

Abstract
Tauroursodeoxycholic acid (TUDCA), an endogenous bile acid, modulates cell death by interrupting classic pathways of apoptosis. Amyloid-β (Aβ) peptide has been implicated in the pathogenesis of Alzheimer’s disease, where a significant loss of neuronal cells is thought to occur by apoptosis. In this study, we explored the cell death pathway and signaling mechanisms involved in Aβ-induced toxicity and further investigated the anti-apoptotic effect(s) of TUDCA. Our data show significant induction of apoptosis in isolated cortical neurons incubated with Aβ peptide. Apoptosis was associated with translocation of pro-apoptotic Bax to the mitochondria, followed by cytochrome c release, caspase activation, and DNA and nuclear fragmentation. In addition, there was almost immediate but weak activation of the serine/threonine protein kinase Akt. Inhibition of the phosphatidylinositide 3′-OH kinase (PI3K) pathway with wortmannin did not markedly affect Aβ-induced cell death, suggesting that this signaling pathway is not crucial for Aβ-mediated toxicity. Notably, co-incubation with TUDCA significantly modulated each of the Aβ-induced apoptotic events. Moreover, wortmannin decreased TUDCA protection against Aβ-induced apoptosis, reduced Akt phosphorylation, and increased Bax translocation to mitochondria. Together, these findings indicate that Aβ-induced apoptosis of cortical neurons proceeds through a Bax mitochondrial pathway. Further, the PI3K signaling cascade plays a role in regulating the anti-apoptotic effects of TUDCA.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the selective loss of synapses and neurons and the presence of senile plaques, composed primarily of Aβ (1). Aβ is a proteolytic fragment of the large amyloid precursor protein (2) and has been suggested to play a critical role in the pathogenesis of AD. Previous studies have shown that Aβ-induced toxicity involves oxidative stress, inflammation, and perturbation of calcium homeostasis (3). In addition, both necrotic and apoptotic processes are thought to occur in primary neurons and neuronal cell lines after exposure to Aβ as well as in AD brains (4–7). However, the precise intracellular signaling pathways by which Aβ peptide triggers cell death, especially in primary neurons, are not fully understood.
Apoptosis plays a critical role in sculpting the developing rat brain and has a potentially important function in neurodegenerative diseases (8). The regulation of mitochondrial membrane integrity and the release of apoptogenic factors from mitochondria are key components of the apoptosis repertoire, and are tightly controlled by the Bcl-2 family of proteins (9,10). The anti-apoptotic members, such as Bcl-2 and Bcl-xL, localize predominantly at the mitochondrial membrane and promote cell survival; whereas pro-apoptotic Bax, Bid, and Bad reside in the cytoplasm and induce apoptosis (9). In addition, the subcellular localization of Bad is further regulated by phosphorylation. Survival signals induce its phosphorylation, thereby sequestering it from its mitochondrial targets. In contrast, loss of survival signals reduces levels of Bad phosphorylation, thus promoting its translocation to mitochondria (11–13).
Several studies have demonstrated that Aβ induces the cell-intrinsic death machinery, in which the pro-apoptotic protein Bax might play a crucial role (14–16). In fact, there is good evidence to support the mitochondrial pathway of apoptosis in Aβ-induced neurotoxicity. For example, intracellular Aβ aggregates or granules have been detected in the brains of patients with AD (17). Microinjection of Aβ peptide rapidly induces cell death of primary human neurons through the p53-Bax apoptotic pathway (18). Finally, mitochondrial dysfunction and DNA damage are triggered in cells exposed to Aβ (19–21). We have shown that Aβ peptide can induce cytochrome c release in neuronal cells via mitochondrial membrane permeabilization (22), which appears to be associated with direct changes on membrane lipid and protein structure (23).
Recent data have implicated phosphatidylinositide 3′-OH kinase (PI3K) in promoting survival downstream of apoptosis-inducing stimuli. Once activated, PI3K can activate a growing number of cellular intermediates, including the serine/threonine protein kinase Akt, that are capable of suppressing apoptosis. In fact, it was determined that Akt can induce phosphorylation of pro-apoptotic Bad at Ser-136, resulting in 14-3-3 binding and sequestration from Bcl-xL (11,12). In contrast, the loss of these survival signals promotes dephosphorylation of Bad and its translocation to mitochondria (13). In this regard, Akt activation and Bad phosphorylation were detected in brains from transgenic mice overexpressing a mutant form of human amyloid precursor protein, perhaps as a protective mechanism against increased levels of Aβ peptide (24). In addition, Akt and the extracellular signal-regulated kinase were both activated in a neuroblastoma cell line exposed to Aβ peptide (25).
Akt can also act as an important signaling intermediate upstream of survival gene expression that is dependent on the nuclear factor κB (NF-κB) (26). Within the nucleus, NF-κB binds to specific response elements of target genes, such as proinflammatory cytokines, but also anti-apoptotic proteins that stabilize mitochondria (27). Aβ was reported to be a potent inducer of NF-κB in primary neurons and also in neurons surrounding early plaques from patients with AD (28). In fact, the inhibition of NF-κB was shown to potentiate Aβ-mediated neuronal apoptosis (29).
The role of apoptosis in Aβ-induced toxicity suggests that its modulation may slow the neurodegenerative process. The endogenous hydrophilic bile acid ursodeoxycholic acid (UDCA) and its taurine-conjugate tauroursodeoxycholic acid (TUDCA) increase the apoptotic threshold in several cell types (30). Further, we have demonstrated that TUDCA interrupts classic apoptotic pathways by inhibiting mitochondrial membrane depolarization and channel formation (31–33). TUDCA is also a potent suppressor of Aβ-induced changes of lipid/protein fluidity and oxidative status in the membranes of isolated mitochondria (23). In addition, UDCA may significantly modulate the mitogen-activated protein kinase survival pathway (34). Finally, TUDCA is neuro-protective in a transgenic mouse model of Huntington’s disease (35) and in rat models of ischemic and hemorrhagic stroke (36,37).
In this study, we characterized the cell signaling pathways associated with Aβ injury to isolated rat cortical neurons, and further investigated alternative mechanisms by which TUDCA may protect against Aβ-induced cell death. Our results indicate that Aβ is a weak activator of Akt but a strong inducer of the Bax pro-apoptotic mitochondrial pathway. Further, TUDCA modulates Aβ-induced apoptosis by activating a PI3K-dependent pathway in association with Akt phosphorylation, which in turn suppresses translocation of Bax.
MATERIALS AND METHODS
Isolation and Culture of Rat Cortical Neurons
Primary cultures of rat cortical neurons were prepared from 17- to 18-day-old fetuses of Wistar rats as previously described (38) with minor modifications. In short, pregnant rats were ether-anesthetized and decapitated. The fetuses were collected in Hank’s balanced salt solution (HBSS-1; Invitrogen, Grand Island, NY, USA) and rapidly decapitated. After removal of meninges and white matter, the brain cortex was collected in Hank’s balanced salt solution without Ca2+ and Mg2+ (HBSS-2). The cortex was then mechanically fragmented, transferred to a 0.025% trypsin in HBSS-2 solution, and incubated for 15 min at 37 °C. Following trypsinization, cells were washed twice in HBSS-2 containing 10% fetal calf serum and resuspended in Neurobasal medium (Invitrogen), supplemented with 0.5 mM l-glutamine, 25 μM l-glutamic acid, 2% B-27 supplement (Invitrogen), and 12 mg/mL gentamicin. Neurons were then plated on tissue culture plates, precoated with poly-d-lysine at 1 × 106 cells/mL, and maintained at 37 °C in a humidified atmosphere of 5% CO2. All experiments were performed on cells cultured for 2 to 3 d in fresh medium without glutamic acid and B-27 supplement. Cells were characterized by phase contrast microscopy and indirect immunocytochemistry for neurofilaments and glial fibrillary acidic protein. Neuronal cultures were >95% pure. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985).
Induction of Apoptosis
Isolated rat neurons were cultured as described above and then incubated with either 25 μM Aβ peptide 25–35 or reverse Aβ peptide 35–25 as control (Bachem AG, Bubendorf, Switzerland) for 5 min to 24 h, with or without 100 μM TUDCA (Sigma Chemical, St. Louis, MO, USA), or no addition. In co-incubation experiments, TUDCA was added to neurons 12 h prior to incubation with Aβ peptide. Wortmannin (Calbiochem, San Diego, CA, USA), an inhibitor of PI3K phosphorylation, was added to cells 1 h prior to TUDCA at a final concentration of 200 nM. Attached cells were fixed for morphologic analysis of apoptotic changes or processed for viability. In addition, total, cytosolic, and mitochondrial protein fractions were extracted for immunoblotting and caspase activity; and total RNA was obtained for Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) studies.
Measurement of Cell Death
Cell death was measured by trypan blue exclusion. In addition, Hoechst labeling of cells was used to detect apoptotic nuclei. In brief, neurons were fixed with 4% formaldehyde in phosphate-buffered saline (PBS), pH 7.4, for 10 min at room temperature, incubated with Hoechst dye 33258 (Sigma Chemical) at 5 mg/mL in PBS for 5 min, washed with PBS, and mounted using PBS:glycerol (3:1, v/v). Fluorescence was visualized using an Axioskop fluorescence microscope (Carl Zeiss GmbH, Jena, Germany). Fluorescent nuclei were scored and categorized according to the condensation and staining characteristics of chromatin. Normal nuclei showed noncondensed chromatin dispersed over the entire nucleus. Apoptotic nuclei were identified by condensed and fragmented chromatin contiguous to the nuclear membrane, as well as nuclear fragmentation and apoptotic bodies. Three random microscopic fields per sample of approximately 250 nuclei were counted and mean values expressed as the percentage of apoptotic nuclei. Finally, apoptosis was confirmed by assessment of DNA laddering. Cells were digested overnight at 55 °C in buffer containing 25 mM Tris HCl, pH 7.5, 10 mM EDTA, 100 mM NaCl, 0.5% sodium dodecyl sulfate (SDS), and 1 mg/mL proteinase K. After phenol/chloroform extraction, DNA was precipitated in ethanol, centrifuged at 4 °C for 15 min, and resolved by 1.5% agarose gel electrophoresis.
N-acetyl-Asp-Glu-Val-Asp-Specific Caspase Activity
Caspase activity was determined in cytosolic protein extracts after cells were harvested and homogenized in isolation buffer containing 10 mM Tris HCl buffer, pH 7.6, 5 mM MgCl2, 1.5 mM KAc, 2 mM dithiothreitol (DTT), and protease inhibitor cocktail tablets (Complete; Roche Applied Science, Mannheim, Germany). General caspase-3–like activity was evaluated by enzymatic cleavage of chromophore p-nitroanilide (pNA) from the substrate N-acetyl-Asp-Glu-Val-Asp-pNA (DEVD-pNA; Sigma Chemical). The proteolytic reaction was carried out in isolation buffer containing 50 μg cytosolic protein and 50 μM DEVD-pNA. The reaction mixtures were incubated at 37 °C for 1 h, and the formation of pNA was measured at 405 nm using a 96-well plate reader. Protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s specifications.
Bax Translocation and Cytochrome c Release
Cellular distribution of Bax and cytochrome c in neuronal cells was determined using mitochondrial and cytosolic protein extracts. Briefly, cells were collected by centrifugation at 600g for 5 min at 4 °C. The pellets were washed once in ice-cold PBS and resuspended with 3 volumes of isolation buffer containing 20 mM HEPES/KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, supplemented with complete protease inhibitor cocktail tablets, in 250 mM sucrose. After chilling on ice for 15 min, cells were disrupted by 40 strokes of a glass homogenizer, and homogenates were centrifuged twice at 2500g for 10 min at 4 °C to remove unbroken cells and nuclei. The mitochondrial fraction was centrifuged at 12000g for 30 min at 4 °C, and the pellet resuspended in isolation buffer and frozen at −80 °C. For cytosolic proteins, the 12000g supernatants were removed, filtered sequentially through 0.2 μm and 0.1 μm Ultrafree MC filters (Millipore, Bedford, MA, USA), to remove other cellular organelles, and frozen at −80 °C. Typically, 100 μg of mitochondrial and 50 μg of cytosolic proteins were separated on a 15% SDS-polyacrylamide gel for detection of cytochrome c and Bax. Mitochondrial contamination of the cytosolic protein extracts was determined with cytochrome c oxidase (subunit II).
Immunoblotting Analysis
Bcl-2 family, NF-κB, IκB, p-Akt1, and p-Bad protein levels, as well as Bax and cytochrome c distribution were determined by Western blot analysis. Briefly, following SDS-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred onto nitrocellulose membranes. Immunoblots were then incubated with 15% H2O2 for 15 min at room temperature and sequentially incubated overnight at 4 °C with 5% milk-blocking solution; primary mouse monoclonal antibodies reactive to Bcl-2, NF-κB (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and cytochrome c (PharMingen, San Diego, CA, USA); or primary rabbit polyclonal antibodies reactive to Bcl-x, Bax, IκB, p-Akt1 (Ser-473), and p-Bad (Ser-136) (Santa Cruz Biotechnology). Finally, immunoblots were incubated with secondary antibodies conjugated with horseradish peroxidase (Bio-Rad Laboratories) for 3 h at room temperature and processed for specific protein detection using the Super-Signal™ substrate (Pierce, Rockford, IL, USA). β-Actin and total Akt1/2 were used as controls.
RNA Isolation and RT-PCR
Changes in Bcl-2 family transcript expression were determined by semiquantitative RT-PCR. Total RNA was extracted from rat primary hepatocytes using the TRIZOL reagent (Invitrogen). For RT-PCR, 5 μg of total RNA was reverse-transcribed using oligo(dT) (Integrated DNA Technologies, Coralville, IA, USA) and SuperScript II reverse transcriptase (Invitrogen). Specific oligonucleotide primer pairs were incubated with cDNA template for PCR amplification using the Expand High Fidelity PCR System from Roche Applied Sciences. The following sequences were used as primers: Bcl-2 sense primer, 5′-CTG GTGGCAACATCGCTCTG-3′ and Bcl-2 antisense primer, 5′-GGTCTGCTGACCTCACTTGTG-3′; Bax sense primer, 5′-TGGTTGCCCTTTTCTACTT-TG-3′ and Bax antisense primer, 5′-GAAGTAGGAAAGGAGGCCATC-3′; Bcl-x sense primer, 5′-AGGTCGGCGATGAGTTTGAA-3′ and Bcl-x antisense primer, 5′-CGGCTCTCGGCTGCTGCATT-3′; and β-actin sense primer, 5′-TGCCCATCTATGAGGGTTACG-3′ and β-actin antisense primer, 5′-TAGAAGCATTTGCGGTGCACG-3′. The product of constitutively expressed β-actin mRNA served as control. The amplified PCR products were detected by agarose gel electrophoresis and ethidium bromide staining.
Densitometry and Statistical Analysis
The relative intensities of protein and nucleic acid bands were analyzed using the ImageMaster 1D Elite densitometric analysis program (Amersham Pharmacia Biotech, Uppsala, Sweden). All data were expressed as mean ±SEM from at least 3 separate experiments. Statistical analysis was performed using GraphPad InStat version 3.00 for Windows 95 (Graph-Pad Software, San Diego, CA, USA) for ANOVA and Bonferroni’s multiple comparison tests. Values of P < 0.05 were considered significant.
RESULTS
TUDCA Inhibits Aβ-Induced Apoptosis in Cortical Neurons
Several studies have demonstrated that the Aβ peptide in concentrations of 1 to 50 μM is highly toxic to a variety of primary neurons and neuronal cell lines. In addition, the 25–35 fragment is the biological active fragment of Aβ that has previously been shown to mimic the effects of the natural occurring Aβ 1–40 (4). Because TUDCA prevents cell death by different agents in a variety of cell types, we tested whether TUDCA could modulate Aβ toxicity. Thus, primary cultures of rat cortical neurons were exposed to 25 μM Aβ 25–35, or the control reverse peptide for 24 h, with or without pretreatment with 100 μM TUDCA for 12 h. The Aβ peptide–induced apoptosis in cortical neurons was assessed by nuclear fragmentation, DNA laddering, and caspase-3 processing (Figure 1A). In contrast, the control reverse peptide was not toxic to cortical neurons (data not shown). Lower concentrations of Aβ and shorter incubations of 1 and 8 h were less effective at inducing apoptosis (data not shown). Notably, pretreatment of cortical neurons with TUDCA reduced the Aβ-induced nuclear fragmentation by almost 65% (P < 0.01) (Figure 1B). Finally, general caspase activity was determined by the DEVD-specific caspase activation assay. Cytosolic proteins were extracted from cortical neurons and incubated with DEVD-pNA, a preferred substrate for caspase-3-like enzymes. As expected, caspase activity increased more than 2-fold in Aβ-treated neurons (P < 0.01), and co-incubation with TUDCA reduced caspase activation by almost 70% (P < 0.05) (Figure 1B). Lower doses of TUDCA and shorter incubation periods resulted in less inhibition of apoptosis (data not shown). Taken together, these findings indicate that cortical neurons undergo Aβ-induced apoptosis, which in turn can be markedly prevented by TUDCA.
Figure 1.

Aβ-induced apoptosis in rat cortical neurons is inhibited by TUDCA. Cells cultured for 3 d were incubated either with vehicle (control), 25 μM Aβ fragment 25–35, 100 μM TUDCA, or a combination of Aβ plus TUDCA for 24 h. In co-incubation experiments, cells were pretreated with TUDCA for 12 h and the bile acid was left in the culture medium with Aβ. A: Morphological and biochemical characteristics of apoptosis in rat cortical neurons incubated with Aβ peptide. Fluorescence microscopy of Hoechst staining (top) shows condensed or fragmented nuclei indicative of apoptosis. DNA laddering (middle) and caspase-3 processing (bottom) are also characteristic of apoptotic cell death. B: TUDCA prevents Aβ-induced apoptosis in rat cortical neurons. Histograms show mean ±SEM values of nuclear fragmentation (top) and caspase-3–like activity (bottom) for at least 5 independent experiments. *P < 0.01 from control; †P < 0.01 and ‡P < 0.05 from Aβ peptide.
TUDCA Abrogates Release of Cytochrome c by Aβ
The release of cytochrome c from mitochondria has been repeatedly shown to activate procaspase-9, followed by recruitment and activation of procaspase-3, which in turn cleaves several death substrates. Therefore, to examine the involvement of the mitochondrial pathway of apoptosis in Aβ-induced toxicity and the mechanism by which TUDCA prevents cell death, we determined cytochrome c release in intact cells. Control cortical neurons exhibited almost undetectable levels of cytosolic cytochrome c (Figure 2). In marked contrast, exposure of cells to Aβ resulted in almost an 8-fold increase of cytochrome c in the cytosol (P < 0.01), whereas mitochondrial levels were markedly reduced (P < 0.05). TUDCA prevented Aβ-induced release of cytochrome c, thus reducing cytosolic levels by >50% (P < 0.05). The results support a previous observation using isolated mitochondria that TUDCA prevents the observed apoptosis, in part by inhibiting the mitochondrial pathway (22).
Figure 2.
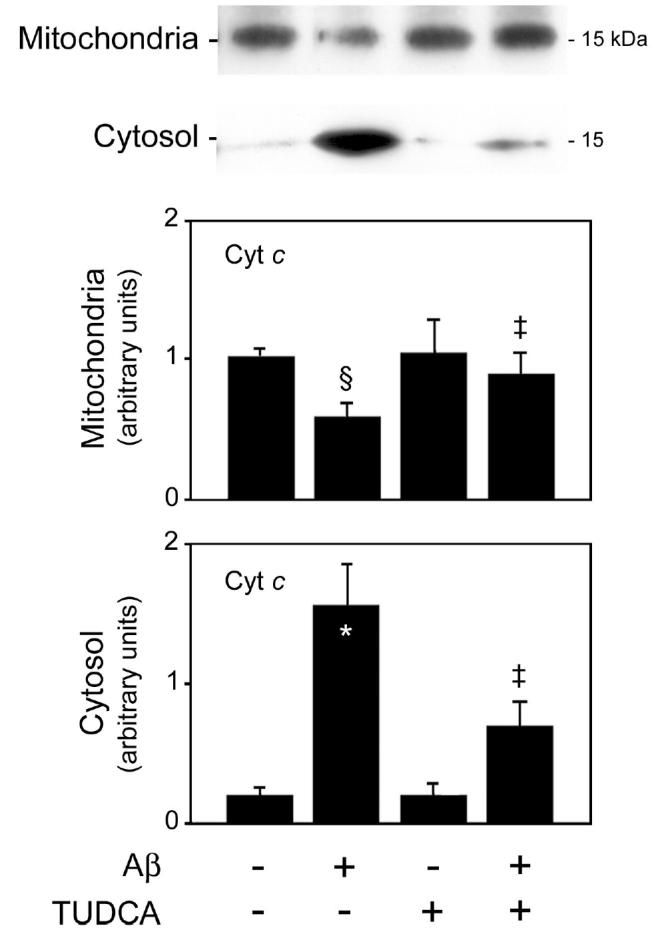
TUDCA inhibits Aβ-induced cytochrome c release in rat cortical neurons. Cells were incubated with either vehicle (control), 25 μM Aβ fragment 25–35, 100 μM TUDCA, or a combination of Aβ plus TUDCA for 24 h as described in Materials and Methods. Mitochondrial (top) and cytosolic (bottom) proteins were processed for Western blot analysis. Following SDS-PAGE and transfer, the nitrocellulose membranes were incubated with a monoclonal antibody to cytochrome c (Cyt c). Histograms are mean ±SEM for at least 3 different experiments. §P < 0.05 and *P < 0.01 from control; ‡P < 0.05 from Aβ.
TUDCA Prevents Aβ-Induced Translocation of Bax
Bax cellular distribution was evaluated by immunoblot analysis to determine whether TUDCA prevents Aβ-induced cytochrome c release by altering Bax translocation from cytosol to mitochondria. The results revealed a >2-fold increase in mitochondrial levels of Bax in neurons exposed to Aβ (P < 0.05) (Figure 3A). Co-incubation with TUDCA reduced mitochondrial Bax to almost control levels (P < 0.05). Translocation of Bax induced by Aβ, however, did not coincide with a decrease in cytosolic levels of Bax protein. In fact, cytosolic Bax was largely increased during incubation of cortical neurons with Aβ (P < 0.05), suggesting that translocation is accompanied by increased protein expression. Nevertheless, TUDCA reduced cytosolic Bax by ~70% (P < 0.05).
Figure 3.
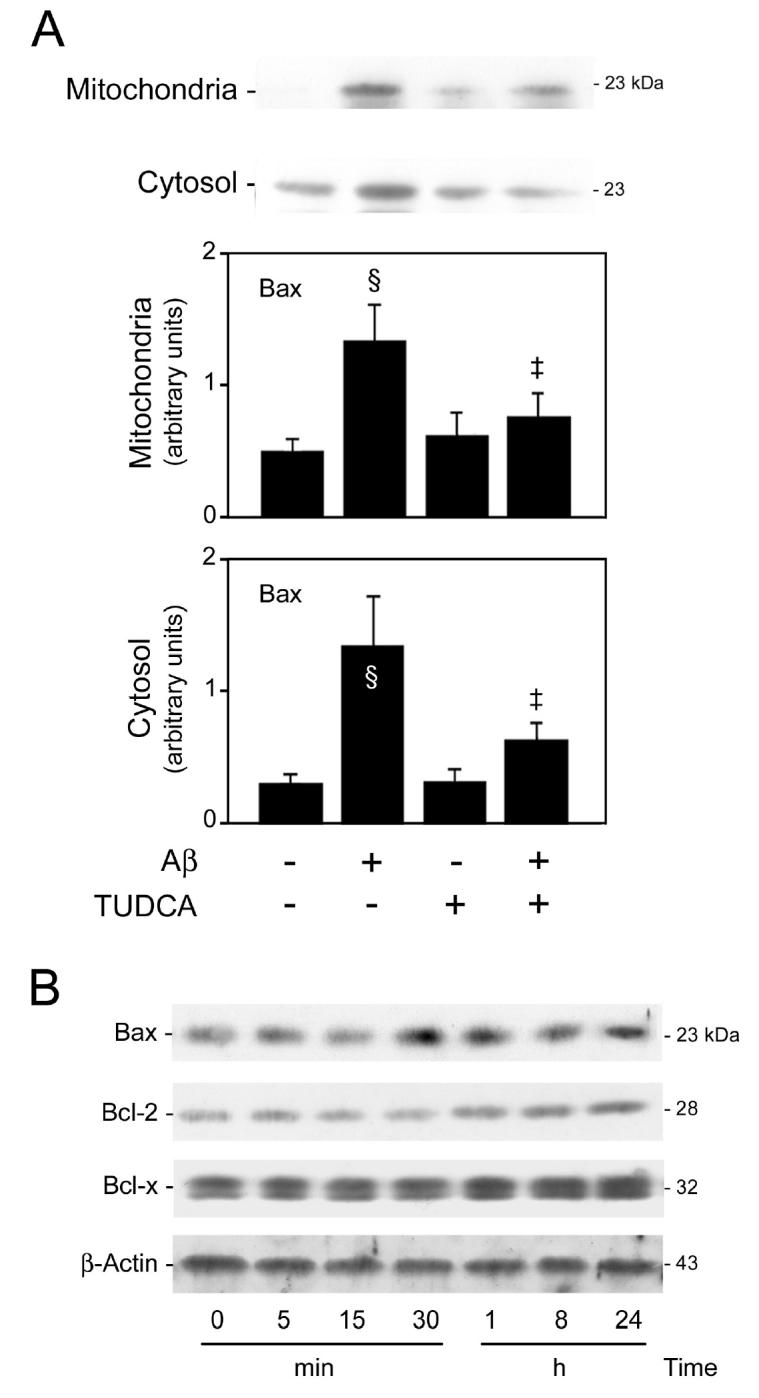
Aβ-induced translocation of Bax and modulation of Bcl-2 proteins in rat cortical neurons. Mitochondrial, cytosolic, and total proteins were processed for Western blot analysis. Following SDS-PAGE and transfer, the nitrocellulose membranes were incubated with a monoclonal antibody to Bcl-2 and polyclonal antibodies to Bax and Bcl-xL. A: TUDCA inhibits Aβ-induced translocation of Bax. Neuronal cells were incubated with vehicle (control), 25 μM Aβ fragment 25–35, 100 μM TUDCA, or a combination of Aβ plus TUDCA for 24 h as described in Materials and Methods. Representative Western blots are shown for mitochondrial (top) and cytosolic (bottom) fractions. Histograms are mean ±SEM for at least 3 different experiments. §P < 0.05 from control; ‡P < 0.05 from AβB: Aβ markedly upregulates Bax expression but only slightly induces Bcl-2 expression. Cells cultured for 3 d were incubated with either vehicle (control), or 25 μM Aβ fragment 25–35 for 5, 15, and 30 min and 1, 8, and 24 h. Representative Western blots are shown for Bax, Bcl-2, Bcl-xL, and β-actin proteins.
To determine the modulation of Bax by Aβ, both mRNA and protein levels were evaluated by RT-PCR and Western blot analysis, respectively. The results showed a pronounced increase in both bax mRNA and protein levels (P < 0.01) up to 24 h of incubation with Aβ (Figure 3B). These results suggested that both posttranslational and transcriptional mechanisms are involved in controlling the activity of pro-apoptotic Bax during Aβ-induced cell death. We also investigated the expression of anti-apoptotic members of the Bcl-2 family, such as Bcl-2 and Bcl-xL, in cortical neurons exposed to Aβ (Figure 3B). Both gene and protein levels of Bcl-2 were slightly increased late in the time course, whereas Bcl-xL was relatively unchanged. Thus, the balance between pro- and anti-apoptotic members of the Bcl-2 family appears to benefit Bax translocation and, consequently, the mitochondrial pathway of apoptosis.
Inhibition of the PI3K/Akt-Dependent Pathway Suppresses TUDCA Anti-apoptotic Effects
We next investigated whether signaling through Akt was involved in either Aβ-induced cell death or TUDCA protection. Thus, lysates from cells incubated with Aβ or TUDCA were subjected to Western blot analysis using anti-phospho-Akt1 antibody to the activated kinase. Blots were also probed with an antibody that recognizes total Akt1/2 levels, and the data expressed relative to those results. Aβ induced a weak, rapid, and transient activation of Akt (Figure 4A). Phosphorylation of Akt was observed within 5 min, increasing up to 40% (P < 0.05), but remained above control levels for only 15 min. In fact, exposure to Aβ for 24 h resulted in significant levels of apoptosis, but no detectable Akt phosporylation. We then investigated whether NF-κB activation and Bad phosphorylation, 2 possible downstream effectors of Akt (39), were required during activation of this survival pathway. The results indicated that IκB levels were reduced at 5 min of Aβ incubation (P < 0.05), remained significantly low for 30 min (P < 0.01), and then returned to control values. This is consistent with strong activation of NF-κB and may explain the slight increase in Bcl-2 protein expression observed at later time points. Finally, Bad phosphorylation was only slightly increased in cortical neurons immediately after Akt activation (P < 0.05). The results suggested that this level of Akt activation was largely ineffective in protecting against Aβ-induced apoptosis. In contrast, TUDCA increased Akt activity as early as 15 min, and this was sustained for at least 24 h (Figure 4B). After 1 h of TUDCA incubation, Akt phosphorylation was increased > 2-fold compared with controls (P < 0.01). Interestingly, the addition of TUDCA had no effect on IκB levels; and phosphorylated Bad was only slightly increased (data not shown).
Figure 4.
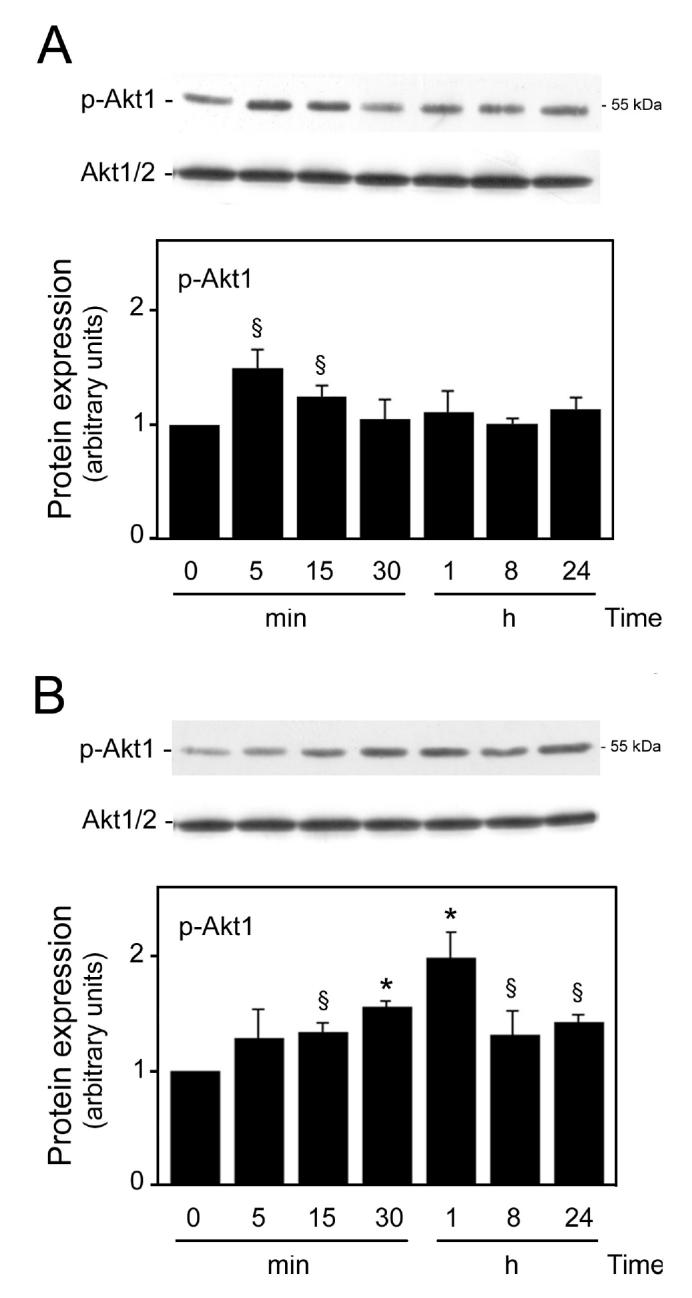
TUDCA and Aβ modulate Akt phosphorylation in rat cortical neurons. Cells were incubated with vehicle (control), 25 μM Aβ fragment 25–35, or 100 μM TUDCA for 5, 15, and 30 min and 1, 8, and 24 h as described in Materials and Methods. After incubation with Aβ (A) or TUDCA (B), total proteins were processed for Western blot analysis. Following SDS-PAGE and transfer, the nitrocellulose membranes were incubated with a polyclonal antibody for p-Akt1. Results were normalized to total Akt1/2 protein expression. Histograms are mean ±SEM for at least 3 different experiments. §P < 0.05 and *P < 0.01 from respective control.
Akt appears to be an important factor in the PI3K survival signal (39). To address the role of the PI3K pathway in TUDCA protection, neurons were preincubated with the PI3K inhibitor wortmannin. As assessed by the trypan blue exclusion assay and Hoechst staining, both control and TUDCA-treated cells were significantly less viable after exposure to wortmannin (P < 0.01) (Figure 5A). In addition, pretreatment with the inhibitor slightly increased Aβ-induced cell death compared with Aβ alone. More importantly, wortmannin suppressed the protective effect of TUDCA in modulating Aβ-induced apoptosis and blocked the Akt phosphorylation associated with TUDCA (Figure 5B). Further, Aβ-mediated Bax translocation, normally inhibited by TUDCA was no longer responsive after inhibition of PI3K signaling (Figure 5C). Similar results were obtained in cytochrome c release studies (data not shown). Interestingly, wortmannin significantly increased apoptosis and Bax translocation induced by TUDCA alone, consistent with a pro-apoptotic bile acid effect following inhibition of the PI3K pathway (34). These data strongly suggest that TUDCA-induced PI3K activation is necessary for protection from Aβ-induced cell death.
Figure 5.
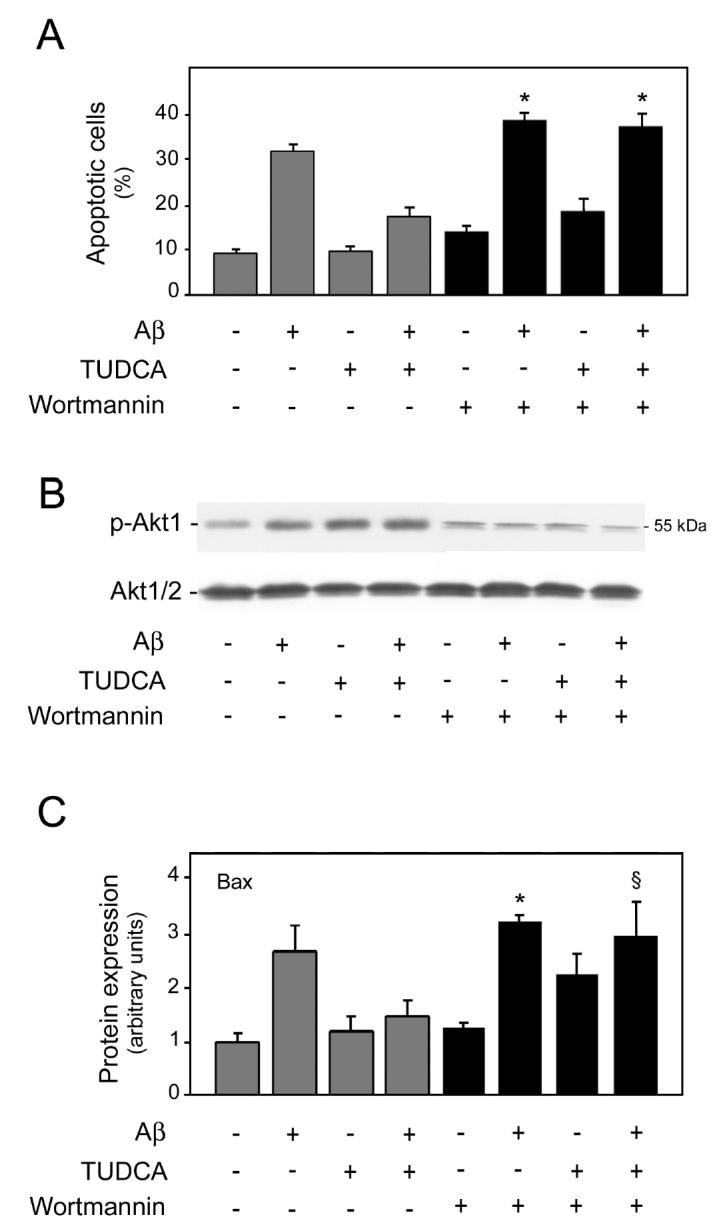
TUDCA prevents Aβ peptide–induced apoptosis through a PI3K-dependent pathway. Cultured neurons were incubated with either vehicle (control), 25 μM Aβ fragment 25–35, 100 μM TUDCA, or a combination of Aβ plus TUDCA, as described in Materials and Methods. Cells were pretreated with 200 nM wortmannin for 1 h prior to adding TUDCA, and the PI3K inhibitor was left in the culture medium during incubation with both Aβ and TUDCA. A: Wortmannin slightly increased Aβ toxicity at 24 h, but almost completely abolished the inhibitory effect of TUDCA in Aβ-induced apoptosis. Histograms show mean ±SEM values of nuclear fragmentation for at least 5 different experiments. *P < 0.01 from controls treated with wortmannin. B: PI3K inhibition was associated with reduced Akt phosphorylation by Aβ at 5 min, TUDCA alone, or a combination of Aβ plus TUDCA. After incubation with wortmannin, total proteins were processed for Western blot analysis. Following SDS-PAGE and transfer, the nitrocellulose membranes were incubated with polyclonal antibodies for p-Akt1 and total Akt1/2. Representative Western blots are shown for Akt phosphorylation. C: Wortmannin almost completely abolished the inhibitory effect of TUDCA on Aβ-mediated Bax translocation from cytosol to mitochondria at 24 h. After incubation with wortmannin, total, cytosolic, and mitochondrial proteins were processed for Western blot analysis. Following SDS-PAGE and transfer, the nitrocellulose membranes were incubated with antibodies for p-Bad and Bax. Histograms are mean ±SEM for at least 3 independent experiments. §P < 0.05 and *P < 0.01 from controls treated with wortmannin.
DISCUSSION
The accumulation of Aβ in the brain has been implicated as a potential cause for the neuronal loss that occurs in AD. Cell death from Aβ-induced toxicity is a complex process that is thought to involve a number of different pathways, including oxidative stress, perturbation of calcium homeostasis, mitochondrial dysfunction, and activation of caspases. In the present study, we showed that Aβ peptide is a strong inducer of the Bax pro-apoptotic mitochondrial pathway and a weak activator of Akt phosphorylation in rat primary cortical neurons. We further demonstrated that TUDCA modulates Aβ-induced apoptosis by activating a PI3K survival pathway, thereby suppressing Bax translocation.
The role of apoptosis in Aβ-induced neurotoxicity is not fully understood. Nevertheless, several groups have shown that apoptosis is selectively increased in primary neuronal cultures exposed to Aβ and also augmented in brain tissue derived from AD patients (40). In this regard, many components of the mitochondrial apoptotic cascade appear to be involved in the neuronal toxicity of Aβ peptides. Aβ can upregulate pro-apoptotic Bax expression or require Bax to mediate neurotoxicity (14–16). In addition, Bax protein levels have only inconsistently been reported to be increased in AD brain (41–44). We show here that cytochrome c is significantly depleted from mitochondria of rat cortical neurons in response to incubation with Aβ. As expected, the release of cytochrome c was accompanied by caspase-3 activation and subsequent DNA degradation and nuclear fragmentation. During Aβ-induced apoptosis, Bax protein levels increased in mitochondria, thus providing a mechanism for cytochrome c release. Bax translocation was also associated with an increased synthesis of the pro-apoptotic protein. The p53 protein is one possible candidate for the upregulation of Bax, as it is highly expressed in cells after accumulation of Aβ and in the brains of AD patients (18,45,46). In addition, p53 inhibitors can prevent neuronal cell death induced by Aβ (16).
We also examined the effects of Aβ on a kinase-survival pathway. In rat cortical neurons, Aβ was a weak and transient activator of Akt. Also, Akt phosphorylation was not required for Aβ-induced cell death, as wortmannin completely inhibited Akt activation but only slightly increased the number of apoptotic cells. Activation of Akt protects cells from apoptotic signals, such as growth factor withdrawal, cell-cycle disruption, and cell detachment (47,48). The role of Akt activation after Aβ treatment, although ineffective, may represent a cellular response to counteract Aβ neurotoxicity. In addition, the rapid Akt activation observed in this study strongly suggests that this is a receptor-mediated response. Recent reports have demonstrated the interaction of Aβ with cell surface receptors such as the 75-kDa neurotrophin receptor (49), the cell surface amyloid precursor protein (50), and a novel β-amyloid peptide binding protein (51).
UDCA and TUDCA are nontoxic, endogenous bile acids that can act as anti-apoptotic agents. They directly inhibit reactive oxygen species production, collapse of the transmembrane potential, and disruption of the outer mitochondrial membrane (23). Additionally, both bile acids mitigate mitochondrial insufficiency and toxicity by inhibiting Bax translocation from cytosol to mitochondria (31,32). Membrane stability inhibits cytochrome c release, thereby reducing downstream events such as caspase activation and substrate cleavage. In addition to inhibiting mitochondrial membrane depolarization and channel formation, TUDCA may also play key regulatory roles in signal transduction by modulating calcium levels and gene expression (52).
We have recently shown that administration of TUDCA to a transgenic mouse model of Huntington’s disease significantly reduced striatal degeneration and improved locomotor and sensorimotor deficits (35). Further, TUDCA significantly reduced apoptosis, infarct volumes, and neurobehavioral impairment in ischemic and hemorrhagic models of stroke (36,37). The present study extends our previous reports and shows a neuroprotective effect of TUDCA in rat cortical neurons exposed to Aβ peptide. TUDCA significantly reduced Bax translocation, thus inhibiting cytochrome c release, caspase activation, and DNA and nuclear fragmentation. In addition, we showed that TUDCA activated the PI3K-dependent survival pathway. Specific inhibitors of PI3K blocked Akt activation and almost completely abolished the protective effect of TUDCA against Aβ-induced cell death. Moreover, previous investigations have reported that inhibition of the mitogen-activated protein kinase and PI3K pathways results in significant apoptosis induced by certain bile acids, including UDCA (34).
Recent studies have demonstrated that Akt regulates apoptosis at multiple sites by inactivating pro-apoptotic proteins such as Bad, caspase-9, and the Forkhead family of transcription factors. In addition, it activates anti-apoptotic proteins such as NF-κB and the cAMP-response element-binding protein (39). Moreover, the PI3K/Akt pathway has been shown to inhibit cytochrome c release, as well as effectively suppress Bax translocation to the mitochondria (53,54). Our results also suggest that PI3K/Akt activation by TUDCA in isolated neurons is apparently sufficient to retain Bax in the cytoplasm after Aβ treatment. These data provide additional mechanisms of action for TUDCA, where the bile acid is shown to interfere with alternate molecular targets upstream of the mitochondrial commitment, suggesting a more complex mechanism for the protective role of TUDCA. However, our results do not exclude the possibility that other mechanisms may also mediate TUDCA protection from Aβ-induced apoptosis, including signaling pathways such as the c-Jun NH2-terminal kinase pathway. Interestingly, cholesterol-lowering drugs may lower the risk for developing AD, by reducing Aβ accumulation (55).
In conclusion, these data show that TUDCA can significantly reduce the neuronal apoptosis associated with Aβ peptide. TUDCA appears to inhibit cell death by interfering with the mitochondrial pathway of apoptosis in a PI3K-dependent manner that suppresses Bax translocation. The marked anti-apoptotic properties of TUDCA together with its lack of toxicity make it an attractive candidate in the treatment of neurodegenerative disorders, such as AD, where increased levels of apoptosis may contribute to the pathogenesis of the disease.
Acknowledgments
The authors thank Paulo Ribeiro for skillful technical assistance. This work was supported by grant POCTI/BCI/44929/2002 from Fundação para a Ciência e a Tecnologia (FCT), Lisbon, Portugal (to CMPR), and PhD fellowships SFRH/BD/4823/2001 and SFRH/BD/12655/2003 (to SS and REC, respectively) from FCT.
REFERENCES
- 1.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang J, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev Apr. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 4.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–82. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 5.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–5. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behl C, Davis JB, Klier FG, Schubert D. Amyloid β peptide induces necrosis rather than apoptosis. Brain Res. 1994;645:253–64. doi: 10.1016/0006-8993(94)91659-4. [DOI] [PubMed] [Google Scholar]
- 7.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport. 1994;5:2529–33. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 8.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–9. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 9.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 10.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–9. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 11.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 12.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 13.Wang HG, et al. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–43. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 14.Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A. Amyloid β peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J Neurosci. 1996;16:7533–9. doi: 10.1523/JNEUROSCI.16-23-07533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selznick LA, Zheng TS, Flavell RA, Rakic P, Roth KA. Amyloid β-induced neuronal death is bax-dependent but caspase-independent. J Neuropathol Exp Neurol. 2000;59:271–9. doi: 10.1093/jnen/59.4.271. [DOI] [PubMed] [Google Scholar]
- 16.Culmsee C, et al. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid β-peptide. J Neurochem. 2001;77:220–8. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- 17.Gouras GK, et al. Intraneuronal Aβ42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. Selective cytotoxicity of intracellular amyloid β peptide1-42 through p53 and Bax in cultured primary human neurons. J Cell Biol. 2002;156:519–29. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruce-Keller AJ, et al. Bcl-2 protects isolated plasma and mitochondrial membranes against lipid peroxidation induced by hydrogen peroxide and amyloid β-peptide. J Neurochem. 1998;70:31–9. doi: 10.1046/j.1471-4159.1998.70010031.x. [DOI] [PubMed] [Google Scholar]
- 20.Xu J, Chen S, Ku G, Ahmed SH, Xu J, Chen H, Hsu CY. Amyloid β peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J Cereb Blood Flow Metab. 2001;21:702–10. doi: 10.1097/00004647-200106000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L. β-amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. 2002;10:258–67. doi: 10.1006/nbdi.2002.0516. [DOI] [PubMed] [Google Scholar]
- 22.Rodrigues CMP, Solá S, Silva R, Brites D. Bilirubin and amyloid-β peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol Med. 2000;6:936–46. [PMC free article] [PubMed] [Google Scholar]
- 23.Rodrigues CMP, Solá S, Brito MA, Brondino CD, Brites D, Moura JJ. Amyloid β-peptide disrupts mitochondrial membrane lipid and protein structure: protective role of tauroursodeoxycholate. Biochem Biophys Res Commun. 2001;281:468–74. doi: 10.1006/bbrc.2001.4370. [DOI] [PubMed] [Google Scholar]
- 24.Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–8. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei W, Wang X, Kusiak JW. Signaling events in amyloid β-peptide-induced neuronal death and insulin-like growth factor I protection. J Biol Chem. 2002;277:17649–56. doi: 10.1074/jbc.M111704200. [DOI] [PubMed] [Google Scholar]
- 26.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr Biol. 1999;9:601–4. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 27.Mattson MP, Camandola S. NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–54. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-κB is activated in primary neurons by amyloid β peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:2642–7. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-κB potentiates amyloid β-mediated neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:9409–14. doi: 10.1073/pnas.96.16.9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodrigues CMP, Fan G, Ma X, Kren BT, Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest. 1998;101:2790–9. doi: 10.1172/JCI1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodrigues CMP, Ma X, Linehan-Stieers C, Fan G, Kren BT, Steer CJ. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999;6:842–54. doi: 10.1038/sj.cdd.4400560. [DOI] [PubMed] [Google Scholar]
- 32.Rodrigues CMP, Stieers CL, Keene CD, Ma X, Kren BT, Low WC, Steer CJ. Tauroursodeoxycholic acid partially prevents apoptosis induced by 3-nitropro-pionic acid: evidence for a mitochondrial pathway independent of the permeability transition. J Neurochem. 2000;75:2368–79. doi: 10.1046/j.1471-4159.2000.0752368.x. [DOI] [PubMed] [Google Scholar]
- 33.Rodrigues CMP, Solá S, Sharpe JC, Moura JJG, Steer CJ. Tauroursodeoxycholic acid prevents Bax-induced membrane perturbation and cytochrome c release in isolated mitochondria. Biochemistry. 2003;42:3070–80. doi: 10.1021/bi026979d. [DOI] [PubMed] [Google Scholar]
- 34.Qiao L, et al. Inhibition of the MAPK and PI3K pathways enhances UDCA-induced apoptosis in primary rodent hepatocytes. Hepatology. 2002;35:779–89. doi: 10.1053/jhep.2002.32533. [DOI] [PubMed] [Google Scholar]
- 35.Keene CD, Rodrigues CMP, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc Natl Acad Sci USA. 2002;99:10671–6. doi: 10.1073/pnas.162362299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodrigues CMP, Spellman SR, Solá S, Grande AW, Linehan-Stieers C, Low WC, Steer CJ. Neuroprotection by a bile acid in an acute stroke model in the rat. J Cereb Blood Flow Metab. 2002;22:463–71. doi: 10.1097/00004647-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigues CMP, Solá S, Nan Z, Castro RE, Ribeiro PS, Low WC, Steer CJ. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc Natl Acad Sci USA. 2003;100:6087–92. doi: 10.1073/pnas.1031632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hip-pocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–76. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 39.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 40.Shimohama S. Apoptosis in Alzheimer’s disease—an update. Apoptosis. 2000;5:9–16. doi: 10.1023/a:1009625323388. [DOI] [PubMed] [Google Scholar]
- 41.MacGibbon GA, et al. Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimer’s disease hippocampus. Brain Res. 1997;750:223–34. doi: 10.1016/s0006-8993(96)01351-0. [DOI] [PubMed] [Google Scholar]
- 42.Nagy ZS, Esiri MM. Apoptosis-related protein expression in the hippocampus in Alzheimer’s disease. Neurobiol Aging. 1997;18:565–71. doi: 10.1016/s0197-4580(97)00157-7. [DOI] [PubMed] [Google Scholar]
- 43.Su JH, Deng G, Cotman CW. Bax protein expression is increased in Alzheimer’s brain: correlations with DNA damage, Bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol. 1997;56:86–93. doi: 10.1097/00005072-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 44.Engidawork E, Gulesserian T, Seidl R, Cairns N, Lubec G. Expression of apoptosis related proteins in brains of patients with Alzheimer’s disease. Neurosci Lett. 2001;303:79–82. doi: 10.1016/s0304-3940(01)01618-4. [DOI] [PubMed] [Google Scholar]
- 45.LaFerla FM, Hall CK, Ngo L, Jay G. Extracellular deposition of β-amyloid upon p53-dependent neuronal cell death in transgenic mice. J Clin Invest. 1996;98:1626–32. doi: 10.1172/JCI118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun. 1997;232:418–21. doi: 10.1006/bbrc.1997.6301. [DOI] [PubMed] [Google Scholar]
- 47.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 48.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–8. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 49.Kuner P, Schubenel R, Hertel C. β-amyloid binds to p57NTR and activates NF-κB in human neuroblastoma cells. J Neurosci Res. 1998;54:798–804. doi: 10.1002/(SICI)1097-4547(19981215)54:6<798::AID-JNR7>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 50.Lorenzo A, et al. Amyloid β interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer’s disease. Nat Neurosci. 2000;3:460–4. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- 51.Kajkowski EM, et al. β-Amyloid peptide-induced apoptosis regulated by a novel protein containing a G protein activation module. J Biol Chem. 2001;276:18748–56. doi: 10.1074/jbc.M011161200. [DOI] [PubMed] [Google Scholar]
- 52.Guicciardi ME, Gores GJ. Ursodeoxycholic acid cytoprotection: dancing with death receptors and survival pathways. Hepatology. 2002;35:971–3. doi: 10.1053/jhep.2002.32931. [DOI] [PubMed] [Google Scholar]
- 53.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsuruta F, Masuyama N, Gotoh Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem. 2002;277:14040–7. doi: 10.1074/jbc.M108975200. [DOI] [PubMed] [Google Scholar]
- 55.Golde TE, Eckman CB. Cholesterol modulation as an emerging strategy for the treatment of Alzheimer’s disease. Drug Discov Today. 2001;6:1049–55. doi: 10.1016/s1359-6446(01)01965-1. [DOI] [PubMed] [Google Scholar]