

Abstract
Epigallocatechin-3-gallate (EGCG) is the most prominent catechin in green tea. EGCG has been shown to modulate numerous molecular targets in the setting of inflammation and cancer. These molecular targets have also been demonstrated to be important participants in reperfusion injury, hence this study examines the effects of EGCG in myocardial reperfusion injury. Male Wistar rats were subjected to myocardial ischemia (30 min) and reperfusion (up to 2 h). Rats were treated with EGCG (10 mg/kg intravenously) or with vehicle at the end of the ischemia period followed by a continuous infusion (EGCG 10 mg/kg/h) during the reperfusion period. In vehicle-treated rats, extensive myocardial injury was associated with tissue neutrophil infiltration as evaluated by myeloperoxidase activity, and elevated levels of plasma creatine phosphokinase. Vehicle-treated rats also demonstrated increased plasma levels of interleukin-6. These events were associated with cytosol degradation of inhibitor κB-α, activation of IκB kinase, phosphorylation of c-Jun, and subsequent activation of nuclear factor-κB and activator protein-1 in the infarcted heart. In vivo treatment with EGCG reduced myocardial damage and myeloperoxidase activity. Plasma IL-6 and creatine phosphokinase levels were decreased after EGCG administration. This beneficial effect of EGCG was associated with reduction of nuclear factor-κB and activator protein-1 DNA binding. The results of this study suggest that EGCG is beneficial for the treatment of reperfusion-induced myocardial damage by inhibition of the NF-κB and AP-1 pathway.
INTRODUCTION
Reperfusion injury following an acute coronary occlusion event is influenced by a host of complex interactive factors that determine the final outcome of the injury. It has been demonstrated that myocardial damage induced by ischemia-reperfusion is caused by reactive oxygen species (1–3). Recent experimental reports have suggested that the dysfunction is triggered by the endothelial and myocyte generation of a large burst of oxidant molecules and amplified by accumulation of neutrophils into injured tissue (4–6).
These proinflammatory stimuli activate a number of protein kinases, which have the capacity to modulate nuclear factor-κB (NF-κB) or activator protein-1 (AP-1) activity. NF-κB is usually present in the cytoplasm of the cell in an inactive state bound to a related inhibitory protein known as inhibitor κB-α (IκB-α). A common pathway for the activation of NF-κB occurs when its inhibitor protein IκB-α is phosphorylated by IκB kinase (IKK), specifically at the serine-32 and -36 residues of IκB-α (7–10). Phosphorylated IκB-α is targeted for rapid ubiquitination, and then degraded by the 26S proteasome. Degradation of IκB-α unmasks the nuclear translocation sequence of NF-κB, allowing NF-κB to enter the nucleus and direct transcription of target genes (10). AP-1 is a collective term referring to dimeric transcription factors composed of Jun, Fos, or ATF (activating transcription factor) subunits that bind to the AP-1 binding site on the several proinflammatory genes including the IL-6 promoter (11).
Epidemiological evidence suggests that tea consumption may have a strong effect on cardiovascular disease (12,13), but there is scanty information available about the molecular mechanisms involved in the cardioprotection. Epigallocatechin-3-gallate (EGCG) is a prominent catechin present in green tea. Several experimental studies have reported beneficial effects of EGCG in inflammation and cancer (14–16). The molecular mechanisms of EGCG appear to include pleiotropic effects. It has been demonstrated that ingestion of green and black tea significantly increased human plasma antioxidant capacity in vivo (17). EGCG has been demonstrated to dramatically inhibit chemokine-induced neutrophil chemotaxis in vitro (18). EGCG possesses antithrombotic activities primarily due its antiplatelet activities (19). Tea polyphenols have also been noted to induce apoptosis and cell cycle arrest in a wide array of cell lines (20–22). In vitro studies demonstrated that cellular targets of EGCG may account for its anti-inflammatory properties including nitric oxide (23), protein kinase C (20,24), activation of extracellular mitogen-activated protein kinases (25), and STAT-1 (26).
Previously our laboratory has demonstrated that EGCG is a potent inhibitor of IL-8 gene expression in human respiratory epithelial cells. The proximal mechanism of this effect involves, in part, inhibition of IKK (27). The purpose of this study was to investigate the effect of EGCG in a rat model of myocardial ischemia and reperfusion injury and to determine its mechanism of action in vivo. An understanding of potential molecular mechanisms targeted by EGCG is essential to design better therapeutic strategies for cardioprotection in reperfusion injury.
MATERIALS AND METHODS
Myocardial Ischemia and Reperfusion
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by U.S. National Institutes of Health (NIH Publication No. 85-23 revised 1996) and with the approval of the Institutional Animal Care and Use Committee. Male Wistar rats (Charles River) were anesthetized with thiopentone sodium (70 mg/kg intraperitoneally). The right common carotid artery was cannulated to measure mean arterial blood pressure and heart rate by a pressure transducer connected to a Maclab A/D converter (AD Instruments, Milford, MA, USA). The right jugular vein was also cannulated. The trachea was cannulated and artificial respiration was provided by a respirator with a FiO2 of 0.80 at a frequency of 60 strokes/min and a tidal volume of 1 mL/100 g, to maintain normal PaO2, PaCO2, and pH. Myocardial ischemia and reperfusion injury was performed as previously described (28). After opening the chest at the 4th intercostal space, the left main coronary artery was occluded for 30 min by a 4.0 silk suture over a 3-mm air balloon, which was placed on top of the vessel. The occlusion was released after ischemia and reperfusion was allowed for 120 min. Rectal temperature was monitored with a rectal probe and was maintained within 36.5 °C and 37 °C. In a group of rats, vehicle (PBS 1 mL/kg) or EGCG (10 mg/kg) were administered intravenously at reperfusion. These rats then also received 10 mg/kg/h of EGCG during the reperfusion period. In another set of experiments, different groups of rats were sacrificed before the occlusion of the coronary artery (time 0), at the end of 30 min ischemia, and at various time points after reperfusion (30, 60, and 120 min). Hearts were rapidly harvested. The atria, right ventricles, and major vessels were removed from the hearts, and the left ventricles were used for histological and biochemical studies. A group of rats underwent the above surgical procedure with the exception of artery occlusion and reperfusion and served as sham control group. Saline solution (2 to 3 mL) was administered intraperitoneally to all groups to replace the loss of fluid that occurred during the surgical procedure.
Histopathological Analysis and Damage Score
Tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Sections were stained with hematoxylin and eosin for histological evaluation of tissue damage. To have a quantitative estimation of the histological cardiac damage, sections (n = 6 to 8 for each experimental group) were scored by 2 independent observers blinded to the experimental protocol. The following morphological criteria were considered: score 0, no damage; score 1 (mild), interstitial edema and focal necrosis; score 2 (moderate), diffuse myocardial cell swelling and necrosis; score 3 (severe), necrosis with presence of contraction bands and neutrophil infiltrate; score 4 (highly severe), widespread necrosis with presence of contraction bands, neutrophil infiltrate, and hemorrhage.
Myeloperoxidase Activity
Myeloperoxidase activity was determined as an index of neutrophil accumulation, as previously described (28). Cardiac tissues, collected 120 min after reperfusion, were homogenized in a solution containing 0.5% hexa-decyl-trimethylammonium bromide dissolved in 10 mM potassium phosphate buffer (pH 7) and centrifuged for 30 min at 20000g at 4 °C. An aliquot of the supernatant was allowed to react with a solution of tetra-methyl-benzidine (1.6 mM) and 0.1 mM H2O2. The rate of change in absorbance was measured by spectrophotometry at 650 nm. Myeloperoxidase activity was defined as the quantity of enzyme degrading 1 μmol of hydrogen peroxide/min at 37 °C and expressed in units per 100 mg weight of tissue.
Plasma Creatine Phosphokinase Activity
Plasma levels of creatine phosphokinase were evaluated as an index of cardiac cellular damage by using a commercial kit (Sigma, St. Louis, MO, USA).
Subcellular Fractionation and Nuclear Protein Extraction
Tissue samples were homogenized with a poltroon homogenizer in a buffer containing 0.32 M sucrose, 10 mM Tris-HCl (pH 7.4), 1 mM ethylene glycol tetraacetic acid (EGTA), 2 mM ethylenediaminetetraacetic acid (EDTA), 5 mM NaN3, 10 mM β- mercaptoethanol, 20 μM leupeptin, 0.15 μM pepstatin A, 0.2 mM phenylmethanesulfonyl fluoride (PMSF), 50 mM NaF, 1 mM sodium orthovanadate, and 0.4 nM microcystin. The homogenates were centrifuged (1000g, 10 min) and the supernatant (cytosol + membrane extract) collected for evaluation of IκBα content and IKK activity as described below. The pellets were solubilized in Triton buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl (pH 7.4), 1 mM EGTA, 1 mM EDTA, 0.2 mM sodium orthovana-date, 20 μM leupeptin A, 0.2 mM PMSF). The lysates were centrifuged (15000g, 30 min, 4 °C), and the supernatant (nuclear extract) was collected for evaluation of phosphorylated c-Jun and DNA binding of NF-κB and AP-1 (29).
Western Blot Analysis
Western blot analysis was used to identify the cellular content of IκB-α in cytosol extracts and phosphorylated c-Jun in nuclear extracts. Cytosol or nuclear lysates were boiled in loading buffer (125 mM Tris-HCl (pH 6.8), 4% sodium dodecyl sulfate, 20% glycerol, and 10% 2-mercaptoethanol), and 50 μg protein was then loaded onto a 8% to 16% Tris-glycine gradient gel. Proteins were separated electrophoretically and transferred to a nitrocellulose membrane. For immunoblotting, membranes were blocked with 5% nonfat dried milk in Tris-buffered saline for 1 h and then incubated with primary antibody specific for IκBα or phosphorylated c-Jun. The membrane was washed in Tris-buffered saline with 0.1% Tween 20 and incubated with secondary peroxidase-conjugated antibodies. Immunoreaction was visualized by chemiluminescence.
Kinase Activity Assays
IKK activity was determined by immune complex kinase assay and was estimated as the ability to phosphorylate glutathion-S-transferase–IκB-α (29). After immunoprecipitation of lysate with specific antibody directed to IKKγ, the immunoprecipitate was incubated for 30 min at 30 °C in 40 μL of reaction buffer (25 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (pH 7.6), 20 mM MgCl2, 20 mM glycerolphosphate, 0.1 mM sodium ortho-vanadate, 2 mM dithiothreitol, 25 μM adenosine triphosphate (ATP), and 5 μCi of [γ-32P]ATP. Glutathion-S-transferase–IκBα (4 μg) was used as substrate for IKK complex. Reaction products were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and visualized by autoradiography.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assays (EMSA) were performed as previously described (30). An oligonucleotide probe corresponding to AP-1 consensus sequence (5′-CGCTTGATGACTCAGC-CGGAA-3′) and the NF-κB consensus sequence (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) was labeled with γ-(32P)ATP using T4 polynucleotide kinase and purified in Bio-Spin chromatography columns (BioRad, Hercules, CA, USA). Ten micrograms of nuclear protein were preincubated with EMSA buffer (12 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (pH 7.9), 4 mM Tris-HCl (pH 7.9), 25 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, 50 ng/mL poly (d[I-C]), 12% glycerol (v/v), and 0.2 mM PMSF on ice for 10 min before addition of the radiolabeled oligonucleotide for an additional 10 min. Excess of unlabeled oligonucleotide was added in some samples for competition to verify the specificity of NF-κB and AP-1 binding (data not shown). Protein-nucleic acid complexes were resolved using a nondenaturing polyacrylamide gel consisting of 5% acrylamide (29:1 ratio of acrylamide:bisacrylamide) and run in 0.5X TBE (45 mM Tris-HCl, 45 mM boric acid, 1 mM EDTA) for 1 h at constant current (30 mA). Gels were transferred to Whatman 3M paper, dried under a vacuum at 80 °C for 1 h, and exposed to photographic film at −70 °C with an intensifying screen.
Materials
The antibodies directed to IκBα, IKKγ, and the oligonucleotide probe for NF-κB consensus, and AP-1 were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The antibody for phosphorylated c-Jun (phosphorylated at serine 63 and 73) was obtained from Cell Signaling Technology (Beverly, MA, USA). EGCG was purchased from Sigma Aldrich (St. Louis, MO, USA); the drug was dissolved in PBS solution. All other chemicals were from Sigma/Aldrich.
Data Analysis
All values in the figures and text are expressed as mean ± SEM of n observations, where n represents the number of rats (n = 3 to 8 animals for each group). The results were examined by analysis of variance followed by the Bonferroni’s correction post hoc t test. Statistical analysis of scores was performed using the Mann-Whitney U test. A P value less than 0.05 was considered significant.
RESULTS
Effect of In Vivo Treatment with EGCG on Myocardial Injury
Severe myocardial tissue infarction was observed in vehicle-treated rats subjected to occlusion (for 30 min) and reperfusion (for 120 min) of the left coronary artery. Histological examination of the hearts revealed marked necrosis of the tissue with development of contraction bands. (Figure 1B) The degree of damage (on a scale of 0 to 4) in vehicle-treated rats averaged 3.60 ± 0.12. EGCG-treated rats were treated with a bolus dose of EGCG 10 mg/kg at the end of the ischemia period followed by 10 mg/kg/h intravenous infusion for the entire period of reperfusion. Myocardial histology of EGCG-treated rats demonstrated mild architectural alterations characterized by interstitial edema and localized necrotic areas. This attenuation in architectural derangement was observed throughout the distribution area of the left coronary artery and not confined to the border zone of the ischemic region (see Figure 1C for representative sections). The damage score was significantly reduced when compared with vehicle-treated rats (2.25 ± 0.15; P < 0.05).
Figure 1.
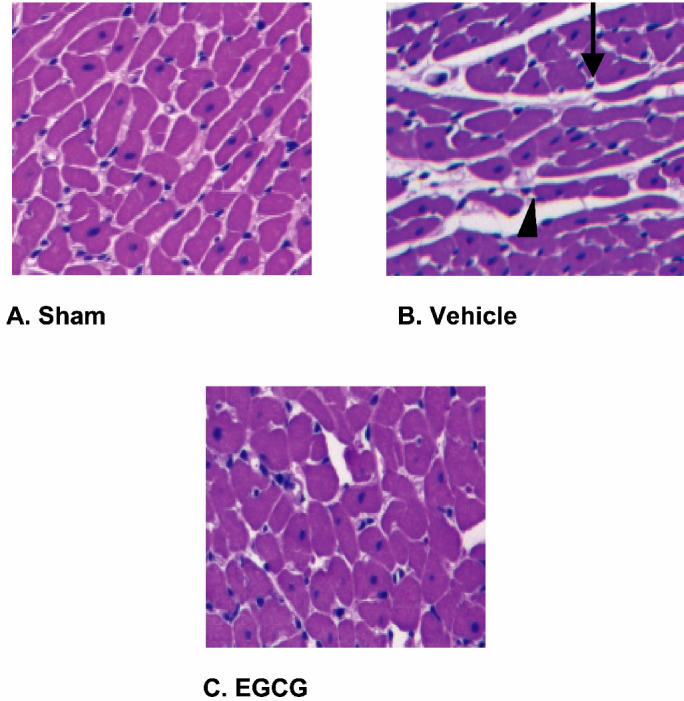
Effect of in vivo treatment with EGCG on myocardial histology. A: Representative cardiac section from a sham-operated rat displays normal tissue structure. B: After 30 min occlusion and 120 min reperfusion of the left coronary artery (I-R), a marked disruption of the myocardial structure characterized by appearance of extensive necrosis, contraction bands (arrow), and thinning of myofibrils (arrow head) was demonstrated in cardiac sections of a vehicle-treated rat. C: In vivo treatment with EGCG ameliorated myocardial damage. EGCG-treated rats received a bolus dose of EGCG 10 mg/kg at the end of the ischemia period followed by 10 mg/kg/h intravenous infusion for the entire period of reperfusion. Magnification × 100. A similar pattern was seen in 6 to 8 different tissue sections in each experimental group.
The derangement of myocardial histology in vehicle-treated rats was accompanied with elevated levels of plasma creatine phosphokinase (1100 ± 112.58 U/L) compared with sham control value of (117.6 ± 31 U/L; P < 0.05; Figure 2). On the contrary, EGCG-treated rats demonstrated a significant attenuation of myocardial injury as evidenced by reduced plasma creatine phosphokinase activity (399 ± 40.23 U/L; P < 0.05) when compared with vehicle-treated rats (see Figure 2).
Figure 2.
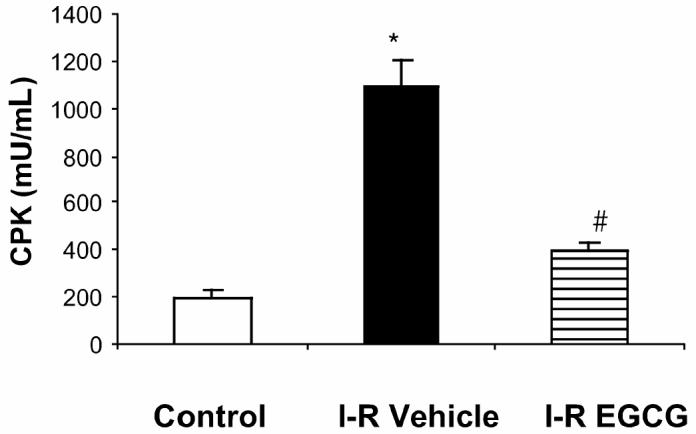
Effect of in vivo treatment with EGCG on plasma levels of creatine phosphokinase in rats subjected to myocardial ischemia and reperfusion (I-R). Each data point represents the mean ± SEM of 6 animals for each group. EGCG-treated rats received a bolus dose of EGCG 10 mg/kg at the end of the ischemia period followed by 10 mg/kg/h intravenous infusion for the entire period of reperfusion. *P < 0.05 compared with sham operated rats control); #P < 0.05 versus vehicle-treated rats subjected to myocardial ischemia and reperfusion (I-R + vehicle).
Effect of In Vivo Treatment with EGCG on Plasma Interleukin-6 Levels
Interleukin (IL)-6 is an inflammatory marker that is derived from hypoxic myocytes and from ischemic-reperfused myocardium (31). As expected, IL-6 levels were significantly elevated after myocardial ischemia and reperfusion in vehicle-treated rats (1312.83 ± 224.6 pg/mL) when compared with sham control animals (132 ± 21 pg/mL; P < 0.05). Treatment with EGCG reduced plasma IL-6 levels (494 ± 92.3 pg/mL; P < 0.05) indicating decreased cytokine response after ischemia and reperfusion (Figure 3).
Figure 3.
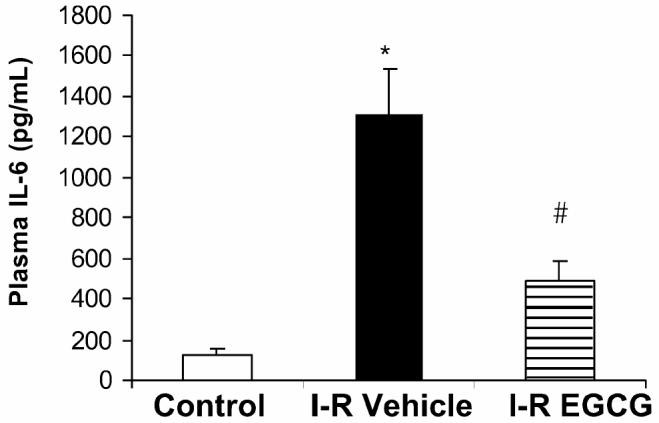
Effect of in vivo treatment with EGCG on plasma IL-6 levels in rats subjected to myocardial ischemia and reperfusion (I-R). Each data point represents the mean ± SEM of 6 animals for each group. EGCG-treated rats received a bolus dose of EGCG 10 mg/kg at the end of the ischemia period followed by 10 mg/kg/h intravenous infusion for the entire period of reperfusion. *P < 0.05 compared with control; #P < 0.05 compared with vehicle-treated rats subjected to myocardial ischemia and reperfusion (I-R + vehicle).
Effect of In Vivo Treatment with EGCG on Neutrophil Infiltration
Reperfusion after ischemia is characterized by the accumulation of neutrophils in the myocardial tissue. Myeloperoxidase activity, an enzyme specific to granulocyte lysosomes, correlates to the number of neutrophils and hence to the degree of reperfusion injury. Myeloperoxidase activity was significantly elevated after myocardial ischemia and reperfusion in vehicle-treated rats (29.30 ± 2.29 U/100 mg tissue) when compared with sham control animals (2.96 ± 1.13 U/100 mg tissue). Treatment with EGCG reduced myeloperoxidase activity (18.73 ± 1.8 U/100 mg tissue), thus suggesting a reduction in neutrophil infiltration (Figure 4).
Figure 4.
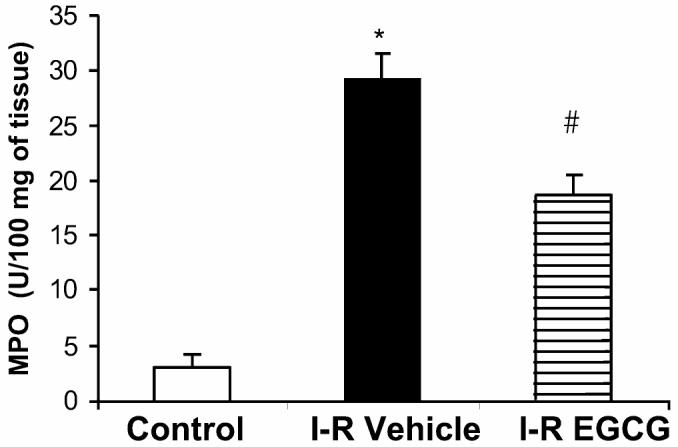
Effect of in vivo treatment with EGCG on MPO activity in rats subjected to myocardial ischemia and reperfusion (I-R). Tissue MPO activity was measured in myocardial tissue in vehicle-treated and EGCG-treated rats and compared with control rats. Each data point represents the mean ± SEM of 6 animals for each group. EGCG-treated rats received a bolus dose of EGCG 10 mg/kg at the end of the ischemia period followed by 10 mg/kg/h intravenous infusion for the entire period of reperfusion.*P < 0.05 compared with control; #P < 0.05 compared with vehicle-treated rats subjected to myocardial ischemia and reperfusion (I-R + vehicle).
Effect of In Vivo Treatment with EGCG on Pressure-Rate Index and Hemodynamic Parameters
The pressure-rate index was measured as an approximation of myocardial oxygen consumption. In vehicle-treated rats, the pressure-rate index declined shortly after occlusion of the left coronary artery and was still decreased upon reperfusion (Figure 5). The reduction of the pressure-rate index reflected a reduction of mean arterial blood pressure at the end of the ischemia and the reperfusion period when compared with basal control values, whereas heart rate was not significantly modified (data not shown). Rats treated with EGCG also showed a similar decrease in the pressure-rate index at the end of ischemia and at the end of reperfusion (see Figure 5). Measurements of hemodynamic parameters indicated that mean arterial blood pressure and heart rate were comparable to vehicle-treated rats.
Figure 5.
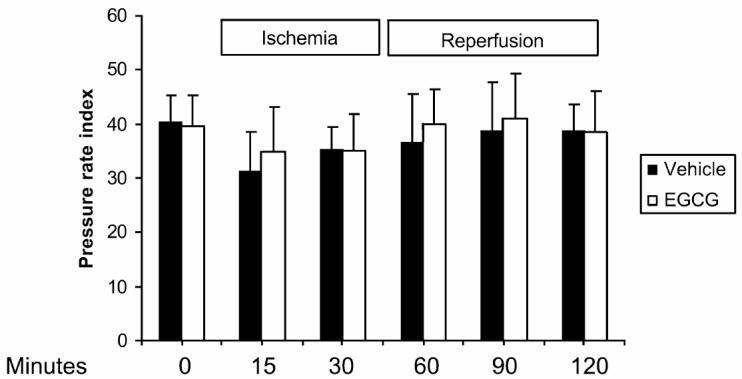
Effect of in vivo treatment with EGCG on pressure-rate index in rats subjected to myocardial ischemia and reperfusion. Data represent pressure-rate index as recorded before occlusion of the left main coronary artery (basal) after 30 min of ischemia (end of ischemia) and after 120 min of reperfusion (end of reperfusion). Pressure rate index is calculated by the following formula (mean blood pressure [mmHg] × beats/min/1000). Values are means ± SEM of 5 to 6 rats per group.
Effect of In Vivo Treatment with EGCG on Activation of NF-κB
To investigate the cellular mechanisms by which treatment with EGCG may attenuate reperfusion-induced injury, we evaluated the activation of NF-κB, a transcription factor centrally involved in the signal transduction of the inflammatory process. In vehicle-treated rats reperfusion after myocardial ischemia resulted in the activation of NF-κB. Increased DNA binding of NF-κB was present at 60 min after reperfusion in vehicle-treated rats (Figure 6A). This binding of DNA was significantly abrogated in rats treated with EGCG (see Figure 6A).
Figure 6.

Effect of in vivo treatment with EGCG on the activation of NF-κB, IKK activation, and IκB-α degradation in rats subjected to myocardial ischemia and reperfusion. EGCG-treated rats received a bolus dose of EGCG 10 mg/kg at the end of the ischemia period followed by 10 mg/kg/h intravenous infusion for the entire period of reperfusion. A: Representative autoradiograph of electrophoretic mobility shift assay for NF-κB binding hearts of vehicle-treated rats at 0 (sham value), 30, 60, and 120 min after reperfusion. B: Representative Western blot analysis demonstrating the effect of vehicle or EGCG on IκB-α degradation at 0 (sham value), 30, 60, and 120 min after reperfusion. Western blot analysis is representative of 3 separate experiments performed at different times. Fold increase was calculated compared with respective sham value (time 0) set to 1.0. Results are representative of 3 separate time-course experiments. *P < 0.05 compared with respective sham value (time 0); #P < 0.05 compared with vehicle-treated rats subjected to myocardial ischemia and reperfusion. C: Representative of autoradiograph of kinase assay demonstrating the effect of EGCG or vehicle treatment on activation of the IKK complex at 0 (sham value), 30, 60, and 120 min after reperfusion. The autoradiograph is representative of 3 experiments with similar results. Fold increase was calculated compared with respective sham value (time 0) set to 1.0. Results are representative of 3 separate time-course experiments. *P < 0.05 compared with respective sham value (time 0); #P < 0.05 versus vehicle-treated rats subjected to myocardial ischemia and reperfusion.
Effect of In Vivo Treatment with EGCG on Degradation of IκBα and Activity of IKK
The common pathway for activation of NF-κB involves phosphorylation of its inhibitor protein IκB-α by IKK. A Western blot analysis showed that the cytosolic content of IκB-α was reduced at 30 min in hearts of vehicle-treated rats. IκB-α content further declined at 60 min after reperfusion thus indicating the occurrence of degradation. IκB-α content was then replenished at 120 min after reperfusion in vehicle-treated rats (see Figure 6B). On the contrary, a marked attenuation of IκB-α degradation was present in rats treated with EGCG. This attenuation was marked at 30 and 60 min after onset of reperfusion (see Figure 6B). We next determined the effect of EGCG on IKK activation and IκB-α degradation. A time-course analysis showed that IKK was activated in a time-dependent manner after reperfusion in vehicle-treated rats (see Figure 6C). Consistent with this data, EGCG also inhibited the increase of IKK activity after reperfusion (see Figure 6C).
Effect of In Vivo Treatment with EGCG on Activation of AP-1 and Phosphorylation of c-Jun
AP-1 activity has been implicated as playing an important role in the inflammatory response by modulating gene expression of several inflammatory mediators including IL-6 transcription. We then examined DNA binding of AP-1 after myocardial ischemia reperfusion. In vehicle-treated rats, a marked increase in DNA binding of AP-1 occurred at 120 min while there was minimal activation of AP-1 at 30 and 60 min. Treatment with EGCG significantly reduced AP-1 activation (Figure 7A). Because phosphorylation of c-Jun is a prerequisite of AP-1 dimerization and activation (32), we next examined the content of phosphorylated c-Jun in nuclear extract after myocardial ischemia reperfusion. A weak phosphorylation of c-Jun was observed in sham control animal (that is, at time 0). However, in vehicle-treated rats phosphorylation of c-Jun was increased in a time-dependent manner after reperfusion (see Figure 7B). In rats treated with EGCG phosphorylation of c-Jun was markedly attenuated at all time points (see Figure 7B).
Figure 7.
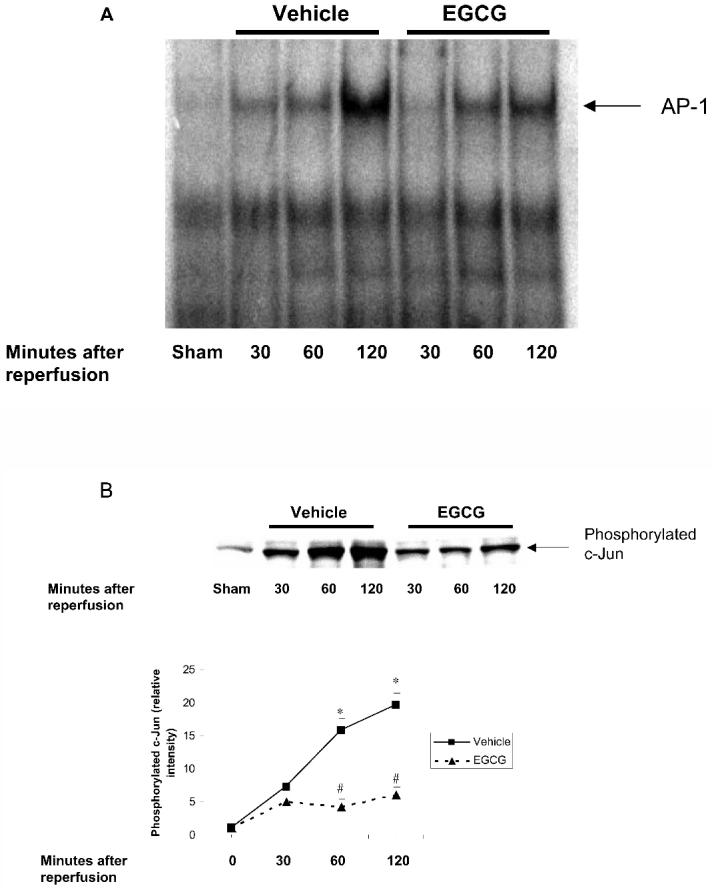
Effect of in vivo treatment with EGCG on activation of AP-1 and phosphorylation of c-Jun in rats subjected to myocardial ischemia and reperfusion. EGCG-treated rats received a bolus dose of EGCG 10 mg/kg at the end of the ischemia period followed by 10 mg/kg/h intravenous infusion for the entire period of reperfusion. A: Representative autoradiograph of electrophoretic mobility shift assay for AP-1 in vehicle-treated rats and EGCG-treated rats at 0 (sham value), 30, 60, and 120 min after reperfusion. The autoradiograph is representative of 3 separate experiments performed at different times. B: Western blot analysis demonstrating the effect of vehicle or EGCG on treatment phosphorylation of c-Jun at 0 (sham value), 30, 60, and 120 min after reperfusion. Western blot analysis is representative of separate experiments performed at different times. Fold increase was calculated compared with respective sham value (time 0) set to 1.0. Results are representative of 3 separate time-course experiments. *P < 0.05 compared with respective sham value (time 0); #P < 0.05 compared with vehicle-treated rats subjected to myocardial ischemia and reperfusion.
DISCUSSION
After myocardial ischemia, the 1st few minutes of reperfusion are of critical importance as lethal myocardial injury commences at this point. There is substantial evidence that neutrophils are recruited to the ischemic reperfused myocardium, where they release reactive oxygen and nitrogen species (33,34). These species can have deleterious effect on the myocytes and the coronary vascular endothelial cells thus causing myocardial injury. In addition, ischemic-reperfused myocytes release cytokines, that is, IL-6 and TNF-α, that can further activate neutrophils, thus enhancing reactive species generation and extending the infarcted area. The overwhelming production of proinflammatory mediators can lead to arrhythmia, contractile dysfunction, and cardiomyocyte death (4,6). In our study, we demonstrated that EGCG markedly attenuates the myocardial injury after ischemia and reperfusion. This cardioprotection was also associated with decreased IL-6 production and decreased neutrophilic infiltration as measured by myeloperoxidase (MPO) assay.
It is difficult in vivo experiments to rule out a direct effect of EGCG on MPO and to establish the exact mechanisms by which EGCG inhibits neutrophil infiltration. Our data are supported by previous in vitro findings that demonstrated that this catechin inhibits leukocyte infiltration as evaluated by histology and MPO activity assay in human skin exposed to ultraviolet rays (35,36).
Activation of NF-κB is a very early regulatory event during reperfusion and precedes the occurrence of oxidative damage, infiltration of neutrophils, and architectural alteration in the myocardium. Because EGCG also reduced reperfusion-induced activation of IKK, degradation of IκB-α, and activation of NF-κB, our results suggest that EGCG exerts cardioprotective effects by inhibiting the inflammatory process at the early event of the transcription mediated by the NF-κB pathway.
Several in vitro studies have proposed that activation of IKK complex is an essential step for NF-κB activation because the kinase phosphorylates IκB-α allow its degradation (37). Our study demonstrates that EGCG is an effective inhibitor of IKK activity in vivo. This is consistent with our in vitro data demonstrating that EGCG inhibits TNF-α–mediated IKK activation in human epithelial cells (27). Similarly, Yang and colleagues showed that EGCG in concentrations of 50 to 200 μM inhibited IKK activity in an intestinal epithelial cell line (38). The presence of a gallate group in EGCG appeared to confer the specificity to blocking IKK activation. Antioxidants, such as pyrrolidinedithio-carbamate and N-acetylcysteine, inhibit IKK activation by deactivating reactive oxygen species that may serve as intermediates upstream to IKK (39,40). Thus it is possible that EGCG, which is an antioxidant (41), may also block the activation of IKK by scavenging the reactive oxygen species. In summary, although it is difficult to establish the exact in vivo mechanisms of action of EGCG, our data taken together with the previous studies suggests that EGCG may inhibit IKK/NF-κB pathway by either inhibiting IKK directly and/or indirectly by modifying the redox state of the inflammatory site.
Activation of the nuclear transcription factor AP-1 has been reported to have an important role in myocardial ischemia and reperfusion injury. AP-1 activity is controlled by signaling through the JNK family of MAP kinases (29). It has been demonstrated that during reperfusion oxidative stress leads to activation and translocation of JNK to the nucleus, where phosphorylation of transcription factors, such as c-Jun occurs (30,42). Several studies support the hypothesis of a positive synergy between NF-κB and AP-1 (43). In our study we demonstrated that both AP-1 and NF-κB are increased in areas of infarction. A similar activation of NF-κB and AP-1 occurs during reperfusion in other previously ischemic organs and tissues, such as brain and liver (44,45). While more studies are needed to clarify the role of AP-1 in myocardial ischemia/reperfusion injury, it is conceivable that the beneficial effects of EGCG may be also mediated by inhibition of the AP-1 pathway.
Our data is in agreement with other studies that have demonstrated that EGCG affects several signaling mechanisms in inflammation. Menegazzi and colleagues showed that interferon-γ–induced STAT-1 activation in carcinoma-derived cell lines of non-gut origin was blocked by EGCG (46). In another study, Watson and colleagues demonstrated that EGCG significantly reduced INF-γ–induced STAT1 activation in T84 epithelia and THP-1 monocytes/macrophages (47).
Epidemiological studies have suggested the preventive effect of green tea consumption against atherosclerosis and coronary heart disease (48–51). There is scanty evidence about the amount of tea that needs to be consumed to confer protection against myocardial ischemia reperfusion injury. In a prospective study, Mukamal and colleagues examined tea consumption in 1900 patients in the Myocardial Infarction Onset Study, a study that examined patients with myocardial infarction presenting to community hospitals in the United States (52). In this study, tea consumption was assessed by questionnaire and patients were followed for 3.8 y. This study demonstrated that mortality was lower among moderate tea drinkers (<14 cups per week) and heavy tea drinkers (>14 cups per week) (52). Another randomized crossover trial showed that short-and long-term black tea consumption could reverse endothelial dysfunction in patients with documented coronary heart disease (13). The current study provides a plausible mechanism as to how tea is beneficial in cardiovascular disease.
On the other hand, there are no reports of clinical toxicity on consumption of green tea in large quantities, as is prevalent in some cultures. Although there are no data available regarding the pharmacokinetics following intravenous green tea injection, Chow and colleagues have reported that it is safe to take green tea polyphenol products in amounts equivalent to the EGCG content in 8 to 16 cups of green tea once a day or in divided doses twice a day for 4 wk (53). Nevertheless, further investigation is required to establish the optimal concentrations of catechins and/or their metabolites for potential therapeutics or prophylactic use.
In conclusion, we have demonstrated that EGCG protects against myocardial reperfusion injury in vivo by a specific inhibition of the IKK/NF-κB and AP-1 pathway. Thus, EGCG and related compounds may present a novel therapeutic strategy for modulating the inflammatory processes that occur during the reestablishment of blood flow after myocardial infarction.
Acknowledgments
Supported by NIH grant T 32 GM08478 (RA), RO1GM061723 (HRW), and NIH RO1HL 60730 (BZ).
REFERENCES
- 1.Hearse DJ. Reperfusion-induced injury: a possible role for oxidant stress and its manipulation. Cardiovasc. Drugs Ther. 5 Suppl. 1991;2:225–35. doi: 10.1007/BF00054745. [DOI] [PubMed] [Google Scholar]
- 2.Gross GJ, Farber NE, Hardman HF, Warltier DC. Beneficial actions of super-oxide dismutase and catalase in stunned myocardium of dogs. Am. J. Physiol. 1986;250:H372–7. doi: 10.1152/ajpheart.1986.250.3.H372. [DOI] [PubMed] [Google Scholar]
- 3.Poltronieri R, Cevese A, Sbarbati A. Protective effect of selenium in cardiac ischemia and reperfusion. Cardioscience. 1992;3:155–60. [PubMed] [Google Scholar]
- 4.Ferrari R et al. (1998) Oxidative stress during myocardial ischaemia and heart failure. Eur. Heart J 19 Suppl B:B2-11. [PubMed]
- 5.Grisham MB, Granger DN, Lefer DJ. Modulation of leukocyte-endothelial interactions by reactive metabolites of oxygen and nitrogen: relevance to ischemic heart disease. Free Radic. Biol. Med. 1998;25:404–33. doi: 10.1016/s0891-5849(98)00094-x. [DOI] [PubMed] [Google Scholar]
- 6.Ambrosio G, Tritto I. Reperfusion injury: experimental evidence and clinical implications. Am. Heart J. 1999;138:S69–75. doi: 10.1016/s0002-8703(99)70323-6. [DOI] [PubMed] [Google Scholar]
- 7.Mukaida N, Okamato S, Ishikawa Y, Matsushima K. Molecular mechanisms of interleukin-8 gene expression. Leukoc. Biol. J. 1994;56:554–8. [PubMed] [Google Scholar]
- 8.DiDonato JA, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 9.Mercurio F, Zhu H, Murray BW. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–6. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 10.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination:the control of NF-kB activity. Annu. Rev. Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 11.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995;270:16483–6. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 12.Duffy SJ, Vita JA, Holbrook M, Swerdloff PL, Keaney JF., Jr Effect of acute and chronic tea consumption on platelet aggregation in patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2001;21:1084–9. doi: 10.1161/01.atv.21.6.1084. [DOI] [PubMed] [Google Scholar]
- 13.Duffy SJ, et al. Short- and long-term black tea consumption reverses endothelial dysfunction in patients with coronary artery disease. Circulation. 2001;104:151–6. doi: 10.1161/01.cir.104.2.151. [DOI] [PubMed] [Google Scholar]
- 14.Lin JK, Liang YC, Lin-Shiau SY. Cancer chemoprevention by tea polyphenols through mitotic signal transduction blockade. Biochem. Pharmacol. 1999;58:911–5. doi: 10.1016/s0006-2952(99)00112-4. [DOI] [PubMed] [Google Scholar]
- 15.Pan MH, Lin-Shiau SY, Ho CT, Lin JH, Lin JK. Suppression of lipopolysaccharide-induced nuclear factor-kappaB activity by theaflavin-3,3′-digallate from black tea and other polyphenols through down-regulation of IkappaB kinase activity in macrophages. Biochem. Pharmacol. 2000;59:357–67. doi: 10.1016/s0006-2952(99)00335-4. [DOI] [PubMed] [Google Scholar]
- 16.Wang ZY, et al. Inhibitory effects of black tea, green tea, decaffeinated black tea, and decaffeinated green tea on ultraviolet B light-induced skin carcinogenesis in 7,12-dimethylbenz[a]anthracene-initiated SKH-1 mice. Cancer Res. 1994;54:3428–35. [PubMed] [Google Scholar]
- 17.Serafini M, Ghiselli A, Ferro-Luzzi A. In vivo antioxidant effect of green and black tea in man. Eur. J. Clin. Nutr. 1996;50:28–32. [PubMed] [Google Scholar]
- 18.Dona M, et al. Neutrophil restraint by green tea: Inhibition of inflammation, associated angiogenesis, and pulmonary fibrosis. J. Immunol. 2003;170:4335–41. doi: 10.4049/jimmunol.170.8.4335. [DOI] [PubMed] [Google Scholar]
- 19.Kang WS, et al. Antithrombotic activities of green tea catechins and (–)-epigallocatechin gallate. Thromb. Res. 1999;96:229–37. doi: 10.1016/s0049-3848(99)00104-8. [DOI] [PubMed] [Google Scholar]
- 20.Levites Y, Amit T, Youdim MB, Mandel S. Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol (–)-epigallocatechin 3-gallate neuroprotective action. J. Biol. Chem. 2002;277:30574–80. doi: 10.1074/jbc.M202832200. [DOI] [PubMed] [Google Scholar]
- 21.Levites Y, Youdim MB, Maor G, Mandel S. Attenuation of 6-hydroxy-dopamine (6-OHDA)-induced nuclear factor-kappaB (NF-kappaB) activation and cell death by tea extracts in neuronal cultures. Biochem. Pharmacol. 2002;63:21–9. doi: 10.1016/s0006-2952(01)00813-9. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad N, Feyes DK, Nieminen AL, Agarwal R, Mukhtar H. Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J. Natl. Cancer Inst. 1997;89:1881–6. doi: 10.1093/jnci/89.24.1881. [DOI] [PubMed] [Google Scholar]
- 23.Chan MM, Fong D, Ho CT, Huang HI. Inhibition of inducible nitric oxide synthase gene expression and enzyme activity by epigallocatechin gallate, a natural product from green tea. Biochem. Pharmacol. 1997;54:1281–6. doi: 10.1016/s0006-2952(97)00504-2. [DOI] [PubMed] [Google Scholar]
- 24.Levites Y, Amit T, Mandel S, Youdim MB. Neuroprotection and neurorescue against Abeta toxicity and protein kinase C–dependent release of non-amyloidogenic soluble precursor protein by green tea polyphenol (–)-epigallo-catechin-3-gallate. FASEB J. 2003;17:952–4. doi: 10.1096/fj.02-0881fje. [DOI] [PubMed] [Google Scholar]
- 25.Kim HS, et al. EGCG blocks tumor promoter-induced MMP-9 expression via suppression of MAPK and AP-1 activation in human gastric AGS cells. Anticancer Res. 2004;24:747–53. [PubMed] [Google Scholar]
- 26.Tedeschi E, Suzuki H, Menegazzi M. Antiinflammatory action of EGCG, the main component of green tea, through STAT-1 inhibition. Ann. N.Y. Acad. Sci. 2002;973:435–7. doi: 10.1111/j.1749-6632.2002.tb04678.x. [DOI] [PubMed] [Google Scholar]
- 27.Chen PC, et al. A green tea–derived polyphenol, epigallocatechin-3-gallate, inhibits IkappaB kinase activation and IL-8 gene expression in respiratory epithelium. Inflammation. 2002;26:233–41. doi: 10.1023/A:1019718718977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zingarelli B, Cuzzocrea S, Zsengeller Z, Salzman AL, Szabo C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc. Res. 1997;36:205–15. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- 29.Mizukami Y, Yoshioka K, Morimoto S, Yoshida K. A novel mechanism of JNK1 activation. Nuclear translocation and activation of JNK1 during ischemia and reperfusion. J. Biol. Chem. 1997;272:16657–62. doi: 10.1074/jbc.272.26.16657. [DOI] [PubMed] [Google Scholar]
- 30.Zingarelli B, Hake PW, Yang Z, O’Connor M, Denenberg A, Wong HR. Absence of inducible nitric oxide synthase modulates early reperfusion-induced NF-kappaB and AP-1 activation and enhances myocardial damage. FASEB J. 2002;16:327–42. doi: 10.1096/fj.01-0533com. [DOI] [PubMed] [Google Scholar]
- 31.Nose P. Cytokines and reperfusion injury. J. Card. Surg. 1993;8:305–8. doi: 10.1111/j.1540-8191.1993.tb01329.x. [DOI] [PubMed] [Google Scholar]
- 32.Wisdom R. AP-1: one switch for many signals. Exp. Cell. Res. 1999;253:180–5. doi: 10.1006/excr.1999.4685. [DOI] [PubMed] [Google Scholar]
- 33.Mitsos SE, et al. Protective effects of N-2-mercaptopropionyl glycine against myocardial reperfusion injury after neutrophil depletion in the dog: evidence for the role of intracellular-derived free radicals. Circulation. 1986;73:1077–86. doi: 10.1161/01.cir.73.5.1077. [DOI] [PubMed] [Google Scholar]
- 34.Duilio C, et al. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H2649–57. doi: 10.1152/ajpheart.2001.280.6.H2649. [DOI] [PubMed] [Google Scholar]
- 35.Katiyar SK, Mukhtar H. Green tea polyphenol (–)-epigallocatechin-3-gallate treatment to mouse skin prevents UVB-induced infiltration of leukocytes, depletion of antigen-presenting cells, and oxidative stress. J. Leukoc. Biol. 2001;69:719–26. [PubMed] [Google Scholar]
- 36.Katiyar SK, Matsui MS, Elmets CA, Mukhtar H. Polyphenolic antioxidant (–)-epigallocatechin-3-gallate from green tea reduces UVB-induced inflammatory responses and infiltration of leukocytes in human skin. Photochem. Photobiol. 1999;69:148–53. [PubMed] [Google Scholar]
- 37.Zandi E, Karin M. Bridging the gap: composition, regulation, and physiological function of the IkappaB kinase complex. Mol. Cell Biol. 1999;19:4547–51. doi: 10.1128/mcb.19.7.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang F, et al. The green tea polyphenol (–)-epigallocatechin-3-gallate blocks nuclear factor-kappa B activation by inhibiting I kappa B kinase activity in the intestinal epithelial cell line IEC-6. Mol. Pharmacol. 2001;60:528–33. [PubMed] [Google Scholar]
- 39.Suzuki YJ, Packer L. Inhibition of NF-kappa B activation by vitamin E derivatives. Biochem. Biophys. Res. Commun. 1993;193:277–83. doi: 10.1006/bbrc.1993.1620. [DOI] [PubMed] [Google Scholar]
- 40.Beauparlant P, Hiscott J. Biological and biochemical inhibitors of the NF-kappa B/Rel proteins and cytokine synthesis. Cytokine Growth Factor Rev. 1996;7:175–90. doi: 10.1016/1359-6101(96)00020-2. [DOI] [PubMed] [Google Scholar]
- 41.L’Allemain G. Multiple actions of EGCG, the main component of green tea. Bull. Cancer. 1999;86:721–4. [PubMed] [Google Scholar]
- 42.Zingarelli B, et al. Differential regulation of activator protein-1 and heat shock factor-1 in myocardial ischemia and reperfusion injury: role of poly(ADP-ribose) polymerase-1. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H1408–15. doi: 10.1152/ajpheart.00953.2003. [DOI] [PubMed] [Google Scholar]
- 43.Stein B, et al. Cross-coupling of the NF-kappa B p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 1993;12:3879–91. doi: 10.1002/j.1460-2075.1993.tb06066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP. (1997) Global ischemia activates nuclear factor-kappa B in forebrain neurons of rats. Stroke 28:1073–80; discussion 1080–71. [DOI] [PubMed]
- 45.Zwacka RM, et al. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation. Nat. Med. 1998;4:698–704. doi: 10.1038/nm0698-698. [DOI] [PubMed] [Google Scholar]
- 46.Menegazzi M, et al. Anti-interferon gamma action of epigallocatechin-3-gallate mediated by specific inhibition of STAT1 activation. FASEB J. 2001;15:1309–11. doi: 10.1096/fj.00-0519fje. [DOI] [PubMed] [Google Scholar]
- 47.Watson JL et al. (2004) The green tea polyphenol epigallocatechin gallate blocks epithelial barrier dysfunction provoked by IFNγ but not by IL-4. Am. J. Physiol. Gastrointest. Liver Physiol Jul 1[Epub ahead of print]. [DOI] [PubMed]
- 48.Sesso H, Gaziano J, Buring J, Hennekens C. Coffee and tea intake and the risk of myocardial infarction. Am. J. Epidemiol. 1999;149:162–7. doi: 10.1093/oxfordjournals.aje.a009782. [DOI] [PubMed] [Google Scholar]
- 49.Cheng TO. Tea is good for the heart. Arch. Intern. Med. 2000;160:2397. doi: 10.1001/archinte.160.15.2397. [DOI] [PubMed] [Google Scholar]
- 50.Geleijnse JM, Launer LJ, Hofman A, Pols HAP, Witteman JCM. Tea flavonoids may protect against atherosclerosis: The Rotterdam study. Arch. Intern. Med. 1999;159:2170–4. doi: 10.1001/archinte.159.18.2170. [DOI] [PubMed] [Google Scholar]
- 51.Hakim IA AM, Alduwaihy M, Al-Rubeaan K, Al-Nuaim AR, Al-Attas OS. Tea consumption and the prevalence of coronary heart disease in Saudi adults: results from a Saudi national study. Prev. Med. 2003;36((1)):64–70. doi: 10.1006/pmed.2002.1130. [DOI] [PubMed] [Google Scholar]
- 52.Mukamal KJ, Maclure M, Muller JE, Sherwood JB, Mittleman MA. Tea consumption and mortality after acute myocardial infarction. Circulation. 2002;105:2476–81. doi: 10.1161/01.cir.0000017201.88994.f7. [DOI] [PubMed] [Google Scholar]
- 53.Chow HH, et al. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin. Cancer Res. 2003;9:3312–9. [PubMed] [Google Scholar]