenic and peripheral action. J Neurosci. 1994;14:2377–82. doi: 10.1523/JNEUROSCI.14-04-02377.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Catania A, Suffredini AF, Lipton JM. Endotoxin causes release of α-melanocyte-stimulating hormone in normal human subjects. Neuroimmunomodulation. 1995;2:258–62. doi: 10.1159/000097204. [DOI] [PubMed] [Google Scholar]
- 60.Janson L, Holmdahl R. Estrogen-mediated immunosupression in autoimmune diseases. Inflamm Res. 1998;47:290–301. doi: 10.1007/s000110050332. [DOI] [PubMed] [Google Scholar]
- 61.Behl C. Oestrogen as a neuroprotective hormone. Nature Rev Neurosci. 2002;3:433–42. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- 62.Da Silva JA, Colville-Nash P, Spector TD, Scott DL, Willoughby DA. Inflammation-induced cartilage degradation in female rodents. Protective role of sex hormones. Arthritis Rhem. 1993;36:1007–15. doi: 10.1002/art.1780360719. [DOI] [PubMed] [Google Scholar]
- 63.Deshpande R, Khalili H, Pergolizzi RG, Michael SD, Chang MD. Estradiol down-regulates LPS-induced cytokine production and NF-κB activation in murine macrophages. Am J Reprod Immunol. 1997;38:46–54. doi: 10.1111/j.1600-0897.1997.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 64.Srivastava S, Weitzmann MN, Cenci S, Ross FP, Adler S, Pacifici R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and Jun D. J Clin Invest. 1999;104:503–13. doi: 10.1172/JCI7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strom TB, Deisseroth A, Morganroth J, Carpenter CB, Merrill JP. Alteration of the cytotoxic action of sensitized lymphocytes by cholinergic agents and activators of adenylate cyclase. Proc Natl Acad Sci USA. 1972;69:2995–9. doi: 10.1073/pnas.69.10.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Antonica A, Magni F, Mearini L, Paolocci N. Vagal control of lymphocyte release from rat thymus. J Auton Nerv Syst. 1994;48:187–97. doi: 10.1016/0165-1838(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 67.Niijima A, Hori T, Katafuchi T, Ichijo T. The effect of interleukin-β on the efferent activity of the vagus nerve to the thymus. J Auton Nerv Syst. 1995;54:137–44. doi: 10.1016/0165-1838(95)00003-g. [DOI] [PubMed] [Google Scholar]
- 68.Hori T, Katafuchi T, Take S, Shimizu N, Niijima A. The autonomic nervous system as a communication channel between the brain and the immune system. Neuroimmunomodulation. 1995;2:203–15. doi: 10.1159/000097198. [DOI] [PubMed] [Google Scholar]
- 69.Borovikova LV, et al. Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton Neurosci. 2000;85:141–7. doi: 10.1016/S1566-0702(00)00233-2. [DOI] [PubMed] [Google Scholar]
- 70.Caulfield P, Birdsall NJM. Intl. union of pharmacology XVII Classification of muscarinic acetylcholine receptors. Pharmacol Reviews. 1998;50:279–90. [PubMed] [Google Scholar]
- 71.Lindstrom J. Nicotinic acetylcholine receptors in health and disease. Mol Neurobiol. 1997;15:193–222. doi: 10.1007/BF02740634. [DOI] [PubMed] [Google Scholar]
- 72.Hiemke C, et al. Expression of alpha subunit genes of nicotinic acetylcholine receptors in human lymphocytes. Neurosci Lett. 1996;214:171–4. doi: 10.1016/0304-3940(96)12908-6. [DOI] [PubMed] [Google Scholar]
- 73.Mita Y, Dobashi K, Suzuki K, Mori M, Nakazawa T. Induction of muscarinic receptor subtypes in monocytic/macrophagic cells differentiated from EoL-1 cells. Eur J Pharmacol. 1996;297:121–7. doi: 10.1016/0014-2999(95)00722-9. [DOI] [PubMed] [Google Scholar]
- 74.Toyabe S, et al. Identification o as thymus in mice. Immunol. 1997;92:201–5. doi: 10.1046/j.1365-2567.1997.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sato KZ, et al. Diversity of mRNA expression for muscarine acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett. 1999;266:17–20. doi: 10.1016/s0304-3940(99)00259-1. [DOI] [PubMed] [Google Scholar]
- 76.Walch L, Brink C, Norel X. The muscarinic receptor subtypes in human blood vessels. Therapie. 2001;56:223–6. [PubMed] [Google Scholar]
- 77.Tayebati SK, El-Assouad D, Ricci A, Amenta F. Immunochemical and immunocytochemical characterization of cholinergic markers in human peripheral blood lymphocytes. J Neuroimmunol. 2002;132:147–55. doi: 10.1016/s0165-5728(02)00325-9. [DOI] [PubMed] [Google Scholar]
- 78.Kawashima K, Fujii T. Extraneural cholinergic system in lymphocytes. Pharmacol Ther. 2000;86:29–48. doi: 10.1016/s0163-7258(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 79.Tracey KJ, et al. Anti-cachectin/TNF-monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–4. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 80.Bernik TR, et al. Pharmacological stimulation of the cholinergic anti-inflammatory pathway. J Exp Med. 2002;195:1–9. [Google Scholar]
- 81.Villa P, et al. Inhibition of multiple pro-inflammatory mediators (TNF, IL-6, and NO) abrogate lethality in a murine mode of polymicrobial sepsis. J Endot Res. 1997;4:197–204. [Google Scholar]
- 82.Martiney JA, et al. Prevention and treatment of experimental autoimmune encephalomyelitis by CNI-1493, a macrophage deactivating agent. J Immunol. 1998;160:5588–95. [PubMed] [Google Scholar]
- 83.Akerlund K, et al. Anti-inflammatory effect of a new TNFα inhibitor (CNI-1493) in collagen-induced arthritis in rats. J Clin Exp Immunol. 1999;115:32–41. doi: 10.1046/j.1365-2249.1999.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bianchi M, et al. Suppression of pro-inflammatory cytokines in monocytes by tetravalent guanylhidrazone. J Exp Med. 1996;83:927–36. doi: 10.1084/jem.183.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang H, Zhang M, Bianchi M, Sherry B, Sama A, Tracey KJ. Fetuin (α-2-HS-Glycoprotein) opsonizes cationic macrophage-deactivating molecules. Proc Natl Acad Sci USA. 1998;95:14429–34. doi: 10.1073/pnas.95.24.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tracey KJ. Suppression of TNF and other pro-inflammatory cytokines by tetravalent guanylhydrazone CNI-1492. Prog Clin Biol Res. 1998;397:335–43. [PubMed] [Google Scholar]
- 87.Meistrell ME, III, et al. TNF is a brain-damaging cytokine in cerebral ischemia. Shock. 1997;8:341–8. [PubMed] [Google Scholar]
- 88.Hoover DB, Hancock JC, DePorter TE. Effect of vagotomy on cholinergic parameters in nuclei of rat medulla oblongata. Brain Res Bull. 1985;15:5–11. doi: 10.1016/0361-9230(85)90054-1. [DOI] [PubMed] [Google Scholar]
- 89.Hyde TM, Gibbs M, Peroutka SJ. Distribution of muscarinic cholinergic receptors in the dorsal vagal complex and other selected nuclei in the human medulla. Brain Res. 1988;447:287–92. doi: 10.1016/0006-8993(88)91131-6. [DOI] [PubMed] [Google Scholar]
- 90.Lawrence AJ, Jarrot B. Neurochemical modulation of cardiovascular control in the nucleus tractus solitarius. Prog Neurobiol. 1996;48:21–53. doi: 10.1016/0301-0082(95)00034-8. [DOI] [PubMed] [Google Scholar]
- 91.Shihara M, Hori N, Hirooka Y, Eshima K, Akaike N, Takeshita A. Cholinergic system in the nucleus of the solitary tract of rats. Amer J Physiol-Regulat Integrat Compar Physiol. 1999;276:R1141–8. doi: 10.1152/ajpregu.1999.276.4.R1141. [DOI] [PubMed] [Google Scholar]
- 92.Sykes RM, Spyer KM, Izzo PN. Demonstration of glutamate immunoreactivity in vagal sensory afferents in the nucleus tractus solitarius of the rat. Brain Res. 1997;762:1–11. doi: 10.1016/s0006-8993(97)00368-5. [DOI] [PubMed] [Google Scholar]
- 93.Mascarucci P, Perego C, Terrazzino S, De Simoni MG. Glutamate release in the nucleus tractus solitarius induced by peripheral lipopolysaccharide and interleukin-1β. Neuroscience. 1998;86:1285–90. doi: 10.1016/s0306-4522(98)00105-5. [DOI] [PubMed] [Google Scholar]
- 94.Hornby P. Receptors and transmission in the brain-gut axis. II Excitatory amino acid receptors in the brain-gut axis. Am J Physiol-Gastrointest Liver Physiol. 2001;280:G1055–60. doi: 10.1152/ajpgi.2001.280.6.G1055. [DOI] [PubMed] [Google Scholar]
- 95.Lin LH, Talman WT. N-methyl-D-aspartate receptors on neurons that synthesize nitric oxide in rat nucleus tractus solitarii. Neuroscience. 2000;100:581–8. doi: 10.1016/s0306-4522(00)00314-6. [DOI] [PubMed] [Google Scholar]
- 96.Chen C-Y, Bonham AC. Non-NMDA and NMDA receptors transmit area postrema input to aortic baroreceptor neurons in NTS. Am J Physiol-Heart Circulat Physiol. 1998;275:H1695–706. doi: 10.1152/ajpheart.1998.275.5.H1695. [DOI] [PubMed] [Google Scholar]
- 97.Ferreira M, et al. Evidence for functional alpha-7 neuronal nicotinic receptor subtype located on motoneurones of the dorsal motor nucleus of the vagus. J Pharmacol Exper Therap. 2001;296:260–9. [PubMed] [Google Scholar]
- 98.Browning KN, Travagli RA. Mechanism of action of baclofen in rat dorsal motor nucleus of the vagus. Am J Physiol-Gastrointest Liver Physiol. 2001;280:G1106–13. doi: 10.1152/ajpgi.2001.280.6.G1106. [DOI] [PubMed] [Google Scholar]
- 99.Lewis MW, Travagli RA. Effects of substance P on identified neurons of the rat dorsal motor nucleus of the vagus. Am J Physiol-Gastrointestin Liver Physiol. 2001;281:G164–72. doi: 10.1152/ajpgi.2001.281.1.G164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blondeau C, Clerc N, Baude A. Neurokinin-1 and neurokinin-3 receptors are expressed in vagal efferent neurons that innervate different parts of the gastro-intestinal tract. Neuroscience. 2002;110:339–49. doi: 10.1016/s0306-4522(01)00452-3. [DOI] [PubMed] [Google Scholar]
- 101.Atkinson L, Batten TF, Deuchars J. P2X(2) receptor immunoreactivity in the dorsal vagal complex and area postrema of the rat. Neuroscience. 2000;99:683–96. doi: 10.1016/s0306-4522(00)00233-5. [DOI] [PubMed] [Google Scholar]
- 102.Steinlein O. New functions for nicotine acetylcholine receptors? Behav Brain Res. 1998;95:31–5. doi: 10.1016/s0166-4328(97)00207-6. [DOI] [PubMed] [Google Scholar]
- 103.Marubio LM, Changeux J-P. Nicotinic acetylcholine receptor knockout mice as animal models for studying receptor function. Eur J Pharmacol. 2000;393:113–21. doi: 10.1016/s0014-2999(00)00007-8. [DOI] [PubMed] [Google Scholar]
- 104.Leonard S, Bertrand D. Neuronal nicotinic receptors: from structure to function. Nicotine Tob Res. 2001;3:203–23. doi: 10.1080/14622200110050213. [DOI] [PubMed] [Google Scholar]
- 105.Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 106.Orr-Urtreger A, et al. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack α-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–71. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Korach M, et al. Cardiac variability in critically ill adults: Influence of sepsis. Crit Care Med. 2001;29:1380–5. doi: 10.1097/00003246-200107000-00013. [DOI] [PubMed] [Google Scholar]
- 108.Winchell RJ, Hoyt DB. Spectral analysis of heart rate variability in the ICU: a measure of autonomic function. J Surg Res. 1996;63:11–6. doi: 10.1006/jsre.1996.0214. [DOI] [PubMed] [Google Scholar]
- 109.Chensue SW, Terebuh PD, Remick DG, Scales WE, Kunkel SL. In vivo biological and immunohistochemical analysis of interleukin-1α, β and tumor necrosis factor during experimental endotoxemia. Am J Pathol. 1991;138:395–402. [PMC free article] [PubMed] [Google Scholar]
- 110.Honzikova N, Semrad B, Fiser B, Labrova R. Baroreflex sensitivity determined by spectral method and heart rate variability, and two-years mortality in patients after myocardial infarction. Physiol Res. 2000;49:643–50. [PubMed] [Google Scholar]
- 111.Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274:R577–95. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- 112.Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995;92:1487–93. doi: 10.1161/01.cir.92.6.1487. [DOI] [PubMed] [Google Scholar]
- 113.George MS, et al. Vagus nerve stimulation: a new tool for brain research and therapy. Biol Psychiatry. 2000;47:287–95. doi: 10.1016/s0006-3223(99)00308-x. [DOI] [PubMed] [Google Scholar]
- 114.Valencia I, Holder DL, Helmers SL, Madsen JR, Riviello JJ. Vagus nerve stimulation in pediatric epilepsy: a review. Pediatr Neurol. 2001;25:368–76. doi: 10.1016/s0887-8994(01)00319-8. [DOI] [PubMed] [Google Scholar]
- 115.George MS, et al. Vagus nerve stimulation. A potential therapy for resistant depression? Psychiatr Clin North Am. 2000;23:757–83. doi: 10.1016/s0193-953x(05)70196-9. [DOI] [PubMed] [Google Scholar]
- 116.Handforth A, et al. Vagus nerve stimulation therapy for partial-onset seizures: A randomized active-control trial. Neurology. 1998;51:48–55. doi: 10.1212/wnl.51.1.48. [DOI] [PubMed] [Google Scholar]
- 117.Krahl SE, Clark KB, Smith DC, Browning RA. Locus coeroleus lesions suppress the seizure attenuating effects of vagus nerve stimulation. Epilepsia. 1998;39:709–14. doi: 10.1111/j.1528-1157.1998.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 118.Naritoku DK, Terry WJ, Helfert RH. Regional induction of Fos immunoreactivity in the brain by anticonvulsant stimulation of the vagus nerve. Epilepsy Res. 1995;22:53–62. doi: 10.1016/0920-1211(95)00035-9. [DOI] [PubMed] [Google Scholar]
- 119.Wang H, et al. Nicotine inhibits the release of HMGB1 through post-transcriptional mechanism. Shock. 2002;17(S1):62. [Google Scholar]
- 120.Han JL, et al. Cholinergic suppression of cytokine release from human macrophages. Shock. 2003;19(S1):22. [Google Scholar]