

Abstract
Angiotensin II (AII) contributes to the pathogenesis of many cardiovascular disorders. Oxidant-mediated activation of poly(adenosine diphosphate–ribose) polymerase (PARP) plays a role in the development of endothelial dysfunction and the pathogenesis of various cardiovascular diseases. We have investigated whether activation of the nuclear enzyme PARP contributes to the development of AII-induced endothelial dysfunction. AII in cultured endothelial cells induced DNA single-strand breakage and dose-dependently activated PARP, which was inhibited by the AII subtype 1 receptor antagonist, losartan; the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor, apocynin; and the nitric oxide synthase inhibitor, N-nitro-l-arginine methyl ester. Infusion of sub-pressor doses of AII to rats for 7 to 14 d induced the development of endothelial dysfunction ex vivo. The PARP inhibitors PJ34 or INO-1001 prevented the development of the endothelial dysfunction and restored normal endothelial function. Similarly, PARP-deficient mice infused with AII for 7 d were found resistant to the AII-induced development of endothelial dysfunction, as opposed to the wild-type controls. In spontaneously hypertensive rats there was marked PARP activation in the aorta, heart, and kidney. The endothelial dysfunction, the cardiovascular alterations and the activation of PARP were prevented by the angiotensin-converting enzyme inhibitor enalapril. We conclude that AII, via AII receptor subtype 1 activation and reactive oxygen and nitrogen species generation, triggers DNA breakage, which activates PARP in the vascular endothelium, leading to the development of endothelial dysfunction in hypertension.
INTRODUCTION
Poly(adenosine diphosphate [ADP]–ribose) polymerase-1 (PARP-1), a monomeric enzyme present in eukaryotes, is the major isoform of an expanding family of poly(ADP-ribosyl)ating enzymes (1,2). The main isoform of the family, PARP-1, primarily functions as a DNA damage sensor in the nucleus. Upon binding to damaged DNA, mainly through the 2nd zinc finger domain, PARP-1 forms homodimers and catalyzes the cleavage of nicotinamide dinucleotide (oxidized) (NAD+) into nicotinamide and ADP-ribose and uses the latter to synthesize branched nucleic acid–like polymers poly(ADP-ribose) covalently attached to nuclear acceptor proteins. The biological role of PARP-1 is complex and includes the regulation of DNA repair and maintenance of genomic integrity (overviewed in 1,2).
PARP-1 has been implicated in a variety of pathophysiological processes. PARP is an energy-consuming enzyme, which transfers ADP ribose units to nuclear proteins. As a result of this process, the intracellular NAD+ and adenosine 5′-triphosphate (ATP) levels remarkably decrease, resulting in cell dysfunction and cell death via the necrotic route (overviewed in 1,2).
PARP becomes activated in response to DNA single-strand breaks, which can develop as a response to free radical and oxidant cell injury. Oxidative and nitrosative stress triggers the activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP), which contributes to the pathogenesis of various cardiovascular diseases including myocardial infarction and ischemia-reperfusion and heart failure, as well as diabetic endothelial dysfunction (1,2).
Increased oxidative and nitrosative stress have been proposed as a mechanism through which angiotensin II (AII) contributes to the development of cardiovascular disease (3–6). In endothelial cells incubated with AII, increased production of superoxide has been reported. In addition, the increased oxidative stress has been shown to contribute to the development of AII-induced endothelial dysfunction, as it has been shown to be ameliorated by antioxidants (7,8). One of the key factors in AII-induced oxidant generation in endothelial cells is related to up-regulation of NADPH oxidase (9). Endothelial cells from p47(phox–/–) mice produce less superoxide in response to AII (10).
As oxidative stress is a key trigger of the activation of PARP (1,2), the aim of the present study was to investigate the role of PARP activation in the pathogenesis of AII-mediated endothelial dysfunction.
MATERIALS AND METHODS
AII-Induced Activation of PARP In Vitro
Murine microvascular endothelial cells were plated at a density of 250000 cells per well in a 12-well plate and grown in Ham’s F12 media supplemented with 10% fetal calf serum and 0.03 mg/mL endothelial cell growth supplement. After 24 h, the media was replaced and the cells were treated with increasing concentrations of AII (30, 100, or 300 μM) for 24 h. These concentrations of AII have previously been shown to induce oxidative stress in endothelial cells in vitro (11,12). In subsequent experiments mouse endothelial cells were pretreated for 30 min with increasing concentrations of the PARP inhibitors PJ-34 (13,14) or INO-1001 (15,16) by the angiotensin II subtype 1 receptor antagonist, losartan; the NADPH oxidase inhibitor, apocynin; the nitric oxide synthase (NOS) inhibitor, N-nitro-l-arginine methyl ester (l-NAME); or by the xanthine oxidase inhibitor, allopurinol, followed by the administration of 300 μM AII.
DNA single-strand breakage was measured by terminal deoxynucleotidyltransferase–mediated deoxy-uridine-triphosphate (dUTP)-biotin nick end labeling using a commercially available kit (ApopTag Kit, Oncor Inc, Gaithersburg, MD, USA). For the measurement of cellular PARP activity (13,14), the media was removed and replaced with 0.5 mL N-2-hydroxyethylpiperazine-N′-2′-ethanesulfonic acid (pH 7.5) containing 0.01% digitonin and 3H-NAD (0.5 μCi mL−1), and the cells were incubated for 20 min at 37 °C. The cells were then scraped from the wells and placed in Eppendorf tubes containing 200 μL ice-cold 50% trichloroacetic acid (TCA) (w/v). The tubes were then placed at 4 °C. After 4 h, the tubes were centrifuged at 1800g for 10 min and the supernatant removed. The pellet was washed twice with 500 μL ice-cold 5% TCA. The pellet was solubilized in 250 μL NaOH (0.1 M) containing 2% sodium dodecyl sulfate overnight at 37 °C. The PARP activity was then determined by measuring the radioactivity incorporated using a Wallac scintillation counter (Wallac, Turku, Finland). The solubilized protein (250 μL) was mixed with 5 mL scintillant (ScintiSafe Plus, Fisher, Pittsburgh, PA, USA) before being counted for 10 min. Results are expressed as a percentage of the PARP activity observed in untreated cells. Cell viability was measured by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide assay (13,14) and was unaffected by the pharmacological inhibitors used.
Role of PARP Activation in the Endothelial Dysfunction Developing in Response to Sub-pressor Doses of AII in Rats and Mice
All in vivo investigations in this article conform to the Guide for the Care and Use of Laboratory Animals published by US National Institutes of Health (NIH Publication No. 85–23, revised 1985) and were performed with the approval of the local Institutional Animal Care and Use Committee.
ALZET osmotic mini-pumps (DURECT, Cupertino, CA, USA) were used to provide continuous AII administration for 1 or 2 wk in rats (male Sprague-Dawley, 325 to 350 g). Subcutaneous implantation was conducted during pentobarbital anesthesia using a sterile surgical technique. Human AII was infused at a dose of 200 ng/kg/min. This dose of AII has previously been demonstrated to exert no pressor effects in the animals and has been shown to trigger the generation of reactive nitrogen species in the vascular endothelium (11).
Fourteen rats receiving AII were treated from day 0 (n = 8) or day 7 (n = 6) with the PARP inhibitor PJ-34 orally (30 mg/kg/d in drinking water) for 1 wk and used for vascular measurement on day 7 or 14. An additional 20 rats receiving AII were also treated with vehicle (n = 8) or with the PARP inhibitor INO-1001 from day 0 (n = 8) or day 7 (n = 4) following AII pump implantation. INO-1001 (n = 12) or vehicle (n = 8) was delivered by ALZET osmotic minipumps implanted subcutaneously (5 mg/kg/d). Previous studies have demonstrated that the selected PARP inhibitor treatment regimens are effective in blocking vascular and cardiac PARP activation (13–17). At the end of observation, mean blood pressure was measured as described previously (18). Animals were anesthetized with thiopentone sodium (60 mg/kg, intraperitoneally) and were placed on controlled heating pads. Core temperature, measured via a rectal probe, was maintained at 37 °C. A microtip catheter transducer (SPR-524; Millar Instruments, Houston, TX, USA) was inserted into the right carotid artery, and pressure signal was recorded for 10 min using a MacLab A/D converter (AD Instruments, Mountain View, CA, USA), and stored and displayed on an Apple Macintosh computer (Cupertino, CA, USA). Animals were subsequently sacrificed by exsanguinations, and thoracic aortas were rapidly removed, cleared from periadventitial fat, cut into 3- to 4-mm wide rings using operation microscope, and mounted in organ baths filled with warmed (37 °C), oxygenated (95% O2, 5% CO2) Krebs’ solution (CaCl2 1.6 mM; MgSO4 1.17 mM; EDTA 0.026 mM; NaCl 130 mM; NaHCO3 14.9 mM; KCl 4.7 mM; KH2PO4 1.18 mM; glucose 11 mM). Isometric tension was measured as described (12,13,17) with isometric transducers (Kent Scientific Corporation, Litchfield, CT, USA), digitized using a MacLab A/D converter and stored and displayed on a Macintosh computer. A tension of 1.5 g was applied and the rings were equilibrated for 60 min. This was followed by precontraction of rings with epinephrine (10−6 M) and relaxation to acetylcholine (10−9 to 3 × 10−4 M).
Sections of thoracic aortas were fixed in 10% formalin, and then sliced and embedded in paraffin for immunohistochemical detection of poly(ADP-ribose) formation, an immunohistochemical indicator of the activation of PARP. Immunohistochemical detection of poly(ADP-ribose) was performed in paraffin sections (3 μm) after deparaffinization in xylene and rehydrated in decreasing concentrations (100%, 95%, and 75%) of ethanol followed by a 10-min incubation in phosphate-buffered saline (PBS) (pH 7.4). Sections were treated with 0.3% hydrogen peroxide for 15 min to block endogenous peroxidase activity and then rinsed briefly in PBS. Non–specific binding was blocked by incubating the slides for 1 h in 0.25% Triton/PBS containing 2% horse serum. Poly(ADP-ribose) was detected using a chicken monoclonal anti-poly(ADP-ribose) antibody (Tulip Biolabs, West Point, PA, USA) and isotype-matched control antibody, and applied in a dilution of 1:600 for 1 h at room temperature. After extensive washing (3 × 10 min) with 0.25% Triton/PBS, immunoreactivity was detected with a biotinylated horse antichicken secondary antibody and an avidin-biotin-peroxidase complex, both supplied in the Vector Elite kit (Vector Laboratories, Burlingame, CA, USA). Color was developed using Ni-DAB substrate-kit (Vector Laboratories). Sections were then counterstained with nuclear fast red, dehydrated, and mounted in Permount. Photomicrographs were taken with a Zeiss Axiolab microscope (Carl Zeiss, Göttingen Germany) equipped with a Fuji HC-300C digital camera (Fuji Electriclo Co, Tokyo, Japan). All immunohistochemical samples were coded and examined and graded by an investigator in a blinded fashion.
The role of PARP in the development of AII-induced endothelial dysfunction was also investigated in wild-type and PARP-deficient mice. AII (at the same dose as in the rat study) was infused for 1 wk in 6 wild-type and 6 PARP-1–deficient mice, followed by the evaluation of the endothelium-dependent relaxant responses ex vivo.
Role of AII and PARP in the Development of Endothelial Dysfunction in Spontaneously Hypertensive Rats
Male spontaneous hypertensive rats (SHR) (8 wk old) were purchased from Charles River Laboratories (Wilmington, MA, USA) and were treated with vehicle (n = 10), or the angiotensin-converting enzyme inhibitor enalapril for 8 wk (20 mg/kg/d, n = 10) administered in the drinking water. Food and water were available ad libitum. Matched-weight Wistar-Kyoto (WKY) were purchased from the same supplier and were used as baseline controls. After 8 wk, hemodynamic analysis was conducted in rats anesthetized with intraperitoneal injection of pentobarbital (60 mg/kg). Animals were placed on controlled heating pads and core temperature was measured via a rectal probe and maintained at 36 to 38 °C. A microtip catheter transducer (SPR-524; Millar Instruments) was inserted into the right carotid artery and advanced into the left ventricle under pressure control. After stabilization, the pressure signal was continuously recorded using a MacLab A/D converter (AD Instruments, Mountain View, CA, USA) and stored and displayed on an Apple Macintosh computer. The heart rate, the left ventricular systolic and end-diastolic pressures were measured. The maximal slope of systolic pressure increment (+dP/dt), an index of contractility, was calculated. After these measurements, the catheter was pulled back into the aorta for the measurement of arterial blood pressure. Animals were subsequently sacrificed by exsanguination, and measurements of vascular function were conducted as described in the previous section “Role of PARP Activation in the Endothelial Dysfunction Developing in Response to Sub-pressor Doses of AII in Rats and Mice.” Hearts, kidneys and thoracic aortas were obtained for immunohistochemical analysis of PARP activation, as described in “Role of PARP Activation in the Endothelial Dysfunction Developing in Response to Sub-pressor Doses of AII in Rats and Mice.”
Statistical Analysis
Results are reported as mean ±SEM. Statistical significance between 2 measurements was determined by the two-tailed unpaired Student t test, and among groups it was determined by analysis of variance with Bonferroni’s correction. Probability values of P < 0.05 were considered significant.
Reagents
Reagents were obtained from Sigma/Aldrich (St. Louis, MO, USA), unless indicated otherwise in the Materials and Methods section. The phenanthridinone PARP inhibitor PJ34 and the isoindolinone PARP inhibitor INO-1001 were synthesized as previously described (11–14).
RESULTS
AII Induces PARP Activation in Murine Endothelial Cells In Vitro
Exposure of endothelial cells to increasing concentrations of AII dose-dependently increased PARP activity (Figure 1). Treatment of endothelial cells with 300 μM AII increased PARP activation approximately 3-fold over baseline levels. PARP activity was inhibited dose-dependently by PJ-34 and INO-1001 with IC50 values of approximately 30 nM and 3 nM, respectively. The AII-induced activation of PARP was dose-dependently inhibited by the AII subtype 1 receptor antagonist, losartan; the NADPH oxidase inhibitor, apocynin; and the NOS inhibitor, l-NAME; but was unaffected by the xanthine oxidase inhibitor allopurinol (Figure 2). As DNA single-strand breakage is the obligatory trigger of PARP activation, we have investigated whether AII induces DNA strand breaks in the endothelial cells and whether the pharmacological agents used affect it. We found that AII induced a significant degree of DNA strand breakage, and this was blocked by losartan, apocynin, l-NAME, but not by allopurinol (Figure 3).
Figure 1.

AII induces PARP activation in mouse endothelial cells in vitro. Exposure of endothelial cells to increasing concentrations of AII dose dependently increased PARP activity. Results shown represent mean ±SEM from 3 separate experiments; **P < 0.01 compared with untreated cells.
Figure 2.

Inhibition of AII receptor activation (losartan), NADPH oxidase (apocynin), or NOS (NG-nitro-l-arginine) dose-dependently reduced angiotensin-mediated activation of PARP in endothelial cells. Inhibition of xanthine oxidase by allopurinol had no effect on AII-mediated activation of PARP. Results are expressed as mean ±SEM of the % inhibition of PARP activation following 24-h treatment of mouse endothelial cells with AII (300 μM) with and without various drug concentrations. Statistical significance was determined from 3 separate experiments using Student t test where P < 0.05 was considered significant; *P < 0.05 compared with angiotensin treatment alone.
Figure 3.
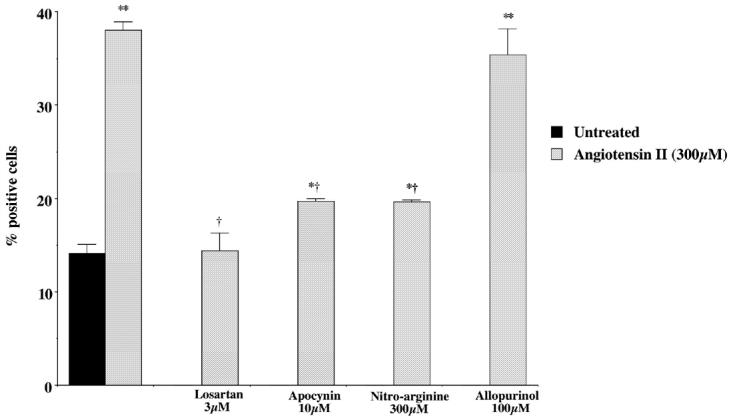
AII increases DNA strand breakage in endothelial cells. Treatment of mouse endothelial cells with AII (300 μM) for 24 h increased the number of cells exhibiting open DNA ends from 14% to 38%. Treatment with the AII receptor antagonist losartan, the NADPH oxidase inhibitor apocynin, the NOS inhibitor NG-nitro-l-arginine, but not the xanthine oxidase inhibitor allopurinol, dose-dependently reduced DNA strand breakage following angiotensin treatment. Results are expressed as mean ±SEM of the % of cells from 3 separate experiments with a total of at least 800 nuclei counted for each treatment. Statistical significance was determined using Student t test where P < 0.05 was considered significant; *P < 0.05, **P < 0.01 compared with untreated cells and †P < 0.05 compared with AII treatment alone.
AII Infusion Triggers PARP Activation and Induces the Development of Endothelial Dysfunction In Vivo
Infusion of AII to Sprague-Dawley rats (at a sub-pressor dose of 200 ng/kg/min) for 7 d induced the development of endothelial dysfunction, as assessed by the endothelium-dependent relaxant responses to acetylcholine in the thoracic aorta ex vivo (Figure 4). This endothelial dysfunction was sustained when AII was infused at the same rate for an additional 1-wk period (see Figure 4). The PARP inhibitors PJ34 (see Figure 4) or INO-1001 (not shown) at doses of 20 mg/kg/d, starting from day 1, prevented the development of the endothelial dysfunction. Administration of the PARP inhibitors for the 2nd wk only, restored normal endothelial function. Infusion of AII failed to induce a pressor response and there were no differences in mean arterial blood pressure values between any of the experimental groups (not shown). Immunohistochemical detection of poly(ADP-ribose) confirmed that the PARP inhibitors block endothelial PARP activation in the AII-treated animals (Figure 5). The role of PARP in the development of endothelial dysfunction was also confirmed using wild-type and PARP-1–deficient mice. Whereas 1 wk of AII infusion induced a significant loss of endothelium-dependent relaxations in wild-type mice, there was no detectable loss of endothelium-dependent relaxations in the PARP-1–deficient mice (Figure 6).
Figure 4.

Pharmacological inhibition of PARP with PJ34 prevents (A) or improves (B) the AII-induced endothelial dysfunction. Acetylcholine-induced endothelium-dependent relaxations are shown. Each point of the curve represents mean ±SEM of 6 to 8 experiments in vascular rings. *P < 0.05 compared with control (CO) and †P < 0.05 compared with AII. Animals receiving AII were treated with PJ34 (30 mg/kg/d) from day 0 (A: n = 8) or d 7 (B: n = 6) following AII-releasing minipump implantation for 1 wk.
Figure 5.

Evidence of poly(ADP-ribose) formation in the rat aorta after AII infusion. Immunohistochemical staining for poly(ADP-ribose) formation, an indicator of PARP activation, in AII-infused (AII) and INO1001-treated AII-infused rats (AII + INO1001). The evidence of poly(ADP-ribose) accumulation can be seen as dark nuclear staining, indicated by arrows in the endothelial cells. Treatment with INO1001 (right panel) markedly reduced AII-induced PARP activation in the endothelial cells. Similar immunohistochemical profiles were seen in 4 to 5 aortas per group. There was no detectable PARP activation in the control vessels (not shown).
Figure 6.

The absence of PARP-1 prevents the AII-induced endothelial dysfunction. Acetylcholine-induced endothelium-dependent relaxations are shown. Each point of the curve represents mean ±SEM of 6 experiments in vascular rings. *P < 0.05 denotes significant loss of endothelium-dependent relaxations in the AII-treated wild-type mice, when compared with the response in vehicle-treated wild-type mice.
Enalapril Normalizes Endothelial and Cardiovascular Functions and Prevents the Activation of PARP in SHR
There was no significant difference in body weights among the control group (Wistar Kyoto rats), the SHR group, and the enalapril-treated group (not shown). The arterial blood pressure was almost doubled in the SHR group compared with the control group (209 ± 5 mmHg and 109 ± 6 mmHg, respectively). Enalapril reduced the arterial blood pressure to 138 ± 23 mmHg (P < 0.01). The left ventricular systolic pressure was also markedly increased in the SHR group, which was normalized in the presence of enalapril (Figure 7). Similarly, +dP/dt values were higher in the SHR animals and were normalized by enalapril treatment (see Figure 7). There was a marked impairment in the endothelium-dependent relaxant function of the SHR animals, which was normalized with enalapril treatment (Figure 8). Previous studies have demonstrated that pharmacological inhibition of PARP also normalizes the endothelial dysfunction associated with hypertension (19). There was a marked increase in the degree of poly(ADP-ribose) staining (a marker of PARP activation) in the heart, kidney, and aorta of SHR animals, which was abolished in the enalapril-treated SHR animals (Figure 9).
Figure 7.
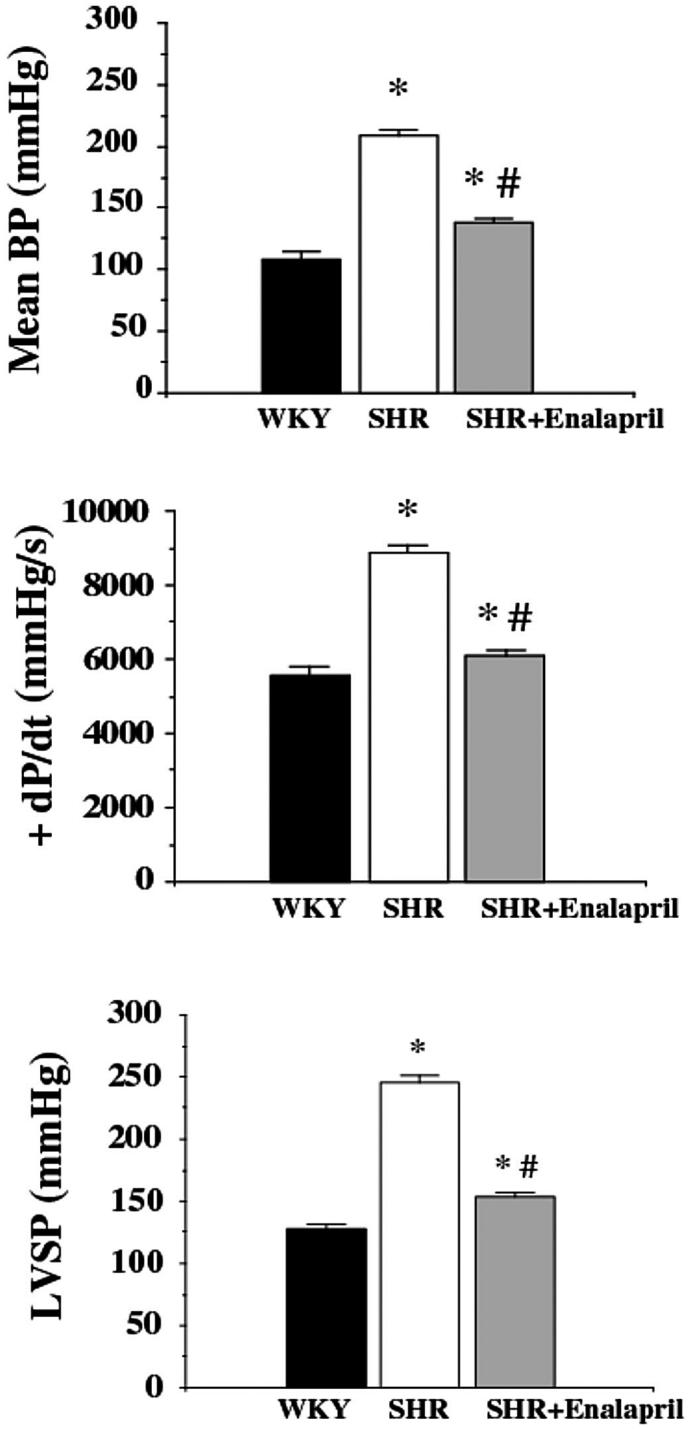
Reversal of hypertension-induced increased systolic function and blood pressure by enalapril treatment in SHR. Effect of hypertension (SHR) and enalapril (SHR + enalapril) on mean blood pressure (mean BP), left ventricular +dP/dt, and left ventricular systolic pressure. Data are means ± SEM. *P < 0.05 compared with controls; #P < 0.05 compared with SHR.
Figure 8.
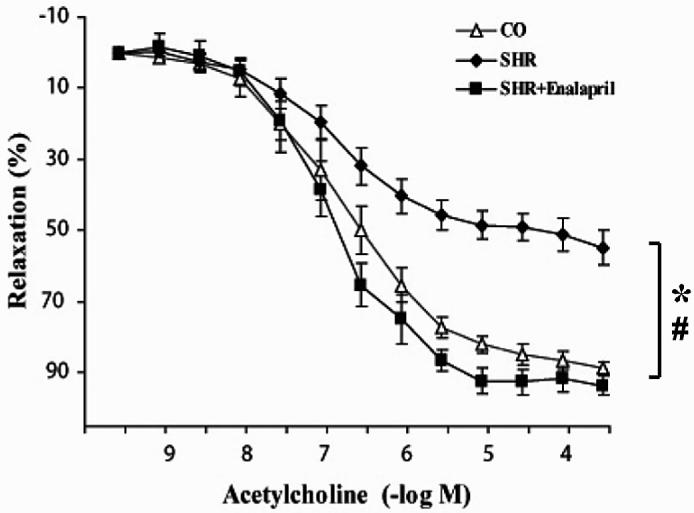
Reversal of hypertension-induced endothelial dysfunction by enalapril treatment in spontaneously hypertensive rats (SHR). Data are means ± SEM *P < 0.05 compared with controls; #P < 0.05 compared with SHR.
Figure 9.

Evidence of poly(ADP-ribose) formation in the heart, aorta, and kidneys of SHR and reversal by enalapril treatment. There was no detectable PARP activation in the control sections. The evidence of poly(ADP-ribose) accumulation can be seen as dark nuclear staining in the myocytes (upper left panel), endothelial cells, and vascular smooth muscle (middle left panel), and tubuli and glomeruli (lower left panel) of SHR. Treatment with enalapril (right panels) markedly reduced PARP activation in the myocytes, endothelial and vascular smooth muscle cells, and also in glomeruli and tubuli. Similar immunohistochemical profiles were seen in 4 to 5 tissue samples per group.
DISCUSSION
Multiple reports indicate the importance of PARP activation in the development of endothelial dysfunction associated with endotoxic shock (20), ischemia-reperfusion injury (13,21), chronic heart failure (17), and diabetes mellitus (14,22). This endothelial dysfunction is not necessarily associated with endothelial cell denudation or death, and it can be reversible. As demonstrated in thoracic aortas from rats with diabetes mellitus, the endothelial dysfunction that develops is rapidly reversible by pharmacological inhibition of PARP and is due to the endothelial depletion of NADPH, an essential co-factor of the endothelial NOS (14,22).
We have recently reported that the endothelial dysfunction associated with hypertension and with aging in rats is also dependent on PARP and can be prevented by pharmacological inhibition of PARP with PJ34 (23). As the production of AII is an essential contributor to the cardiovascular alterations associated with hypertension (and plays a central role in the development of endothelial dysfunction) and is an inducer of oxidative and nitrosative stress in endothelial cells (3–10), here we tested whether AII is capable of inducing PARP activation, and whether PARP activation is inhibited by blockade of AII generation in hypertension. The in vitro studies in endothelial cells demonstrated that AII is capable of inducing a dose-dependent PARP activation. The concentrations of AII required to induce PARP activation are higher than the circulating concentrations of AII, but are in the same concentration range as what has been used in multiple previous studies to induce oxidative stress in endothelial cells in vitro (for example, 11,12,23–27).
Previous work has demonstrated that endothelial cells exposed to AII in vitro respond with the generation of multiple oxygen- and nitrogen-derived reactive species through a variety of mechanisms, including superoxide generation via NADPH oxidase, xanthine oxidase, the mitochondrial electron transport chain, and superoxide and nitric oxide generation from the endothelial NOS (3–10, 23–27). Our in vitro pharmacological studies indicate that AII subtype 1 activation, NADPH oxidase–derived (but not xanthine oxidase–derived) superoxide generation, and nitric oxide production (or a nitric oxide–derived reactive species such as peroxynitrite) play roles in the AII-induced DNA single-strand breakage, and the subsequent activation of PARP. Consistently with the current findings, recent in vitro studies demonstrated that the combination of NO and superoxide formation leads to the generation of peroxynitrite in AII-exposed endothelial cells (11,12).
Our subsequent in vivo experiments demonstrated that a low continuous infusion of AII results in the development of endothelial dysfunction. These findings are consistent with recent studies by Bauer and his group (11), who demonstrated that the same regimen of AII infusion induces the formation of nitrotyrosine in the vasculature. The finding that PARP inhibitors (2 different PARP inhibitors of 2 different structural classes) and genetic PARP-1 deficiency normalize the AII-induced endothelial dysfunction in vivo is consistent with the hypothesis that reactive oxygen and nitrogen species are produced in the vicinity of the endothelium in AII-treated animals, which induce DNA strand breakage, PARP activation, and reversible endothelial dysfunction.
Our findings in SHR animals are also consistent with this hypothesis, as we report here that inhibition of angiotensin-converting enzyme with enalapril blocked the activation of PARP in spontaneously hypertensive rats. This observation indicates that not only endogenously infused AII, but also endogenously produced AII is able to activate PARP in the vasculature. The dose of enalapril used in the current study has been shown to provide a full blockade of angiotensin-converting enzyme (ACE) and has been demonstrated to normalize blood pressure and cardiovascular function (28–30). The question may arise as to whether PARP activation in SHR animals is related to AII directly activating oxidative and nitrosative processes in the endothelial cells, or alternatively, to the physical phenomenon of hypertension, which could, in theory, enhance endothelial oxidant and free radical production via increased shear stress (31–33). The finding that AII can activate PARP in endothelial cells in vitro and in vivo—at sub-pressor doses—tends to support a direct mechanism.
PARP is also involved in the regulation of cytokine and chemokine production, in part via activation of nuclear factor kappa B (9,10). It is known that AII can up-regulate inflammatory signal transduction pathways in cardiovascular disease, in part via activation of nuclear factor kappa B (1–5,34,35). Future work is required to determine whether AII-mediated up-regulation of pro-inflammatory signal transduction pathways may also involve the PARP pathway.
In conclusion, the current results demonstrate the first time that AII induces PARP activation in vitro and in vivo. In addition, based on the results of studies conducted with pharmacological inhibitors, we conclude that AII-induced PARP activation is related to reactive species generation from NOS and NADPH oxidase. Based on the results obtained with 2 structurally different potent PARP inhibitors and with PARP-1–deficient animals, we conclude that AII-induced endothelial dysfunction in vivo is mediated, at least in part, by PARP activation. In addition, the current report is the first direct demonstration of PARP activation in hypertension, and the first evidence that the protective effects of ACE inhibitors in spontaneously hypertensive rats are associated with reduced PARP activation. Based on the current results, we put forward the hypothesis that pathophysiological formation of elevated circulating levels of AII triggers oxidative and nitrosative stress in the endothelial cells, which activates PARP, which subsequently triggers the development of endothelial dysfunction (Figure 10). Note, however, that the role of AII in triggering endothelial dysfunction and cardiovascular alterations goes beyond hypertension. AII-mediated oxidative stress and related pathophysiological processes have been implicated in the development of diabetic complications, chronic heart failure, and atherosclerosis (diseases all associated with the activation of PARP). It is conceivable that AII may act as an endogenous trigger of PARP activation in these conditions.
Figure 10.
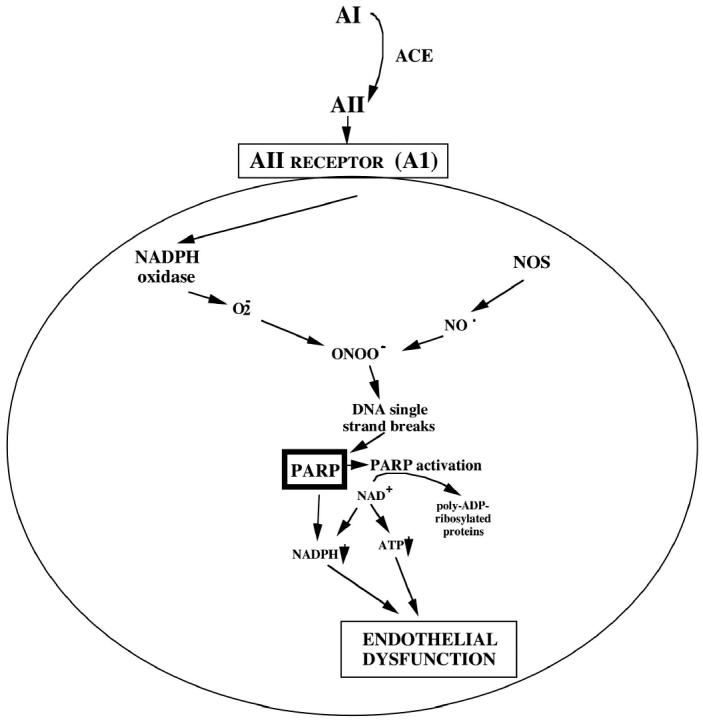
Role of PARP activation in the AII-induced endothelial dysfunction in vivo. ACE produces AII from its precursor, angiotensin I (AI). AII, via its AI AII receptor on the surface of the endothelial cell, activates endothelial cell NADPH oxidase, which produces superoxide (O2−). Superoxide reacts with nitric oxide, produced from the constitutive endothelial NOS, to form peroxynitrite (ONOO). DNA single-strand breaks initiated by ONOO trigger the activation of poly(ADP-ribose) polymerase-1 (PARP), and the resulting intracellular futile metabolic cycle leads to the development of a reversible endothelial dysfunction.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 HL59266) to CS and by a grant from the Hungarian Research Fund (OTKA T047095). Dr Pacher is on leave from the Institute of Pharmacology and Pharmacotherapy, Semmelweis University, Budapest, Hungary. CS and PP contributed equally to this work. The expert technical assistance of Ms L Cannastra is appreciated.
REFERENCES
- 1.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribos) polymerase inhibitors. Pharmacol. Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 2.Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell. Mol. Life Sci. 2002;59:1534–53. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drexler H, Hornig B. Endothelial dysfunction in human disease. J. Mol. Cell. Cardiol. 1999;31:51–60. doi: 10.1006/jmcc.1998.0843. [DOI] [PubMed] [Google Scholar]
- 4.Wolf G. Free radical production and angiotensin. Curr. Hypertens. Rep. 2000;2:167–73. doi: 10.1007/s11906-000-0078-z. [DOI] [PubMed] [Google Scholar]
- 5.Singh BM, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in the therapy of hypertension and coronary heart disease. Arch. Intern. Med. 2003;163:1296–304. doi: 10.1001/archinte.163.11.1296. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert RE, Krum H, Wilkinson-Berka J, Kelly DJ. The renin-angiotensin system and the long-term complications of diabetes: pathophysiological and therapeutic considerations. Diabet. Med. 2003;20:607–21. doi: 10.1046/j.1464-5491.2003.00979.x. [DOI] [PubMed] [Google Scholar]
- 7.Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–93. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, Chen Y, Chabrashvili T, Aslam S, Borrego Conde LJ, Umans JG, Wilcox CS. Role of oxidative stress in endothelial dysfunction and enhanced responses to angiotensin II of afferent arterioles from rabbits infused with angiotensin II. J. Am. Soc. Nephrol. 2003;14:2783–9. doi: 10.1097/01.asn.0000090747.59919.d2. [DOI] [PubMed] [Google Scholar]
- 9.Rueckschloss U, Duerrschmidt N, Morawietz H. NADPH oxidase in endothelial cells: impact on atherosclerosis. Antioxid. Redox. Signal. 2003;5:171–80. doi: 10.1089/152308603764816532. [DOI] [PubMed] [Google Scholar]
- 10.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–5. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wattanapitayakul SK, Weinstein DM, Holycross BJ, Bauer JA. Endothelial dysfunction and peroxynitrite formation are early events in angiotensin-induced cardiovascular disorders. FASEB J. 2000;14:271–8. doi: 10.1096/fasebj.14.2.271. [DOI] [PubMed] [Google Scholar]
- 12.Mihm MJ, Wattanapitayakul SK, Piao SF, Hoyt DG, Bauer JA. Effects of angiotensin II on vascular endothelial cells: formation of receptor-mediated reactive nitrogen species. Biochem. Pharmacol. 2003;65:1189–97. doi: 10.1016/s0006-2952(03)00012-1. [DOI] [PubMed] [Google Scholar]
- 13.Jagtap P, et al. Novel phenanthridinone inhibitors of poly (adenosine 5′-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit. Care Med. 2002;30:1071–82. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 14.Garcia Soriano F, et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001;7:108–13. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 15.Shimoda K, et al. Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;285:240–9. doi: 10.1152/ajplung.00319.2002. [DOI] [PubMed] [Google Scholar]
- 16.Khan TA, et al. Poly(ADP-ribose) polymerase inhibition improves postischemic myocardial function after cardioplegia-cardiopulmonary bypass. J. Am. Coll. Surg. 2003;197:270–7. doi: 10.1016/S1072-7515(03)00538-6. [DOI] [PubMed] [Google Scholar]
- 17.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J. Am. Coll. Cardiol. 2002;40:1006–16. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 18.Pacher P, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 19.Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int. J. Mol. Med. 2002;9:659–64. [PubMed] [Google Scholar]
- 20.Szabo C, Cuzzocrea S, Zingarelli B, O’Connor M, Salzman AL. Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly(ADP-ribose) synthetase by peroxynitrite. J. Clin. Invest. 1997;100:723–35. doi: 10.1172/JCI119585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuzzocrea S, et al. Beneficial effects of 3-aminobenzamide, an inhibitor of poly(ADP-ribose) synthetase in a rat model of splanchnic artery occlusion and reperfusion. Br. J. Pharmacol. 1997;121:1065–74. doi: 10.1038/sj.bjp.0701234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soriano FG, Pacher P, Mabley J, Liaudet L, Szabo C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ. Res. 2001;89:684–91. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, et al. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc. Res. 1999;44:215–22. doi: 10.1016/s0008-6363(99)00183-2. [DOI] [PubMed] [Google Scholar]
- 24.Pueyo ME, et al. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2000;20:645–51. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 25.Mervaala EM, et al. Endothelial dysfunction and xanthine oxidoreductase activity in rats with human renin and angiotensinogen genes. Hypertension. 2001;37:414–8. doi: 10.1161/01.hyp.37.2.414. [DOI] [PubMed] [Google Scholar]
- 26.Cai H, et al. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J. Biol. Chem. 2002;277:48311–7. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- 27.Mollnau H, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 2002;90:58–65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- 28.Goto K, Fujii K, Onaka U, Abe I, Fujishima M. Renin-angiotensin system blockade improves endothelial dysfunction in hypertension. Hypertension. 2000;36:575–80. doi: 10.1161/01.hyp.36.4.575. [DOI] [PubMed] [Google Scholar]
- 29.Raasch W, et al. Regression of ventricular and vascular hypertrophy: are there differences between structurally different ACE inhibitors? J. Hypertens. 2002;20:2495–504. doi: 10.1097/01.hjh.0000042885.24999.e9. [DOI] [PubMed] [Google Scholar]
- 30.Ji Y, Huang Y, Han Y, Xu Y, Ferro A. Cardiac effects of amiloride and of enalapril in the spontaneously hypertensive rat. J Hypertens. 2003;21:1583–9. doi: 10.1097/00004872-200308000-00024. [DOI] [PubMed] [Google Scholar]
- 31.Go YM, et al. Evidence for peroxynitrite as a signaling molecule in flow-dependent activation of c-Jun NH(2)-terminal kinase. Am. J. Physiol. 1999;277:1647–53. doi: 10.1152/ajpheart.1999.277.4.H1647. [DOI] [PubMed] [Google Scholar]
- 32.Silacci P, Desgeorges A, Mazzolai L, Chambaz C, Hayoz D. Flow pulsatility is a critical determinant of oxidative stress in endothelial cells. Hypertension. 2001;38:1162–6. doi: 10.1161/hy1101.095993. [DOI] [PubMed] [Google Scholar]
- 33.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;284:1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 34.Tham DM, et al. Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol. Genomics. 2002;11:21–30. doi: 10.1152/physiolgenomics.00062.2002. [DOI] [PubMed] [Google Scholar]
- 35.Costanzo A, et al. Endothelial activation by angiotensin II through NFkappaB and p38 pathways: Involvement of NFkappaB-inducible kinase (NIK), free oxygen radicals, and selective inhibition by aspirin. J. Cell. Physiol. 2003;195:402–10. doi: 10.1002/jcp.10191. [DOI] [PubMed] [Google Scholar]