

Abstract
Vascular endothelial growth factor (VEGF) plays a central role in the development of ocular neovascularization (NV) and is an excellent target for therapeutic intervention. VEGF acts through several receptors, including VEGF receptor 1, VEGF receptor 2, neuropilin-1 (Npn1), and Npn2, but the exact role of these receptors in the development of retinal NV is unknown. In this study, we investigated the expression of npn2 mRNA during new blood vessel growth in the retina and used npn2 knockout mice to assess the impact of deficiency of Npn2 on retinal NV. The level of npn2 mRNA in the retina increased during retinal vascular development, after exposure to hyperoxia, and after the onset of retinal ischemia. Immunohistochemistry showed colocalization of Npn2 with a vascular marker in retinal NV. Compared with littermate controls, mice deficient in Npn2 had significantly less ischemia-induced retinal NV and very little subretinal NV due to expression of a Vegf transgene. These data suggest that Npn2 facilitates VEGF-induced retinal NV and may constitute a useful target for therapeutic intervention in ocular diseases complicated by NV.
INTRODUCTION
There is compelling evidence indicating that vascular endothelial growth factor (VEGF) plays a central role in the development of retinal and choroidal neovascularization (NV) (for review, see 1). This has led to clinical trials testing the effects of VEGF antagonists in patients with subfoveal choroidal NV due to age-related macular degeneration. Preliminary results suggest that intravitreous injections of an aptamer that binds VEGF165 or an antibody fragment that binds all VEGF isoforms provide benefit, but that there is room for improvement (2–4).
One potential way to achieve greater benefit with VEGF antagonists is to improve delivery. Intravitreous injections at 4- to 6-wk intervals result in high levels of antagonist for brief periods followed by much longer periods during which levels are low and may be subtherapeutic. Improvements are also likely to come from a better understanding of the mechanism by which VEGF stimulates retinal or choroidal NV. In this regard, it is valuable to obtain clues from mechanisms identified in other vascular beds, while at the same time understanding that all inferences must be directly tested in the vascular beds of the eye.
Knockout mice have provided important information regarding the role of VEGF in embryonic vascular development. Deletion of even 1 allele of the VEGF gene results in absence of endothelial cell differentiation absence of blood vessel formation, and early embryonic death (5,6). Overexpression of VEGF in embryos is also catastrophic, indicating that precise control of VEGF expression is needed for vascular development (7). There are 2 VEGF receptors that also play key roles. Absence of VEGF receptor 2 (VEGFR2) results in early embryonic death with a phenotype very similar to absence of VEGF: failure of endothelial cells to differentiate and complete disruption of blood vessel formation (8). Embryos lacking VEGF receptor 1 (VEGFR1) also die early due to disruption of blood vessel formation, but endothelial cells are able to differentiate, indicating that dysfunction occurs at a later stage of development (9). This phenotype can be rescued by expressing a truncated form of VEGFR1 that has an intact extracellular domain, but lacks a tyrosine kinase domain (10). This indicates, that interactions with other extracellular or membrane-bound proteins but not intracellular signaling, are critical for VEGFR1-mediated developmental angiogenesis.
Other lines of evidence have also indicated that VEGFR1 and VEGFR2 do not act alone in the transduction of VEGF signaling; neuropilins play an important role. Neuropilin-1 (Npn1) and neuropilin-2 (Npn2) were 1st identified as receptors for semaphorins, a family of extracellular proteins that function in axon guidance (11–13). But the Npns have short intracellular domains and activate intracellular signaling by forming complexes with plexins, which have intracellular kinase domains (14,15). Similarly, Npn1 and Npn2 form complexes with VEGFR1 and VEGFR2, and thereby act as coreceptors for VEGF ligands that contain a heparin-binding domain (16–20). Targeted disruption of npn1 results in embryonic death at E12.5 to E13.5 with a combination of neural, vascular, and cardiac abnormalities (21). In contrast, Npn2 does not play an essential role in vascular development, because npn2−/− mice are viable (22). However, mice lacking Npn2 show absence or severe reduction of small lymphatics and capillaries, suggesting that Npn2 plays a role in vascular development that if disturbed is compatible with survival (23).
The role of Npns and semaphorins in NV in adults is less clear. However, a potential role of Npns in tumor angiogenesis has been suggested by demonstrations that semaphorins 3F and 3B have tumor suppressor activity, in part due to direct effects on tumor cells, but also due to suppression of tumor vessels (24–26). Therefore, there is good rationale for investigating the role of Npns in other angiogenic disease processes. In this study, we have used npn2−/− mice to explore the role of Npn2 in retinal and subretinal NV.
MATERIALS AND METHODS
Measurement of Npn2 mRNA Levels in Retina
Mice were treated in accordance with the Association for Research in Vision and Ophthalmology guidelines for the use of animals in research. Neonatal C57BL/6 mice (Jackson Labs, Bar Harbor, ME, USA) were euthanized at several time points after birth with or without exposure to 75% oxygen between postnatal day 0 and 21 (P0 and P21). Eyes were removed, retinas were dissected, and total retinal RNA was isolated using RNeasy kits (QUIGEN Inc, Chatsworth, CA, USA). RNA concentration was measured by spectrophotometry (Gen SpecIII, Hitachi, Tokyo, Japan) and after treatment with DNase I (Ambion Inc, Austin, TX, USA), 1 μg of RNA was incubated with reverse transcriptase (SuperScript II, Life Technologies, Gaithersburg, MD, USA) and 5 μM oligo-d(T) primer. Samples of cDNA were aliquoted and stored at −80°C.
Real-time polymerase chain reaction was performed using a Light Cycler rapid thermal cycler system (Roche Applied Bioscience) according to the manufacturer’s instructions. Primers specific for npn2 (forward: 5′-CTG CCC TAT GAT GCC AGC AA-3′and reverse: 5′-ACG CTT CGG TCT TCA GGG AA-3′) and cyclophilin A (forward: 5′-CAG ACG CCA CTG TCG CTT T-3′ and reverse: 5′-TGT CTT TGG AAC TTT GTC TGC AA-3′) were used. Cyclophilin was used as a control for normalization (27). cDNA was amplified in a 20-mL volume containing LightCycler-DNA Master SYBR Green I mix (Roche Diagnostics Ltd), primers (0.5 mM), nucleotides, and MgCl2 (2 to 5 mM). Standard curves generated with purified cDNA were used to calculate copy number according to the Roche absolute quantification technique manual. Values are expressed as copies of npn2 mRNA per 105 copies of cyclophilin A mRNA.
Immunohistochemical Staining for Npn2
Mice with ischemic retinopathy or control mice were euthanized, and eyes were rapidly removed and frozen in optimum cutting temperature embedding compound (Miles Diagnostics, Elkhart, IN, USA). Frozen sections (10 μm) were thawed, air-dried, and fixed in prechilled acetone. Sections were incubated in 10% normal donkey serum followed by overnight incubation at 4 °C in polyclonal rabbit antiserum to mouse Npn2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and biotinylated Griffonia simplicifolia lectin B4 (GSA) (Vector Laboratories, Burlingame, CA, USA), which selectively binds to vascular cells. Signals were visualized using Cy3-conjugated donkey antirabbit antibody (1:100, Jackson Immuno Research Laboratories Inc, West Grove, PA, USA) and avidin conjugated with fluorescein isothiocynate (FITC) (1:100, Biosource International, Camarillo, CA, USA) for 45 min at room temperature. The sections were thoroughly washed with phosphate-buffered saline containing 0.25% Triton X-100 (PBST) between all incubations, and both the primary and secondary antibodies were diluted in PBST containing 1% donkey serum. Fluorescently immunolabeled sections were cover-slipped with antifade medium (0.1% 1,4-phenylenediamine in 45 mL glycerin and 5 mL phosphate-buffered saline). Controls included sections from npn2−/− mice with ischemic retinopathy and sections from npn2+/+ mice with ischemic retinopathy for which the primary antibody was omitted. Sections were examined under a Nikon microscope and captured as digital files with a Nikon Digital Still Camera DXM1200 (Nikon Instruments Inc, NY, USA).
Oxygen-Induced Ischemic Retinopathy in Npn2-Deficient Mice and Littermate Controls
Mice with targeted disruption of the npn2 gene (npn2−/− mice) (22) were generously provided by Alex Kolodkin and David Ginty (Johns Hopkins University, Baltimore, MD, USA). The mice were mated into a C57BL/6 background. Litters containing npn2−/−, npn2+/−, and npn2+/+ mice were exposed to 75% oxygen between P7 and P12, and then placed back in room air and euthanized at P17 (28). Eyes were removed and frozen in optimal cutting temperature embedding compound (Miles Diagnostics). Ocular frozen sections (10 μm) were histochemically stained with biotinylated GSA. Slides were incubated in methanol/H2O2 for 10 min at 4 °C, washed with 0.05 M Tris-buffered saline (TBS), pH 7.6, and incubated for 30 min in 10% normal porcine serum. Slides were incubated 2 h at room temperature with biotinylated GSA and after rinsing with 0.05 M TBS, they were incubated with avidin coupled to alkaline phosphatase (Vector Laboratories) for 45 min at room temperature. After being washed for 10 min with 0.05 M TBS, slides were incubated with diaminobenzidine to give a brown reaction product. Some slides were counterstained with hematoxylin, and all were mounted with Cytoseal.
To perform quantitative assessments, 10 μm serial sections were cut through entire eyes, and starting from the 1st section that contained iris and extending to the last section on the other side of the eye that contained iris, every tenth section was stained with GSA. GSA-stained sections were examined with an Axioskop microscope (Zeiss, Thornwood, NY, USA), and images were digitized using a digital color video camera (IK-TU40A, Toshiba, Tokyo, Japan) and a frame grabber. Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA) was used to delineate GSA-stained cells on the surface of the retina and their area was measured. The mean of all measurements from each eye was used as a single experimental value.
Npn2 Deficiency in Transgenic Mice with Expression of VEGF in Photoreceptors
Transgenic mice in which the rhodopsin promoter drives expression of VEGF in photoreceptors (rho/VEGF mice) (29) were crossed with npn2+/− mice, and npn2+/− offspring that carried a rho/VEGF transgene were crossed with npn2+/− mice. At P21, npn2−/−, npn2+/−, and npn2+/+ mice that carried a rho/VEGF transgene were anesthetized and perfused with fluorescein-labeled dextran (2 × 106 average mw; Sigma, St. Louis, MO, USA) and retinal flat mounts were prepared as previously described (30). Briefly, the eyes were removed, fixed for 1 h in 10% phosphate-buffered formalin, and the cornea and lens were removed. The entire retina was carefully dissected from the eyecup, radially cut from the edge of the retina to the equator in all 4 quadrants, and flat-mounted in Aquamount with photoreceptors facing upward. Flat-mounts were examined by fluorescence microscopy at 400× magnification, which provides a narrow depth of field so that when focusing on NV on the outer edge of the retina, the remainder of the retinal vessels are out of focus allowing easy delineation of the NV (30). The outer edge of the retina, which corresponds to the subretinal space in vivo, is easily identified, and therefore there is standardization of focal plane from slide to slide. Images were digitized using a 3 CCD color video camera (IK-TU40A, Toshiba) and a frame grabber. Image-Pro Plus software was set to recognize fluorescently stained NV and used to delineate each of the lesions throughout the entire retina and calculate the number of lesions per retina, the area of each lesion, and the total area of NV per retina.
RESULTS
Npn2 Expression Is Increased in Retina by Hyperoxia and Ischemia
Compared with cyclophilin mRNA, the level of npn2 mRNA in the retina is low; levels are particularly low during the 1st wk after birth and increase during the 2nd wk to adult levels (Figure 1). This is the time that the deep capillary bed of the retina is developing by physiologic angiogenesis. This could mean that Npn2 plays some role in physiologic angiogenesis, but if so, it does not appear to be essential because npn2–/– mice appear to have normal development of retinal blood vessels and normal retinal circulation as adults (not shown).
Figure 1.
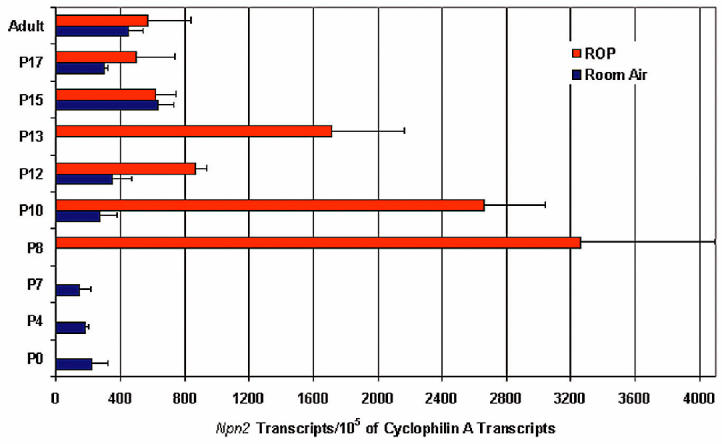
Measurement of Npn2 mRNA during retinal development and in a model of ischemic retinopathy. C57BL/6 mice were euthanized at several time points after birth with or without exposure to 75% oxygen between P7 and P12, and total retinal RNA was isolated. Real-time quantitative reverse transcription–polymerase chain reaction was performed to measure the level of Npn2 and cyclophilin mRNA. Cyclophilin A mRNA is generally modulated very little and serves as a reference for normalization. Data are expressed as the number of copies of Npn2 transcripts per 105 copies of cyclophilin A transcripts, and each bar represents the mean (±SEM) calculated from the retinas of 4 mice. Npn2 expression increased throughout retinal development and reached a stable steady-state level by P15. Exposure to hyperoxia resulted in a significant increase in Npn2 expression (P < 0.01 for difference between P7 room air and P8 hyperoxia), which decreased by the 5th day of oxygen exposure and then briefly increased 1 d after removal from oxygen.
Mice placed in hyperoxia at P7 showed a large increase in npn2 mRNA in the retina at P8, and the level was still markedly elevated at P10, and then decreased substantially after 5 d of hyperoxia. During the 5 d of hyperoxia, development of the retinal vasculature stops and regression of blood vessels occurs, so that when mice are placed in room air at P12, the retina becomes ischemic (28). After 1 d of ischemia, there was another increase in npn2 mRNA levels in the retina, which decreased to the levels seen in untreated mice within 2 d (see Figure 1). In mice with ischemic retinopathy, there was immunostaining for Npn2 in tufts of tissue protruding from the retina (Figure 2). The Npn2 staining colocalized with GSA, demonstrating that the Npn2 was present in retinal NV. There was no staining for Npn2 in npn−/− mice and or for npn+/+ mice with ischemic retinopathy when primary antibody was omitted.
Figure 2.

Immunohistochemical staining for Npn2 is increased in hypoxic retina. Ocular sections from P17 wild type mice or npn2−/− mice with ischemic retinopathy were double–labeled for Npn2 (Cy3, red, 1st column) and the vascular cell marker, Griffonia simplicifolia lectin B4 (GSA; FITC, green, 2nd column). There was staining for Npn2 in tissue extending from the surface of the retina into the vitreous cavity (1st column, top row, arrows). Labeling with GSA (2nd column, top row) showed neovascularization on the surface of the retina (arrows) and retinal vessels within the retina. Merging of the 2 images (3rd column, top row) showed that the Npn2 expression was within the retinal neovascularization on the surface of the retina (arrows). Sections from npn2−/− mice with ischemic retinopathy rarely showed retinal neovascularization, but a section with a tuft of neovascularization that labeled with GSA was identified (2nd column, middle row, arrow). There was no staining for Npn2 within the neovascularization or elsewhere on the section (1st column, middle row). The bottom row shows a section adjacent to the one shown in the top row in which primary anti-Npn2 antibody (1st column) or GSA (2nd column) were omitted, and there is no staining.
Mice Deficient in Npn2 Develop Less Ischemia-Induced Retinal NV Compared with Littermate Controls that Express Npn2
Mice deficient in Npn2 showed no difference from wild-type mice in development of the superficial or deep capillary beds (Figure 3). Littermate npn2−/−, npn2+/−, and npn2+/+ mice were examined in a model of oxygen-induced ischemic retinopathy (28). Npn2−/− mice with ischemic retinopathy had a few small areas of NV above the internal limiting membrane (Figure 4A and 4D), while npn2+/− (see Figure 4B and 4D) and npn2+/+ mice (see Figure 4C and 4E) had more and larger areas of NV (readers unaccustomed to viewing and interpreting retinal NV on retinal sections should refer to Figure 1 in Reference 1, which shows a schematic that assists in the formation of a 3-dimensional mental image). Measurement of the mean area of NV per section by image analysis showed significantly less NV in npn2−/− mice compared with npn2+/+ or npn2+/− mice (see Figure 4G). Although the amount of NV appeared somewhat less in npn2+/− mice compared with npn2+/+ mice, the difference was not statistically significant.
Figure 3.

Mice deficient in Npn2 have normal retinal vessels. Retinal flat mounts from npn−/− mice perfused with fluorescein-labeled dextran show normal superficial and deep retinal capillary beds that appear identical to those seen in npn+/+ mice.
Figure 4.

Mice deficient in Npn2 develop less ischemia-induced retinal NV. Littermate npn2−/− (n = 7), npn2+/− (n = 5), and npn2+/+ mice (n = 5) were exposed to 75% oxygen between P7 and P12 and euthanized on P17. Retinal frozen sections were stained with Griffonia simplicifolia lectin and visualized with DAB, so that vascular cells appear brown. Representative sections from an npn2−/− mouse (A, D) show occasional small areas of retinal neovascularization extending above the internal limiting membrane. In contrast, representative sections from littermate npn2+/− (B, E, arrows) or npn2+/+ mice (C, F, arrows) show many large fronds of retinal neovascularization extending above the internal limiting membrane. Measurement of the area of retinal neovascularization above the internal-limiting membrane on serial sections as described in Materials and Methods demonstrated that npn2−/− mice had significantly less retinal neovascularization than npn2+/− or npn2+/+ (ANOVA with Bonferroni correction for multiple comparisons).
Mice Deficient in Npn2 Develop Less VEGF-Induced Subretinal NV Compared with Littermate Controls that Express Npn2
Transgenic mice in which the rhodopsin promoter drives expression of VEGF in photoreceptors (rho/VEGF mice) develop extensive subretinal NV by P21 (29,30). Mice deficient in Npn2 that carried a rho/VEGF transgene developed significantly less subretinal NV at P21 than littermate controls that carried a rho/VEGF transgene and expressed Npn2 (Figure 5).
Figure 5.

Mice deficient in Npn2 develop less VEGF-induced subretinal neovascularization. Littermate npn2−/−, npn2+/−, and npn2+/+ mice that also carried a rhodopsin promoter (rho)/VEGF transgene were perfused with fluorescein-labeled dextran at P21. Retinal flat mounts were examined by fluorescence microscopy and neovascularization in the subretinal space was measured by image analysis. Representative low power fields of retinas from rho/VEGF- npn2−/− mice (A) showed less neovascularization than those from rho/VEGF- npn2+/− mice (B). This is better seen on high power fields of retinas from rho/VEGF- npn2−/− mice (C) or rho/VEGF- npn2+/− mice (D) in which subretinal neovascularization (arrows) is easily distinguished from retinal vessels that are out of focus in the background. Quantification of the area of subretinal neovascularization by image analysis demonstrated significantly less neovascularization in retinas from mice deficient in Npn2 (n = 23) compared with those that expressed some Npn2 (E, npn2+/−and npn2+/+ mice combined, n = 8).
DISCUSSION
Several lines of evidence suggest that VEGF plays a critical role in the development of retinal NV, but the mechanisms of VEGF signaling are complex and poorly understood. Most studies utilizing in vitro testing suggest that VEGF receptor 2 plays a predominant role in VEGF-induced endothelial cell proliferation and NV. What is the role of the 3 other known VEGF binding partners, VEGFR1, Npn1, and Npn2? The Npns enhance signaling through VEGFR2, but do they simply turn up the gain, or do they play an essential role? We hypothesized that loss of both Npns would probably have a suppressive effect, but that loss of one, particularly Npn2, would probably not have a major impact. Contrary to our expectations, we have found that Npn2 plays a very prominent role in the development of retinal NV. This indicates that Npn2 does more than simply turn up the gain on VEGF signaling in the retina.
Mice deficient in Npn1 die as embryos with neuronal, vascular, and cardiac abnormalities (21). The vascular abnormalities are due to disruption of VEGF signaling through Npn1, the neuronal abnormalities are due to disruption of semaphorin signaling, and perturbation of both pathways contributes to the cardiac abnormalities (31). Mice deficient in Npn2 are viable, indicating that Npn1 is more critical for developmental angiogenesis (22). However, while npn2−/− mice have normal arteries, veins, and large lymphatic vessels, they have a reduction of small lymphatic vessels and capillaries (23). Mice deficient in both Npn1 and Npn2 have a more severe phenotype than mice deficient in either Npn1 or Npn2 (32). The double knockouts die at E8.5 with totally avascular yolk sacs, similar to the phenotype seen in mice deficient in VEGF or VEGFR2 (5,6,8). So, while Npn1 may play a more critical role in vascular development, both Npn1 and Npn2 participate in nonoverlapping fashion, and when both are absent, there is complete absence of vascular development.
If the effect of a protein in embryonic vascular development were completely predictive of its role in retinal NV in adults, one would expect a major role for Npn1 and a minor role for Npn2 in the pathogenesis of retinal NV. Three previous studies aimed at investigating the role of Npns in retinal NV have focused on Npn1. Two of the studies demonstrated increased coexpression of Npn1 and VEGFR2 in new vessels in patients with diabetic retinopathy or mice with ischemic retinopathy (33,34), and the 3rd study showed VEGF-induced upregulation of Npn1, but not Npn2, in cultured endothelial cells (35). The 3rd study also demonstrated suppression of retinal NV by 53% by intravitreous injections of an anti-Npn1 antibody in mice with ischemic retinopathy. The authors concluded that Npn1 plays an important role in the pathogenesis of retinal NV and predicted that Npn2 would likely have a minimal role. Our study demonstrates their prediction to be incorrect. Instead, it appears that both Npn1 and Npn2 participate in the development of retinal NV, but their roles must be different, because the presence of intact Npn1 did little to compensate for the absence of Npn2, which resulted in almost complete elimination of NV.
Npns act as coreceptors for VEGF that enhance signaling through VEGF receptors in an isoform-specific manner. Both Npn1 and Npn2 enhance effects of VEGF165 (VEGF164 in mice), but only Npn2 enhances effects of VEGF145 (VEGF144 in mice) (18). Mice genetically engineered to express only VEGF164 have normal development of retinal blood vessels, whereas mice expressing only VEGF120 or VEGF188 do not, suggesting that VEGF164 plays a critical role (36). VEGF164 has also been postulated to play a primary role in retinal NV, and VEGF144 is a relatively rare isoform. So the selective enhancement of the effects of VEGF144 by Npn2 seems unlikely to account for the lack of complementation by Npn1 in the development of retinal NV. Another possible explanation for lack of complementation is distinct expression patterns. In chick embryos, Npn1 expression is localized preferentially in arteries, whereas Npn2 is preferentially expressed in veins (37). Retinal NV originates from post-capillary venules and perhaps Npn2 enhancement of VEGF signaling in this location is needed for retinal NV to occur. Enhancement of VEGF signaling in arterioles may also contribute to retinal NV and provide the basis for the role of Npn1.
This study highlights the complexity of VEGF receptor functioning. While phosphorylation of tyrosine kinase domains is important, protein-protein interactions also have major impacts. Another unsolved mystery is the role of VEGFR1. It has little effect in vitro when expressed in cells that lack VEGFR2 and has been suggested to be a decoy receptor that down-regulates VEGF signaling (38). But this may be an oversimplification, because the intracellular domain of VEGFR1 contains several phosphorylated residues capable of interacting with SH2-domain proteins (39), and just because phosphorylation and downstream events are difficult to detect in vitro does not mean that they do not occur in vivo. VEGFR1 plays an important role in the pathogenesis of ischemia-induced retinal NV, which is significantly reduced by intravitreous injection of an antibody directed against VEGFR1 or by targeted disruption of the placental growth factor gene, which binds VEGFR1, but not VEGFR2 (40,41). It is conceivable that all 4 VEGF receptors play nonoverlapping, critical roles in the development of retinal NV.
The surprising dependence of retinal NV on intact Npn2 may have important clinical implications. As noted in the introduction, preliminary results from clinical trials suggest that antagonism of VEGF is beneficial in patients with choroidal NV due to age-related macular degeneration, but further improvement is needed. It is difficult for any treatment to be 100% efficient and therefore, it is likely that aptamers or antibodies that bind VEGF reduce, but do not eliminate, excess VEGF. Antagonism of VEGF receptors, including Npn2, may provide a means to further reduce the signaling capacity of the reduced levels of intraocular VEGF and thereby enhance effectiveness. Also, added benefits may be realized because other VEGF ligands, such as placental growth factor, would also be blocked. Additional studies are needed to explore strategies targeting Npns and/or VEGF tyrosine kinase receptors in animal models of ocular NV.
Acknowledgments
The authors thank David Ginty, PhD, and Alex Kolodkin, PhD, for generously providing npn−/− mice. This work was supported by EY05951, EY12609, and core grant P30EY1765 from the National Eye Institute; Research to Prevent Blindness Inc (a Lew R Wasserman Merit Award [PAC]); and Dr and Mrs William Lake. PAC is the George S and Dolores Dore Eccles Professor of Ophthalmology.
REFERENCES
- 1.Campochiaro PA. Retinal and choroidal neovascularization. J. Cell. Physiol. 2000;184:301–10. doi: 10.1002/1097-4652(200009)184:3<301::AID-JCP3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 2.The EyeTech Study Group. . Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina. 2002;22:143–52. doi: 10.1097/00006982-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz SD, et al. Safety of rhuFab V2, an anti-VEGF antibody fragment, as a single intravitreal injection in subjects with neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2001;42((suppl)):S522. [Google Scholar]
- 4.Rosenfeld PJ, Villante N, Feuer WJ, Puliafito CA, McCluskey ER. RhuFav V2 (Anti-VEGF antibody fragment) in neovascular AMD: safety, tolerability, and efficacy of multiple, escalatin dose intravitreal injections. Invest. Ophthalmol. Vis. Sci. 2003;44((suppl)):970. [Google Scholar]
- 5.Ferrara N, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–9. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 7.Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–6. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- 8.Shalaby F, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 9.Fong G-H, Rossant J, Gertsenstein M, Breitman ML. Role of the flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 10.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 1998;95:9349–54. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolodkin AL, et al. Neuropilin is a semaphorin III receptor. Cell. 1997;90:753–62. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- 12.He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell. 1997;90:739–51. doi: 10.1016/s0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Chedotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuroplin family, is a high affinity receptor for the semaphorins sema E and sema IV but not sema III. Neuron. 1997;19:547–59. doi: 10.1016/s0896-6273(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 14.Winberg ML, et al. Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell. 1998;95:903–16. doi: 10.1016/s0092-8674(00)81715-8. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi T, et al. Plexin-neuropilin-1 complexes form functional sema-phorin-3A receptors. Cell. 1999;99:59–69. doi: 10.1016/s0092-8674(00)80062-8. [DOI] [PubMed] [Google Scholar]
- 16.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsburn M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–45. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 17.Migdal M, et al. Neuropilin-1 is a placenta growth factor-2 receptor. J. Biol. Chem. 1998;273:22272–8. doi: 10.1074/jbc.273.35.22272. [DOI] [PubMed] [Google Scholar]
- 18.Gluzman-Poltorak Z, Cohen T, Herzog Y, Neufeld G. Neuropilin-2 and neuropilin-1 are receptors for the 165-amino acid form of vascular endothelial growth factor (VEGF) and of placenta growth factor-2, but only neuropilin-2 functions as a receptor for the 145-amino acid form of VEGF. J. Biol. Chem. 2000;275:18040–5. doi: 10.1074/jbc.M909259199. [DOI] [PubMed] [Google Scholar]
- 19.Gluzman-Poltorak Z, Cohen T, Shibuya M, Neufeld G. Vascular endothelial growth factor receptor-1 and neuropilin-2 form complexes. J. Biol. Chem. 2001;276:18688–94. doi: 10.1074/jbc.M006909200. [DOI] [PubMed] [Google Scholar]
- 20.Mamluk R, et al. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J. Biol. Chem. 2002;277:24818–25. doi: 10.1074/jbc.M200730200. [DOI] [PubMed] [Google Scholar]
- 21.Kawasaki T, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- 22.Giger RJ, et al. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000;25:29–41. doi: 10.1016/s0896-6273(00)80869-7. [DOI] [PubMed] [Google Scholar]
- 23.Yuan L, et al. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002;129:4797–806. doi: 10.1242/dev.129.20.4797. [DOI] [PubMed] [Google Scholar]
- 24.Roche J, et al. Distinct 3p21. 3 deletions in lung cancer, analysis of deleted genes and identification of a new human semaphorin. Oncogene. 1996;12:1289–97. [PubMed] [Google Scholar]
- 25.Sekido Y, et al. Human semaphorins A (V) and (IV) reside in the 3p21. 3 small cell lung cancer deletion region and demonstrate distinct expression patterns. . Proc. Natl. Acad. Sci. U.S.A. 1996;93:4120–5. doi: 10.1073/pnas.93.9.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiang R, et al. Isolation of the human sempahorin III/F gene (SEMA3F) at chromosome 3p21, a region deleted in lung cancer. Genomics. 1996;32:39–48. doi: 10.1006/geno.1996.0074. [DOI] [PubMed] [Google Scholar]
- 27.Shih S-C, et al. Molecular profiling of angiogenesis markers. Am. J. Pathol. 2002;161:35–41. doi: 10.1016/S0002-9440(10)64154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith LEH, et al. Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 1994;35:101–11. [PubMed] [Google Scholar]
- 29.Okamoto N, et al. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am. J. Pathol. 1997;151:281–91. [PMC free article] [PubMed] [Google Scholar]
- 30.Tobe T, et al. Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest. Ophthalmol. Vis. Sci. 1998;39:180–8. [PubMed] [Google Scholar]
- 31.Gu C, et al. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Devel. Cell. 2003;5:45–57. doi: 10.1016/s1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takashima S, et al. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3657–62. doi: 10.1073/pnas.022017899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishida S, et al. Coexpression of VEGF receptors VEGF-R2 and neuropilin-1 in proliferative diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2000;41:1649–56. [PubMed] [Google Scholar]
- 34.Ishihama H, et al. Colocalization of neuropilin-1 and Flk-1 in retinal neovascularization in a mouse model of retinopathy. Invest. Ophthalmol. Vis. Sci. 2001;42:1172–8. [PubMed] [Google Scholar]
- 35.Oh H, et al. Selective induction of neuropilin-1 by vascular endothelial growth factor (VEGF): a mechanism contributing to VEGF-induced angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:383–8. doi: 10.1073/pnas.012074399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stalmans I, et al. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 2002;109:327–36. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herzog Y, Kalcheim C, Kahane N, Reshef R, Neufeld G. Differential expression of neuropilin-1 and neuropilin-2 in arteries and veins. Mech. Dev. 2001;109:115–9. doi: 10.1016/s0925-4773(01)00518-4. [DOI] [PubMed] [Google Scholar]
- 38.Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 1994;269:25646–54. [PubMed] [Google Scholar]
- 39.Igarashi K, et al. Tyrosine-1213 of Flt-1 is a major binding site of Nck and SHP-2. Biochem. Biophys. Res. Comm. 1998;246:95–9. doi: 10.1006/bbrc.1998.8578. [DOI] [PubMed] [Google Scholar]
- 40.Luttun A, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat. Med. 2002;8:831–9. doi: 10.1038/nm731. [DOI] [PubMed] [Google Scholar]
- 41.Carmeliet P, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001;7:575–83. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]