

Abstract
Acute intermittent porphyria (AIP) is a genetic disorder caused by a deficiency of porphobilinogen deaminase (PBGD), the 3rd enzyme in heme synthesis. It is clinically characterized by acute attacks of neuropsychiatric symptoms and biochemically by increased urinary excretion of the porphyrin precursors porphobilinogen (PBG) and 5-aminolevulinic acid (ALA). A mouse model that is partially deficient in PBGD and biochemically mimics AIP after induction of the hepatic ALA synthase by phenobarbital was used in this study to identify the site of formation of the presumably toxic porphyrin precursors and study the effect of enzyme-replacement therapy by using recombinant human PBGD (rhPBGD). After 4 d of phenobarbital administration, high levels of PBG and ALA were found in liver, kidney, plasma, and urine of the PBGD-deficient mice. The administration of rhPBGD intravenously or subcutaneously after a 4-d phenobarbital induction was shown to lower the PBG level in plasma in a dose-dependent manner with maximal effect seen after 30 min and 2 h, respectively. Injection of rhPBGD subcutaneously twice daily during a 4-d phenobarbital induction reduced urinary PBG excretion to 25% of the levels found in PBGD-deficient mice administered with only phenobarbital. This study points to the liver as the main producer of PBG and ALA in the phenobarbital-induced PBGD-deficient mice and demonstrates efficient removal of accumulated PBG in plasma and urine by enzyme-replacement therapy.
INTRODUCTION
Acute intermittent porphyria (AIP) is an autosomal dominant disorder caused by insufficient activity of the 3rd enzyme in the heme biosynthetic pathway, porphobilinogen deaminase (PBGD). It is clinically characterized by acute attacks of neuropsychiatric symptoms (1). Factors known to trigger the attacks are stress, poor nourishment, alcohol or drugs, and chemicals that can induce hepatic ALA synthase (ALAS-1), the 1st and rate-limiting enzyme in heme synthesis (2). The up-regulation of ALAS-1 results in an increased flux of metabolites and overload of the deficient PBGD step with a subsequent accumulation and increased urinary excretion of the porphyrin precursors porphobilinogen (PBG) and 5-aminolevulinic acid (ALA) (1). In patients with symptomatic porphyria, increased levels of porphyrin precursors also have been found in plasma, liver, kidney, and cerebrospinal fluid (3–6). However, despite decades of research, the possible role of the porphyrin precursors in the pathophysiology of AIP symptoms has still not been clarified (7,8).
Current treatment of the acute attack in AIP, based on heme replacement and carbohydrate loading (9), is considered to repress hepatic ALAS-1 and thus the overproduction of PBG and ALA (1). This palliative therapy has improved the prognosis of the disorder but may fail to reverse an established neuropathy (10). Repeated heme administration is often troubled by deteriorated peripheral venous access (11). A new form of treatment, based on PBGD replacement therapy that substitutes the primarily deficient enzyme and thereby avoids the accumulation of the porphyrin precursors, is at present under evaluation (12). Two different isoforms of PBGD, erythroid and housekeeping, exist (13), and the recombinant human PBGD (rhPBGD) is manufactured as the erythroid isoform. The basis for enzyme replacement therapy during an AIP attack is that rhPBGD will remove plasma PBG, resulting in an increased efflux of intracellular PBG to the extracellular compartment and a subsequent removal of this. The hypothesis is that a reduction of intracellular PBG may remove the possible inhibition by high levels of PBG of the 2nd enzyme in heme synthesis, ALA dehydratase (7), and thereby restore ALA metabolism and avoid ALA accumulation.
The aim of this study was to use PBGD-deficient mice, known to biochemically mimic AIP after induction of hepatic ALAS-1 by phenobarbital (14,15), to (a) determine the site of formation of PBG and ALA by studying the accumulation and excretion pattern in tissues, plasma, and urine, and (b) investigate the metabolic effect of both intravenous and subcutaneous administration of recombinant PBGD on PBG and ALA levels in plasma and urine.
MATERIALS AND METHODS
Animals
The animals were kept in facilities at Huddinge University Hospital, Stockholm, Sweden, according to the Swedish regulations and laws for care and use of laboratory animals. Adult female and male PBGD-deficient mice, aged- and sex-matched to a C57BL/6 control strain, were used in the study. The generation of the PBGD-deficient mouse strain was performed as described by Lindberg and others (14). To overload the deficient enzymatic step, the PBGD-deficient mice were intraperitoneally administered with increasing doses (75, 80, 85, 90 mg/kg body weight) of phenobarbital (modified from Lindberg and others [14]) for 4 consecutive days.
Enzyme Administration and Study Design
The recombinant human PBGD (rhPBGD) (HemeBiotech A/S, Lidigö, Sweden) was administered intravenously or subcutaneously. The kinetics of rhPBGD in plasma was determined at different time-points (0, 15, 30, 45, 60, 90, 120, and 180 min) after intravenous injection of 50 μg rhPBGD (2.3 to 2.8 mg/kg) and subcutaneous injection of 300 μg rhPBGD (10 to 15 mg/kg). To investigate if rhPBGD affects the increased plasma and urinary levels of PBG and ALA in phenobarbital-induced PBGD-deficient mice, animals in 1 study were administered intravenously with 3 doses (0.25, 0.5, and 1.0 mg/kg) of rhPBGD, using a single injection in the tail vein at day 4 of the phenobarbital induction. In a 2nd study, they were administered subcutaneously with 3 doses (1, 2, and 5 mg/kg). A control group of PBGD-deficient mice, administered with phenobarbital, was injected with 100 μL of sterile saline. To investigate the effect of repetitive administration of rhPBGD on levels of PBG and ALA in urine, PBGD-deficient mice were treated with rhPBGD during the phenobarbital induction. The mice were administered on days 1 to 4 with phenobarbital and on days 1 to 5 with rhPBGD (20 mg/kg) subcutaneously 2 times daily. As control, the PBGD-deficient mice were administered with phenobarbital on days 8 to 11.
Specimen Collection
Blood was collected from either the saphenous vein or the retro-orbital sinus in MiniCollect© tubes with Li-heparin additive (Greiner Bio-One, Longwood, FL, USA) and immediately centrifuged at 3000g for 10 min. Both plasma and separated blood cells were stored at −80 °C until analysis. Urine was collected protected from light during 24 h by using metabolic cages and stored at −80 °C until analysis. Liver, kidney, spleen, lung, heart, brain, and muscle (thigh) were removed from the anesthetized animal obtained by intraperitoneal injection of 0.2 mL/10 g body weight of a 2.5% tribromoethanol and tert-amyl alcohol (avertin) solution. The tissues were snap-frozen in liquid nitrogen and stored at −80 °C until preparation.
Tissue Preparation
For PBGD activity measurement, a part of the frozen tissue sample (0.1 to 0.4 g) was immersed in 2 mL of 50 mmol/L Tris-HCl buffer (pH 8.2). The sample was homogenized on ice for about 3 min at a speed of 120 rpm using a Potter-Elvehjem glass homogenizer (inner diameter 8.0 mm, frosted wall) fitted with a Teflon pestle (diameter 7.8 mm) and finally centrifuged at 10000g for 10 min at 4 °C. The pellet was discarded and the supernatant was stored at −80 °C until determination of PBGD activity and Western blot analysis. For determination of PBG and ALA concentrations, part of the frozen tissue sample (0.1 to 0.4 g) was homogenized in 0.5 mL 50 mmol/L Tris-HCl buffer (pH 8.2) containing 20 mmol/L citrate using the same procedures as described above. The sample was kept in the dark during the whole procedure. After centrifugation at 3000g for 5 min at 4 °C, the supernatant was transferred to a new tube and centrifuged at 13000g for 10 min at 4 °C. The supernatant was stored at −80 °C until determination of PBG and ALA concentrations.
Determination of PBGD Activity
The PBGD activity in tissue homogenates and blood cells and plasma were assayed by measuring the conversion of PBG to uroporphyrin according to the method of Magnussen and others (16). The protein concentration was analyzed using the Bio-Rad DC Protein Assay (Hercules, CA, USA), and the samples were diluted to a concentration of 0.5 g protein/L with 50 mmol/L Tris-HCl buffer (pH 8.2). A volume of 1.45 mL of the diluted sample was used in the assay, performed as previously described (17). The PBGD activity was expressed in terms of pkat/g protein. One katal equals the conversion of 1 mol of substrate (PBG)/s, and 1 unit/L corresponds to 16.67 nkat/L (18).
Western Blot Analysis of PBGD
A volume of 65 μL of tissue homogenate or blood cells (diluted to 1.0 g protein/L) from control mice, and standards consisting of the erythroid rhPBGD (0.1 g/L) and mouse housekeeping PBGD, produced as reported before (17), was mixed with 25 μL NuPage LDS sample buffer and 10 μL NuPAGE reducing agent (Novex©). The mixture was incubated at 70 °C for 10 min. A volume of 30 μL of the samples and 20 μL of the standards was run in the Western blot assay, performed as previously described (17).
Determination of PBG and ALA Concentrations
PBG and ALA in tissue and plasma were quantified by using a liquid chromatography-mass spectrometry (LC-MS) method developed by HemeBiotech A/S research laboratory (Dr Claes Andersson, HemeBiotech A/S, manuscript in process), and in urine by a quantitative ion exchange column method (19). Homogenized tissue samples and plasma were deproteinized by mixing 100 μL cold acetonitrile with 50 μL sample. After centrifugation at 10000g for 8 min, 50 μL supernatant was transferred to a clean test tube and mixed with 50 μL ethanol and 25 μL triethylamine (ratio 2:2:1). The samples were dried under vacuum. The free amino groups of PBG and ALA were derivatized with phenylisothiocyanate by adding 50 μL phenylisothiocyanate reagent (ethanol:water: triethylamine: phenylisothiocyanate, ratio 7:1:1:1) to each dried sample. The reaction was allowed to proceed for 30 min at room temperature. The samples were dried under vacuum, dissolved in 62.5 μL of 20% ethanol, and centrifuged at 10000g for 5 min. The supernatants were transferred to high-performance liquid chromatography glass vials and kept at 4 °C until analysis. Depending on the concentration of PBG and ALA in the sample, 1 to 50 μL of the supernatant was injected into a Zorbax SB-C18 column (2.1 mm inner diameter × 15 cm) in-line with a Zorbax SB-C8 guard column, which had been equilibrated with95% 25 mmol/L formic acid in water and 5% of 25 mmol/L formic acid dissolved in acetonitrile. The PBG and ALA derivates were eluted with a two-component gradient, which changed from a 2-min isocratic gradient to 40% formic acid in 17 min after sample injection. This ratio was maintained for 1 min before the formic acid was decreased to 20% in 2 min and finally returned to the starting point after an additional 2 min.
The sample was detected by UV absorbance followed by a selected ion monitoring with an electrospray quadropol mass spectrophotometer. A standard curve was run each time with PBG (Sigma, St Louis, MO, USA) and ALA (Bio-Rad), and the concentrations of PBG and ALA in tissue were reported as pmol/mg wet weight tissue, in plasma as μmol/L and in urine as mmol/mol creatinine.
RESULTS
PBGD Activity in Tissues
The basal activity of PBGD in various tissues from the PBGD-deficient mice ranged between 22% to 42% of the activity found in tissues from control mice (Table 1). The highest levels of PBGD activity were observed in spleen, although with a great individual variation. In PBGD-deficient mice the activity ranged between 48 and 756 pkat/g protein (mean 142 ± 195) and in control between 138 and 1270 pkat/g protein (mean 644 ± 430) (Table 1).
Table 1.
Porphobilinogen deaminase (PBGD) activities in tissues from PBGD-deficient and control mice
| PBGD activity (pkat/g protein) (mean ± SD)
|
|||
|---|---|---|---|
| Tissue | PBGD-deficient mice (n = 12) | Control mice (n = 11) | % of control |
| Blood cells | 36.7 ± 19.7 | 126 ± 72.6 | 29 |
| Lung | 35.4 ± 3.3 | 85 ± 14.7 | 42 |
| Kidney | 29.9 ± 6.2 | 91.5 ± 6.4 | 33 |
| Heart | 29.9 ± 2.2 | 78.6 ± 7.2 | 38 |
| Brain | 18.1 ± 3.6 | 64.7 ± 7.5 | 28 |
| Muscle | 13.4 ± 3.6 | 39.2 ± 1.6 | 34 |
| Spleen | 142 ± 195 | 644 ± 430 | 22 |
Molecular Weights of PBGD
The molecular weights of PBGD in spleen and blood cells from control mice were determined by Western blot analysis (Figure 1). In spleen, only the erythroid isoform of PBGD (42 kDa) was found, and in blood cells, both the erythroid and the slightly larger housekeeping isoform of PBGD (44 kDa) were identified. In liver, only a weak band at 44 kDa was detected (data not shown), and no PBGD was detected in the other tissues, i.e. lung, kidney, heart, and brain, suggesting that the amount of PBGD in those tissues was below detection limit of the method.
Figure 1.
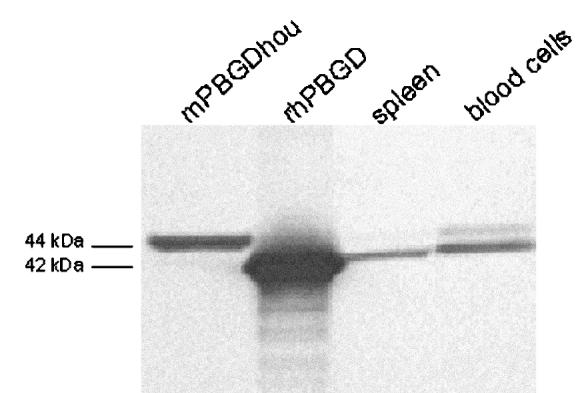
Western blot analysis of PBGD found in spleen and blood cells. The samples (19.5 μg) and standards (1.3 μg) were run on a Bis/Tris acrylamide gel, transferred to a polyvinylidine fluoride membrane, and immunostained using polyclonal rabbit PBGD antiserum. Mouse housekeeping PBGD manufactured as in Johansson and others (17) (mPBGDhou, 44 kDa) and recombinant human erythroid PBGD (rhPBGD, 42 kDa) were used as standards.
PBG and ALA in Plasma, Urine, and Tissues
The concentrations of PBG and ALA were determined in PBGD-deficient mice during the phenobarbital administration. The levels of porphyrin precursors in plasma (Figure 2A) and urine (Figure 2B) from PBGD-deficient mice were gradually increased during the 4-d phenobarbital administration (days 1 to 4). Once the treatment was interrupted the porphyrin precursor levels in urine were reduced to the initial levels within 3 d (Figure 2B). If the procedure was repeated (days 8 to 11), the urinary excretion of PBG and ALA was increased a 2nd time (Figure 2B). Six hours after the 4th phenobarbital injection, high levels of PBG and ALA were found in liver and kidney in the PBGD-deficient mice (Table 2). In the other tissues (that is, spleen, lung, heart and brain), PBG and ALA were under detection limit, <3.5 and <1.0 pmol/mg tissue respectively. Tissue levels of PBG and ALA were also under detection limit in PBGD-deficient mice without phenobarbital administration. No elevated levels of PBG and ALA, except a small increase in plasma, were found in control mice that were administered with phenobarbital.
Figure 2.
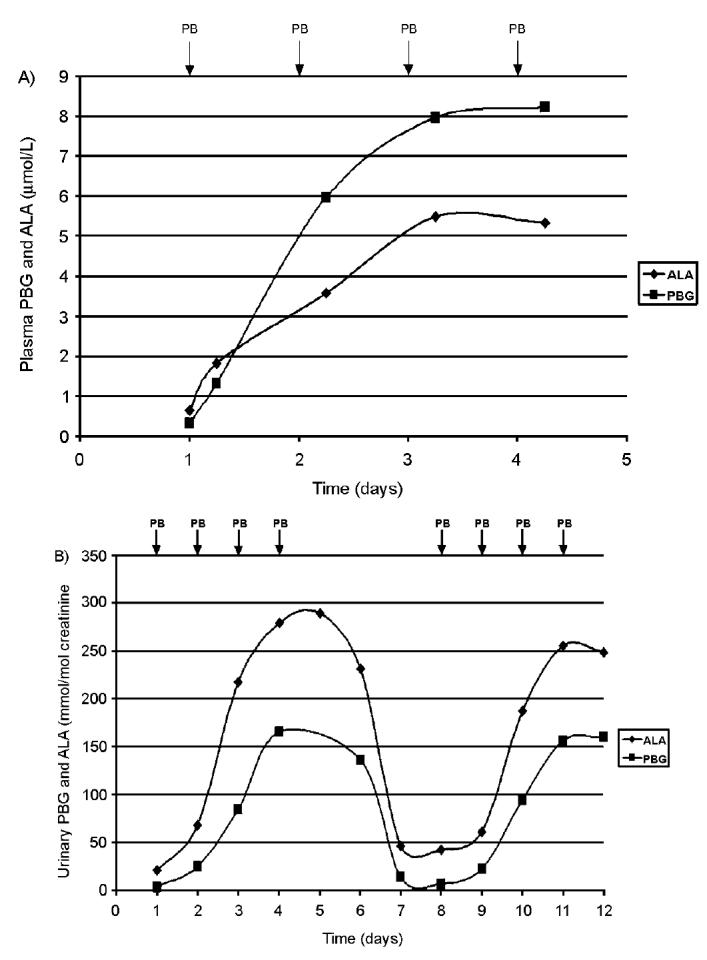
Time-course analysis of levels of PBG and ALA in plasma (A) and urine (B) in PBGD-deficient mice administered with increasing dose of phenobarbital (75, 80, 85, and 90 mg/kg body weight) for 4 d (days 1 to 4). The plasma data represent the mean value from 2 male mice and the urine data from 1 male mouse out of 3 trials. The arrows indicate time of phenobarbital administration (PB). Blood was taken 6 h after each phenobarbital dose and the concentrations of PBG and ALA are expressed as μmol/L. In (B) the results from a 2nd 4-d administration of phenobarbital are presented (days 8 to 11). Urine was collected in 24-h fractions and PBG and ALA concentrations were expressed as mmol/mol creatinine.
Table 2.
5-Aminolevulinic acid (ALA) and porphobilinogen (PBG) concentrations in tissues, plasma, and urine from PBGD-deficient and control mice with or without phenobarbital administration for 4 d
| Sample | PBGD-deficient mice (n = 6) | PBGD-deficient mice + phenobarbital (n = 9) | Control mice (n = 3) | Control mice + phenobarbital (n = 2) |
|---|---|---|---|---|
| ALA concentration (mean ± SD) | ||||
| Liver | UD | 3.8 ± 1.9 | UD | UD |
| Kidney | UD | 5.6 ± 3.5 | UD | UD |
| Spleen | UD | UD | UD | UD |
| Lung | UD | UD | UD | UD |
| Heart | UD | UD | UD | UD |
| Brain | UD | UD | UD | UD |
| Plasma | 0.54 ± 0.45 (n = 64) | 7.0 ± 3.3 (n = 6) | UD | 0.72 ± 0.05 |
| Urine | 19.5 ± 3.9 | 287 ± 24.2 (n = 3) | 15.7 ± 7.4 | 18.4 ± 1.2 (n = 3) |
| PBG concentration (mean ± SD) | ||||
| Liver | UD | 120 ± 40 | UD | UD |
| Kidney | UD | 27 ± 19 | UD | UD |
| Spleen | UD | UD | UD | UD |
| Lung | UD | UD | UD | UD |
| Heart | UD | UD | UD | UD |
| Brain | UD | UD | UD | UD |
| Plasma | 0.32 ± 0.54 (n = 64) | 12.5 ± 3.1 (n = 6) | 0.23 ± 0.08 | 0.63 ± 0.30 |
| Urine | 3.6 ± 1.6 | 168 ± 20.6 (n = 3) | 1.9 ± 1.9 | 2.6 ± 0.9 (n = 3) |
ALA and PBG in tissues are expressed as pmol/mg tissue, in plasma as μmol/L, and in urine as mmol/mol creatinine (UD = under detection limit, i.e. in tissues ALA < 1.0 and PBG < 3.5 pmol/mg tissue, respectively, and in plasma ALA < 0.1 μmol/L). The porphyrin precursors in tissues and plasma were measured 6 h after the 4th injection of phenobarbital. The urine was collected during 24 h after the 4th injection.
Plasma Kinetics of rhPBGD
Recombinant human PBGD (rhPBGD) injected intravenously in mice was cleared from the systemic circulation after 60 min with a plasma half-life of approximately 20 min (Figure 3). A different pattern of rhPBGD in systemic circulation was observed after subcutaneous injection. The highest level of rhPBGD in plasma was found 60 min after injection and the level thereafter declined slowly with a half-life of about 120 min (Figure 3).
Figure 3.
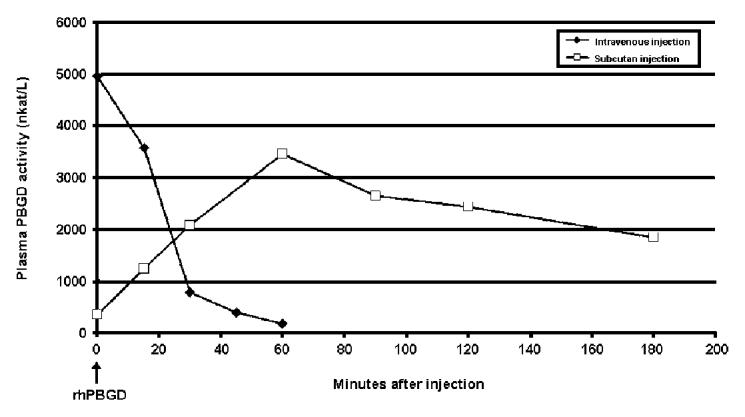
Plasma kinetics for human recombinant porphobilinogen deaminase (rhPBGD) administered intravenously or subcutaneously. For the intravenous administration, 50 μg rhPBGD was injected into the tail vein, and for the subcutaneous administration, 300 μg rhPBGD was injected in the neck. The PBGD activity was expressed as nkat/L. Each data point represents 1 animal.
Effect of rhPBGD on PBG and ALA After 4-d Phenobarbital Induction
Intravenous injection of rhPBGD after the 4th phenobarbital injection resulted in a rapid reduction of plasma PBG levels (Figure 4A). The effect was shown to be dose dependent with an almost complete removal of PBG (6 of 8 mice had undetectable plasma PBG levels) 0.5 h after administration of the highest dose (1 mg/kg). A lowering effect on plasma PBG levels using the highest dose was still present at 2 h following rhPBGD administration (Figure 4A). The subcutaneous injection of rhPBGD resulted in a slower reduction of PBG levels with the best effect seen 2 h after injection (Figure 4B). A dose-response effect was observed with the maximal effect using the highest dose (5 mg/kg). Using this dose the PBG levels, at 2 h after rhPBGD administration, decreased to 19% as compared with 65% in the control group of PBGD-deficient mice that received phenobarbital and saline. Six hours after the intravenous and subcutaneous injections of rhPBGD the PBG levels were returned to or above 100%. No reduction of plasma ALA levels was detected, within the observation time, at any dose of rhPBGD or injection procedure (data not shown).
Figure 4.
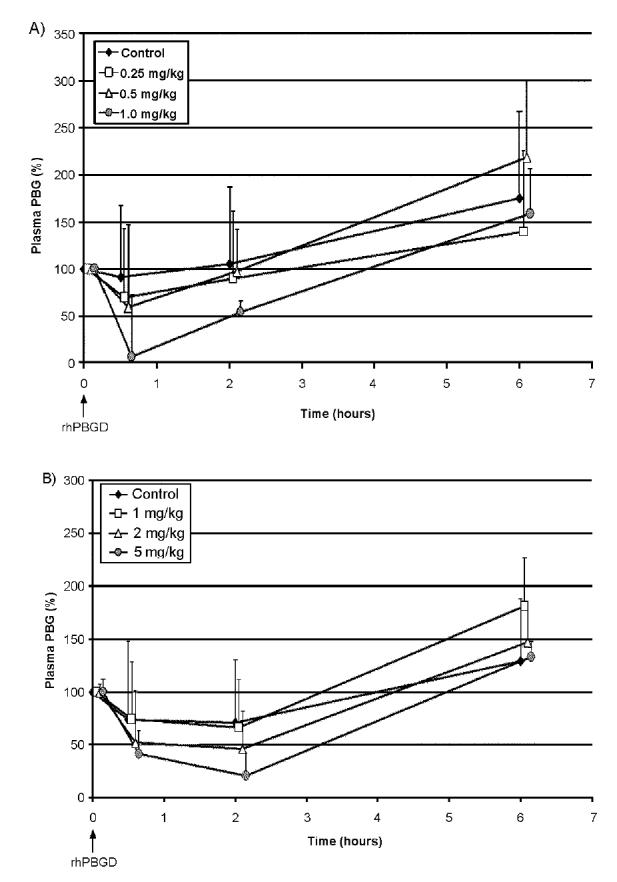
Plasma PBG clearance following administration of recombinant human PBGD (rhPBGD) to PBGD-deficient mice. Eight male PBGD-deficient mice for each dose were 1st administered daily intraperitoneally with increasing doses of phenobarbital (75 to 90 mg/kg) for 4 d to induce plasma PBG levels. At time zero, the animals were administered with rhPBGD. In (A) the mice were intravenously administered with 3 doses of rhPBGD (0.25, 0.5, or 1.0 mg/kg), and in (B) the animals were subcutaneously administered with 1, 2, or 5 mg/kg rhPBGD. Control animals were injected with phenobarbital and saline. Blood was collected after the 4th phenobarbital administration, and subsequently at different time-points (0.5, 2, and 6 h) after rhPBGD administration. The concentration of PBG was determined using an LC-MS method and expressed in terms of relative levels of plasma PBG ± SD, where 100% was defined as the plasma PBG level after 4 d of phenobarbital administration (8.7 ± 4.4 μmol/L, n = 32). To be able to illustrate the standard deviation for each time-point, the values for 0.25 to 1.0 and 1.0 to 5.0 mg/kg rhPBGD have been shifted +0.05 to 0.15 min.
Effect of rhPBGD on PBG and ALA During the 4-d Phenobarbital Induction
The highest PBG concentration in urine found at day 4 in the phenobarbital treated PBGD-deficient mice injected subcutaneously twice daily (day 1 to 5) with rhPBGD was reduced to about 25% of that excreted from control, that is, PBGD-deficient mice injected with phenobarbital and saline (Figure 5). No effect on urinary ALA excretion was observed (data not shown). Later, if the phenobarbital administration was repeated without rhPBGD (day 8 to 11) a similar increase in the urinary excretion of PBG was observed in both groups (Figure 5).
Figure 5.
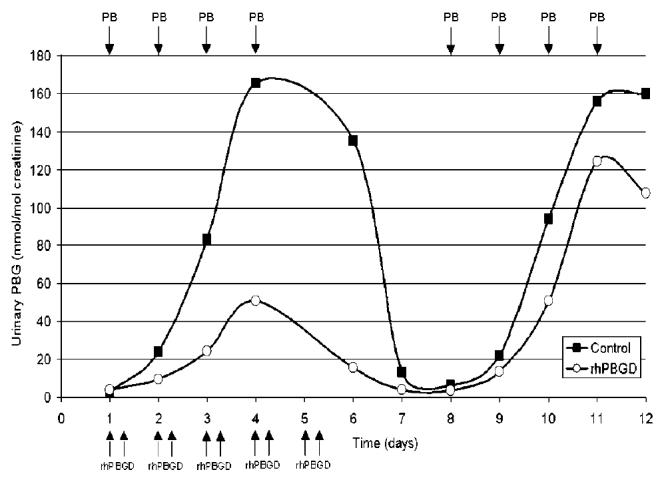
Urinary PBG levels following repeated subcutaneous injections of recombinant human PBGD (rhPBGD). PBGD-deficient mice were administered daily intraperitoneally with increasing doses of phenobarbital (75 to 90 mg/kg) for 4 d at 2 occasions (days 1 to 4 and days 8 to 11). The arrows indicate time of phenobarbital (PB) as well as rhPBGD administration. One mouse was administered subcutaneously with rhPBGD twice daily (20 mg/kg) between days 1 and 5. Urine was collected in 24-h fractions and PBG concentrations were expressed as mmol/mol creatinine.
DISCUSSION
PBG and ALA have been suggested to be involved in the pathophysiology of AIP (1), but their site of formation has not been completely elucidated. In this study we used a mouse model (14) that biochemically mimics AIP after phenobarbital administration to study the accumulation and excretion pattern of the porphyrin precursors. The mouse model was also used to study the effect of rhPBGD on the high levels of PBG and ALA. This study, together with a recent abstract from our group using the same enzyme (12), are the 1st steps to investigate enzyme-replacement therapy for AIP.
The enzymatic characterization of the AIP mouse model showed that the PBGD activity in the different tissues ranged from 22% to 42% of control (Table 1). The value for liver tissue, 28%, is in close agreement with the 30% found in other studies (14,15). The high enzyme activity of the erythroid isoform of PBGD (Table 1 and Figure 1) found in spleen is probably due to the dominance of immature red blood cells. In the adult mouse, both the spleen and bone marrow are hematopoietic organs (20), and the result is therefore consistent with the higher PBGD activity reported for hematopoietic tissues (21).
The low or undetectable levels of PBG and ALA in tissues and plasma from the PBGD-deficient mice without induction of phenobarbital (Table 2) suggest that the biosynthetic pathway operates efficiently in this model in the absence of triggering factors. The induction of hepatic ALAS-1 by phenobarbital in the PBGD-deficient mice resulted in increased concentrations of PBG and ALA in plasma (Table 2 and Figure 2A) and urine (Table 2 and Figure 2B). A 2nd administration of phenobarbital for 4 d caused a similar increased excretion pattern of PBG and ALA in urine (Figure 2B). The concentration of ALA in plasma obtained in this study during the phenobarbital induction was found to be half of that previously reported (15,22) and in urine about 80% lower (14,15,22). This may be due to the use of lower doses of phenobarbital in the present study, because our mice did not survive the higher doses used in the other studies (14,15,22). There are no previous data on plasma and urinary PBG levels in the PBGD-deficient mice. The concentration of ALA in urine from PBGD-deficient mice administered with phenobarbital was twice as high as PBG, which differs from observations in urine from AIP patients with clinical symptoms and asymptomatic patients with high excretion, where PBG values are twice as high as ALA (3,23). The relatively lower PBG concentration found in urine from the PBGD-deficient mice could also be a consequence of the 24-h collection period at room temperature, in which PBG may have been converted nonenzymatically to uroporphyrin (24). In plasma from phenobarbital-provoked PBGD-deficient mice the PBG levels were approximately twice as high as the ALA levels (Table 2 and Figure 2A), which is also found in AIP patients during acute attack and asymptomatic patients with high excretion (3,23).
After 4 d of phenobarbital induction, the liver and kidney tissues of the PBGD-deficient mice contained by far the highest concentrations of PBG and ALA (Table 2). The data support the hypothesis that the hepatic induction of ALAS-1 is primarily responsible for the elevated levels of porphyrin precursors during an acute attack (1). The high values found in kidney could reflect either a site of high synthesis of porphyrin precursors or merely an accumulation during the filtration process. The great dominance of PBG over ALA in the liver (Table 2) could reflect either a higher clearance rate for ALA or that it is readily metabolized to PBG. A high clearance rate could indicate the possibility of a fast distribution of ALA to other organs. However, we did not find high levels of ALA in other organs except liver and kidney. It has previously been reported that ALA accumulates in the extracellular fluid of striatum in PBGD-deficient mice after phenobarbital induction (22), but in the present study, the ALA level was not increased over control value in brain.
The rationale for using enzyme-replacement therapy clinically in the AIP attack is 1st to reduce the high levels of plasma PBG, which will possibly release ALAD inhibition, and thus ALA accumulation as well. The toxicity of the porphyrin precursors is controversial, and so far not elucidated, as there is no clear correlation between the degree of clinical symptoms and the amount of PBG and ALA in the urine (25). During acute AIP attacks PBG and ALA are always increased, but there is also a large number of AIP patients with elevated levels of PBG and ALA in plasma and urine without having any symptoms (23). ALA has been shown to have a number of effects in biological systems (8,26), but has failed to show any effect in vivo when administered to humans (27,28) and rodents (29). Possible toxicity of PBG has been reported (30) but has also been found harmless when injected in vivo (3). The toxicity of PBG has been questioned because patients with ALA dehydratase porphyria and lead poisoning have similar symptoms to AIP, even though only ALA is increased (1).
Intravenous and subcutaneous injections of rhPBGD to phenobarbital-treated PBGD-deficient mice, lowered the high levels of plasma PBG in a dose-dependent manner with maximal effect seen 30 min and 2 h after the administration, respectively (Figure 4). The pattern of plasma PBG reduction is consistent with the different kinetics of rhPBGD observed in plasma after intravenous and subcutaneous injection (Figure 3). Data from the clinical trial showed that intravenous injection of rhPBGD produced an instant, almost complete, removal of plasma PBG (12). Recombinant human PBGD injected intravenously to mice was cleared from the systemic circulation in about 60 min. After the subcutaneous injection, a slower absorption of rhPBGD to plasma was observed with the highest level of rhPBGD found 60 min after administration, which thereafter slowly declined (Figure 3). The rhPBGD dose needed to lower the PBG level in plasma is about 5 times higher using the subcutaneous route as compared with the intravenous route, but the effect on the PBG clearance in plasma seems to be prolonged when using subcutaneous administration of rhPBGD. Six hours after the rhPBGD administration, the levels of PBG in plasma were increased to or even more than 100% (Figure 4). This is probably due to the response of the last phenobarbital injection, given at the same time as rhPBGD. A minor reduction of plasma PBG was also seen in the control group after subcutaneous administration of saline (Figure 4B), which could be explained by a delayed effect of the phenobarbital administration.
The administration of rhPBGD had no effect on ALA levels in the present study. However, the short-term effect seen on the plasma PBG levels after 1 injection of rhPBGD is probably not enough to obtain an effect on ALA levels in plasma. To reduce ALA levels, a prolonged treatment of rhPBGD is probably needed to sustain a decrease in the intracellular level of PBG. The effect of a repeated subcutaneous treatment of rhPBGD during the 4-d phenobarbital induction was studied in 24-h urine fractions from PBGD-deficient mice. A reduction of urinary PBG to about 25% of the level in a control group with PBGD-deficient mice, administered with phenobarbital, was observed (Figure 5), but no effect on ALA was seen. A reduction of ALA levels may be difficult to obtain in the PBGD-deficient mice, because the repeated phenobarbital administrations maintain the induction of ALAS-1 and perpetuate a high synthesis of porphyrin precursors.
In conclusion, this study demonstrates that the major site of formation of PBG and ALA during phenobarbital induction in the PBGD-deficient mice is the liver and possibly also the kidney. High levels of the porphyrin precursors were found in plasma and urine and the injection of rhPBGD, both intravenous and subcutaneous, reduced the plasma and urinary levels of PBG.
Acknowledgments
We wish to thank Associate Professor Stig Thunell for reviewing the manuscript, and Anette Pettersson and Helena Reuterwall at HemeBiotech A/S for technical assistance with the LC-MS method and Western blot. The work was supported by grants from Clas Groschinsky’s Memorial Fund and the Karolinska Institute.
REFERENCES
- 1.Anderson KE, Sassa S, Bishop DF, Desnick RJ. (2001) Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias. In: The Metabolic and Molecular Bases of Inherited Disease (Vol. 2). Scriver CR, Beaudet AL, Valle D, Sly WS (eds.). McGraw-Hill, New York, USA, pp. 2991–3062.
- 2.Granick S. The induction in vitro of the synthesis of delta-aminolevulinic acid synthetase in chemical porphyria: A response to certain drugs, sex hormones, and foreign chemicals. J Biol Chem. 1966;241:1359–75. [PubMed] [Google Scholar]
- 3.Miyagi K, Cardinal R, Bossenmaier I, Watson CJ. The serum porphobilinogen and hepatic porphobilinogen deaminase in normal and porphyric individuals. J Lab Clin Med. 1971;78(5):683–95. [PubMed] [Google Scholar]
- 4.Schmid R, Schwartz S, Watson CJ. Porphyrin content of bone marrow and liver in the various forms of porphyria. AMA Arch Int Med. 1954;93:167–90. doi: 10.1001/archinte.1954.00240260001001. [DOI] [PubMed] [Google Scholar]
- 5.Sweeney VP, Pathak MA, Asbury AK. Acute intermittent porphyria. Increased ALA-synthetase activity during an acute attack. Brain. 1970;93:369–80. doi: 10.1093/brain/93.2.369. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg A, Rimington C. Fate of porphobilinogen in the rat. Relation to acute porphyria in man. Lancet. 1954;2:172–3. doi: 10.1016/s0140-6736(54)90143-9. [DOI] [PubMed] [Google Scholar]
- 7.Thunell S. Porphyrins, porphyrin metabolism and porphyrias. I Update. Scand J Clin Lab Invest. 2000;60(7):509–40. doi: 10.1080/003655100448310. [DOI] [PubMed] [Google Scholar]
- 8.Bechara EJH. Oxidative stress in acute intermittent porphyria and lead poisoning may be triggered by 5-aminolevulinic acid. Braz J Med Biol Res. 1996;29:841–51. [PubMed] [Google Scholar]
- 9.Kauppinen R, Timonen K, Mustajoki P. Treatment of the porphyrias. Ann Med. 1994;26:31–8. doi: 10.3109/07853899409147324. [DOI] [PubMed] [Google Scholar]
- 10.Tenhunen R, Mustajoki P. Acute porphyria: treatment with heme. Semin Liver Dis. 1998;18(1):53–5. doi: 10.1055/s-2007-1007140. [DOI] [PubMed] [Google Scholar]
- 11.Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract. 2002;56(4):272–8. [PubMed] [Google Scholar]
- 12.Sardh E, Rejkjaer L, Harper P, Andersson DE. First clinical trial of i.v rhPBGD in healthy subjects with and without diagnosed manifest acute intermittent porphyria (AIP) Physiol Res. 2003;52:23S. [Google Scholar]
- 13.Grandchamp B, De Verneuil H, Beaumont C, Chretien S, Walter O, Nordmann Y. Tissue-specific expression of porphobilinogen deaminase. Two isoenzymes from a single gene. Eur J Biochem. 1987;162(1):105–10. doi: 10.1111/j.1432-1033.1987.tb10548.x. [DOI] [PubMed] [Google Scholar]
- 14.Lindberg RL, et al. Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria. Nat Genet. 1996;12(2):195–9. doi: 10.1038/ng0296-195. [DOI] [PubMed] [Google Scholar]
- 15.Jover R, Hoffmann F, Scheffler-Koch V, Lindberg RL. Limited heme synthesis in porphobilinogen deaminase-deficient mice impairs transcriptional activation of specific cytochrome P450 genes by phenobarbital. Eur J Biochem. 2000;267(24):7128–37. doi: 10.1046/j.1432-1327.2000.01815.x. [DOI] [PubMed] [Google Scholar]
- 16.Magnussen CR, Levine JB, Doherty JM, Cheesman JO, Tschudy DP. A red cell enzyme method for the diagnosis of acute intermittent porphyria. Blood. 1974;44(6):857–68. [PubMed] [Google Scholar]
- 17.Johansson A, Möller C, Gellerfors P, Harper P. Non-viral mediated gene transfer of porphobilinogen deaminase into mammalian cells. Scand J Clin Lab Invest. 2002;62(2):105–14. doi: 10.1080/003655102753611726. [DOI] [PubMed] [Google Scholar]
- 18.Lentner C. (1984) Physical chemistry composition of blood hematology somatometric data. In: Geigy Scientific Tables (Vol. 3), Ciba-Geigy, pp. 166–7.
- 19.Davis JR, Andelman SL. Urinary delta-aminolevulinic acid (ALA) levels in lead poisoning. I A modified method for the rapid determination of urinary delta-aminolevulinic acid using disposable ion-exchange chromatography columns. Arch Envir Health. 1967;15:53–9. doi: 10.1080/00039896.1967.10664873. [DOI] [PubMed] [Google Scholar]
- 20.Bannerman RM. (1983) Hematology. In: The mouse in medical research (Vol. III). Foster HL, Small JD, Fox JG (eds.). Academic Press, Orlando, FL, USA, pp. 293–312.
- 21.Sassa S. Sequential induction of heme pathway enzymes during erythroid differentiation of mouse friend leukemia virus-infected cells. J Exp Med. 1976;143:305–15. doi: 10.1084/jem.143.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindberg RL, et al. Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J Clin Invest. 1999;103(8):1127–34. doi: 10.1172/JCI5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harper P, Floderus Y, Andersson C, Möller C, Rejkjaer L, Sardh E, Andersson DE. Correlation between plasma and urinary levels of porphobilinogen (PBG) and 5-aminolevulinic acid (ALA) in ten asymptomatic gene carriers of acute intermittent porphyria (AIP) with increased porphyrin precursor excretion. Physiol Res. 2003;52:10S. doi: 10.1373/clinchem.2005.058198. [DOI] [PubMed] [Google Scholar]
- 24.Bossenmaier I, Cardinal R. Stability of delta-aminolevulinic acid and porphobilinogen in urine under varying conditions. Clin Chem. 1968;14:610–4. [PubMed] [Google Scholar]
- 25.Elder GH. Genetic defects in the porphyrias: types and significance. Clin Dermatol. 1998;16(2):225–33. doi: 10.1016/s0738-081x(97)00202-2. [DOI] [PubMed] [Google Scholar]
- 26.Thunell S, et al. Markers for vulnerability in acute porphyria. A hypothesis paper. Eur J Clin Chem Clin Biochem. 1995;33(4):179–94. doi: 10.1515/cclm.1995.33.4.179. [DOI] [PubMed] [Google Scholar]
- 27.Dowdle E, Mustard P, Spong N, Eales L. The metabolism of [5-14C]delta-aminolaevulinic acid in normal and porphyric human subjects. Clin Sci. 1968;34:233–51. [PubMed] [Google Scholar]
- 28.Mustajoki P, Timonen K, Gorchein A, Seppalainen AM, Matikainen E, Tenhunen R. Sustained high plasma 5-aminolaevulinic acid concentration in a volunteer: no porphyric symptoms. Eur J Clin Invest. 1992;22(6):407–11. doi: 10.1111/j.1365-2362.1992.tb01482.x. [DOI] [PubMed] [Google Scholar]
- 29.Edwards SR, Shanley BC, Reynoldson JA. Neuropharmacology of delta-aminolaevulinic acid-I. Effect of acute administration in rodents. Neuropharmacology. 1984;23:477–81. doi: 10.1016/0028-3908(84)90259-4. [DOI] [PubMed] [Google Scholar]
- 30.Feldman DS, Levere RD, Lieberman JS, Cardinal RA, Watson CJ. Presynaptic neuromuscular inhibition by porphobilinogen and porphobilin. Proc Nat Acad Sci USA. 1971;68:383–6. doi: 10.1073/pnas.68.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]