

Abstract
Intercellular adhesion molecule 1 (ICAM-1) and β2 integrins play critical roles in immune responses. ICAM-1 may also participate in regulation of energy balance because ICAM-1–deficient mice become obese on a high-fat diet. We show that mice deficient in these adhesion receptors are unable to respond to fasting by up-regulation of fatty acid oxidation. Normal mice, when fasted, exhibit reduced circulating neutrophil counts and increased ICAM-1 expression and neutrophil recruitment in liver. Mice lacking ICAM-1 or β2 integrins fail to show these responses—instead they become hypoglycemic with steatotic livers. Fasting ICAM-1–deficient mice reduce insulin more slowly than wild-type mice. This produces fasting hyperinsulinemia that prevents activation of adenosine mono-phosphate (AMP)-activated protein kinase in muscles and liver, which results in decreased import of long chain fatty acids into mitochondria. Thus, we show a new role for immune cells and their adhesion receptors in regulating metabolic response to fasting.
INTRODUCTION
Leukocyte adhesion molecules play an essential role in inflammation and immunosurveillance. Intercellular adhesion molecule-1 (ICAM-1) is a member of the immunoglobulin super family of adhesion molecules (1) that is expressed on many cell types including endothelium, hepatocytes, and leukocytes. Two main ligands for ICAM-1 are CD11a/CD18 (LFA-1) and CD11b/CD18 (Mac-1), which are members of the leukocyte-specific β2 integrin (CD18) family that comprises at least 2 other members. Mac-1 is expressed on monocytes, granulocytes, natural killer cells, and a subpopulation of T cells (2), whereas LFA-1 is expressed on all leukocytes (3). Mice deficient in ICAM-1 have impaired immune function and decreased inflammatory response, reflected in impaired activation and migration of leukocytes to places of inflammation, reduced contact hypersensitivity and impaired ability of spleen cells to act as stimulators in mixed lymphocyte responses (4). Mice deficient in CD18 expression showed impaired activation and migration of leukocytes to places of inflammation and impaired T cell proliferation in response to T-cell receptor activation that was more extensive than in ICAM-1–deficient mice (5). In humans, CD18 deficiency causes a severe immunodeficiency condition called Leukocyte Adhesion Deficiency Syndrome type I (LAD I) (6).
Participation in immune and inflammatory response is not the only role for ICAM-1 and CD18 molecules. Our laboratory previously showed that ICAM-1 or CD11b/CD18 deficiency in mice can result in spontaneous obesity in old age when mice are fed normal chow diet or obesity at a young age when mice are fed high-fat diet (7). This suggests a role for ICAM-1 and CD11b/CD18 molecules in regulation of energy balance. Regulation of energy intake does not appear to be affected in either ICAM-1 or CD11b-deficient mice because there was no significant difference in food consumption as compared with wild type, C57Bl/6J mice (WT). The presence of fatty livers in the older ICAM-1–deficient mice (7) suggested that leukocytes and their receptors might be involved in regulation of fat metabolism. Recently, 2 new reports were published describing a close connection between obesity and immune response (8,9). In obese adipose tissue of people and mice, there was significant infiltration by macrophages compared with adipose tissue from lean people and mice. One possibility is that macrophages are attracted by increased lipid content of the cells and try to fight unwanted fat accumulation (10).
During fasting, the basal metabolic rate is decreased and there is a shift in fuel utilization preference from carbohydrates and fat to almost entirely fat. During the switch from fed state to fasting, metabolic response is mediated by decreasing circulating insulin concentration and increasing concentration of counter regulatory hormones, such as glucagon, cortisol, epinephrine, and norepinephrine. This activates glycogenolysis, gluconeogenesis, and fat oxidation (11). The importance of fat metabolism in successful adaptation to fasting was best illustrated in peroxisome-proliferator activated receptor-α (PPARα)-deficient mice (12). PPARα is a nuclear hormone receptor that regulates expression of a number of genes involved in fat oxidation in the liver. Mice deficient in the expression of this master regulator of liver fat metabolism had liver steatosis, severe hypoglycemia, hypoketonemia, hypothermia, and elevated plasma free fatty acid (FFA) levels during fasting. The same mice did not show signs of significant metabolic derangement when they were fed normal chow diet (12).
In this study, we demonstrate that ICAM-1– and CD18-deficient mice have a similar metabolic phenotype to the PPARα-deficient mice. We show that leukocytes and thus the immune system help orchestrate the stress response to starvation by regulating insulin clearance.
MATERIALS AND METHODS
Animals
Mice were housed on a 12-hour dark/12-hour light cycle. Eight- to eleven-week-old males were used and were provided unrestricted access to water and standard lab chow (Prolab 3000, PMI Feeds, Richmond, IN) containing 5.0% (wt/wt) fat, 55% (wt/wt) carbohydrate, and 22% (wt/wt) protein. ICAM-1– (4) and CD18-null mice (4,5) used in the experiments were backcrossed 8 times to C57Bl/6J background. Wild-type mice were C57Bl6/J. Preparations for fasting were usually initiated 24 h before onset of fasting. Metal grids were placed at the bottom of the cages, without changing of actual cages. Mice were given 24 h to adjust to this new environment before the fasting was initiated at 9:00 a.m., and the experiments were performed at 9:00 a.m., 24 h later. Experimental procedures were approved by the Animal Care and Use Committee of the CBR Institute for Biomedical Research.
Respiratory Quotient Measurement
We used an indirect open circuit calorimeter (Oxymax, Columbus Instruments, Columbus, OH). The device consists of 4 mouse chambers where a constant air flow (0.75 liters/min) is pulled through and monitored by a mass sensitive flow meter. The system monitors oxygen and carbon dioxide gases concentrations at the inlet and outlet of the sealed chambers containing the animals. This is used to compute oxygen consumption, carbon dioxide production, and respiratory quotient (RQ).
Metabolite Assays
Blood for glucose measurements was taken from the tail vein. Otherwise, blood was drawn by retro-orbital puncture. Blood glucose was measured using Precision Xtra glucose analyzer (Medisense/Abbott, Abbott Park, IL). Plasma FFAs were determined using Wako NEFA C kit. β-hydroxybutyrate was determined using a kit from Sigma. Liver glycogen was determined by the anthrone method (13). Serum triglycerides were determined using a Wako L-Type TG H assay. Serum insulin was measured using ultrasensitive mouse insulin ELISA (ALPCO Diagnostics, Windham, NH). Serum C-peptide was measured using rat C-peptide radioimmunoassay (RIA) kit (Linco Research, St. Charles, MO). Acylcarnitine profile and total/free carnitine concentration in pooled plasma from fasted WT or ICAM-1–deficient mice was measured using tandem mass spectrometry at Duke University Biochemical Genetics Laboratory.
Staining and Counting of CD11b/CD18+ Cells in Liver Sections
Cryostat sections (10 μ/m) derived from the livers of mice sacrificed at 9 a.m. were fixed in acetone, air-dried, and incubated with rat anti-mouse Mac-1 (Boehringer-Mannheim, Mannheim, Germany) (1/100). The slides were incubated with biotinylated secondary rabbit anti-rat IgG (DakoCytomation, Carpinteria, CA) and visualized using streptavidin-peroxidase and substrate chromogen solution (Zymed, South San Francisco, CA). Numbers of CD11b/CD18+ cells represent average of results obtained by 2 observers blinded to genotype.
Western Blot Analysis
Phosphorylation of AMP-activated protein kinase (AMPK) and acetyl-CoA carboxylase (ACC) was determined using 4% to 15% gradient sodium dodecyl sulfide acrylamide gels (Bio-Rad, Hercules, CA) loaded with either 40 μg of the total muscle protein or 3 μg of the total liver protein. Proteins were transferred to Immobilon-P polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA) and labeled with antibodies against phosphopeptides surrounding Thr 172 of the α-subunit of human AMPK or an affinity-purified polyclonal antibody against AMPK-α (Cell Signalling, Beverly, MA) and Ser 79 of rat ACC (Upstate Biotech, Charlottesville, VA).
Statistical Analysis
Unless stated otherwise, data are presented as mean ± SEM.
RESULTS
ICAM-1 or CD18 Deficiency Diminishes the Physiologic Change in Respiratory Quotient Produced by Fasting.
To evaluate the effects of ICAM-1– or CD18-deficiency on metabolism, we determined respiratory quotient (RQ) by measuring oxygen consumption and carbon dioxide production in the mutant and WT mice. CD18-deficient mice were kept on antibiotics (Baytril) until 7 d prior to RQ measurement because they succumb to infection easily. RQ, the amount of carbon dioxide released per liter of oxygen consumed, depends upon the type of fuel preferentially oxidized in the body. RQ equal to 1 signifies that majority of calories are derived from glucose oxidation, while RQ close to 0.7 or below signifies oxidation of fat (14). In the fed state, there was no significant difference in the RQ between animals of all 3 genotypes (Figure 1A). Three to 4 h after removal of food, WT mice decreased their RQ, consistent with a normal fasting response. However, ICAM-1–deficient mice showed limited ability to change to lipid oxidation as the main source of energy during fasting. The defect was even more pronounced in CD18-deficient mice, showing minimal change in the prefasting RQ value.
Figure 1.
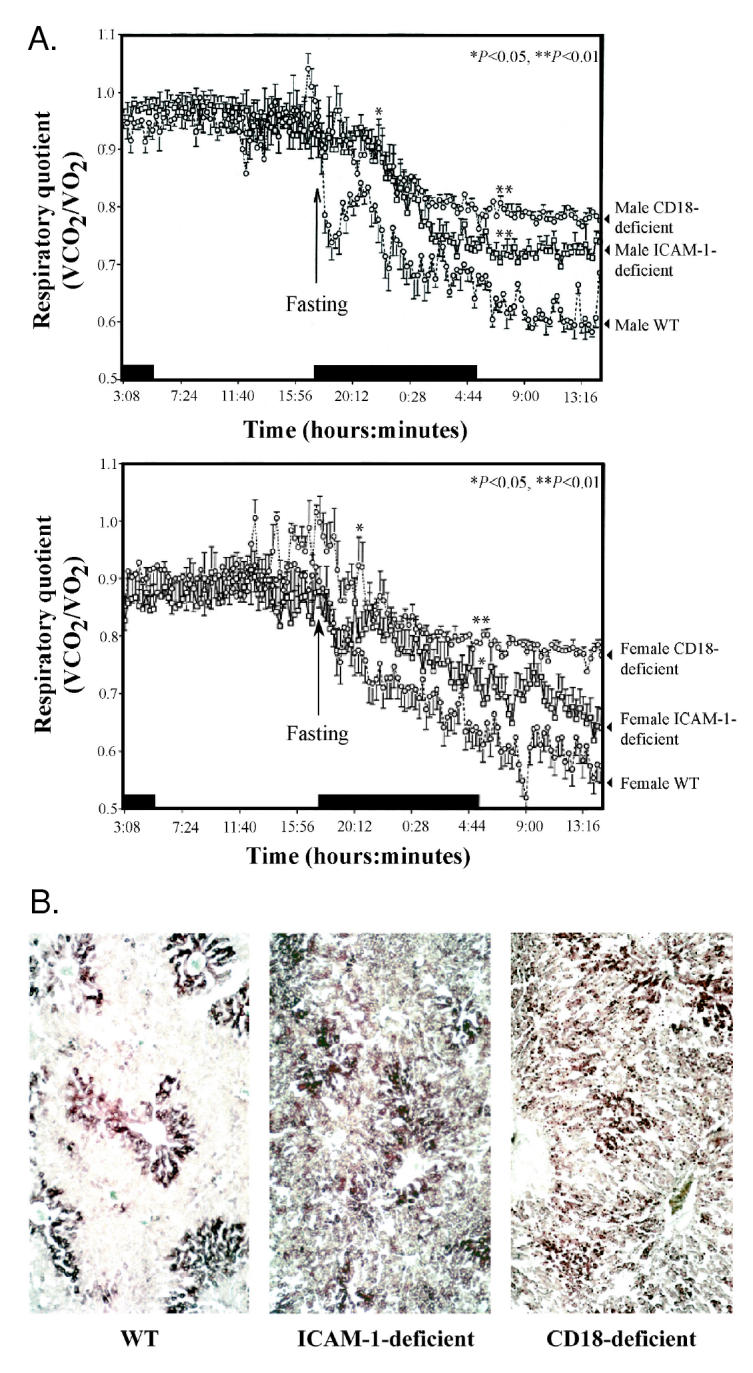
Respiratory quotient and liver fat content of WT, ICAM-1–, and CD18-deficient mice. A: Respiratory quotient was measured in male (top panel) and female (bottom panel) WT, ICAM-1–, and CD18-deficient mice in fed state and upon transition to fasting. ICAM-1– and CD18-deficient mice fail to make a prompt switch to fat oxidation during fasting compared with WT mice. Early after the onset of fasting, the difference in RQ between WT mice and either ICAM-1– or CD18-deficient mice is statistically significant (P < 0.05). After a few hours of fasting, the difference in RQ between WT mice and either ICAM-1– or CD18-deficient mice becomes even greater (P < 0.01). Each experimental group had 4 mice. Data were analyzed using ANOVA followed by Student PLSD (Protected Least Significance). Black bar indicates time periods when animals were in the dark. B: Frozen sections of livers from 24-h fasted male WT, ICAM-1–, and CD18-deficient mice were stained with Oil Red-O to visualize neutral lipids in the liver. Dark staining on the images represents lipid droplets that have retained the dye after destaining procedure. Representative sections are shown.
The mild steatosis that usually occurs with fasting can be explained by the fact that uptake of FFAs into hepatocytes exceeds the capacity for oxidation and export of triacylglycerols (15). ICAM-1–deficient or CD18-deficient mice showed marked micronodular fatty infiltration of the liver after 24-h fasting, while in fasted WT livers, only the expected minor accumulation of lipid droplets was found (see Figure 1B). Thus deletion of either ICAM-1 or CD18 molecules leads to impaired fat metabolism during fasting.
Metabolic Defects Observed in ICAM-1–Deficient Mice
To better understand the metabolic defects caused by deficiency of leukocyte adhesion molecules, we performed detailed analysis of ICAM-1–deficient mice. We chose to work with these mice as they are less immunodeficient than CD18-deficient animals and do not need to be kept on antibiotics. Serum concentration of triglycerides during fasting was significantly higher in ICAM-1–deficient mice compared with WT mice (Table 1). This shows that the ability of ICAM-1–deficient mice to secrete triglycerides from the liver was not impaired and therefore was not likely the cause of steatosis.
Table 1.
Metabolic parameters of fed and fasted WT and ICAM-1–deficient micea
| Fed
|
Fasted
|
|||||
|---|---|---|---|---|---|---|
| Metabolite measurements | WT | ICAM-1–deficient | P | WT | ICAM-1–deficient | P |
| Triglycerides (mg/dL) | 55.8 ± 5.1 | 38.7 ± 5.6 | <0.05 | 29.5 ± 6.2 | 88 ± 8.7 | <0.01 |
| FFA (meq/L) | 0.49 ± 0.04 | 0.63 ± 0.08 | >0.05 | 0.86 ± 0.07 | 1.19 ± 0.09 | <0.01 |
| β-hydroxybutyrate (mg/dL) | 0.7 ± 0.05 | 0.8 ± 0.08 | >0.05 | 25.5 ± 1.6 | 8.8 ± 1.0 | <0.01 |
| Glycogen (mg/100 g of liver tissue) | 4333 ± 135 | 4703 ± 459 | >0.05 | 218.5 ± 49.7 | 37.92 ± 15.5 | <0.01 |
Fasting induces disturbance in serum concentration and liver content of several analytes in ICAM-1–deficient mice. WT or ICAM-1–deficient mice were bled at the end of the dark cycle (9 a.m.). Data were analyzed by unpaired t test followed by normality posttest. Each experimental group had 7 to 8 mice.
Fasting Stimulates Release of FFAs from Fat Stores into Circulation
Serum FFAs were elevated in the fasted mice compared with fed mice. However, FFA level was significantly higher in fasted ICAM-1–deficient mice compared with fasted WT mice (Table 1). Greater FFAs in fasted ICAM-1–deficient mice could be caused by a lower uptake rate of FFAs by tissues and/or by the combination of decreased FFA uptake and decreased fat oxidation in these animals.
β-hydroxybutyrate is an important product of fatty acid oxidation in the liver. Formation of β-hydroxybutyrate is very low in the fed state and its serum level is greatly increased during fasting. Production of β-hydroxybutyrate in ICAM-1–deficient mice during fasting was 3-fold lower than in WT mice (Table 1). Again, this result is most consistent with the inability of ICAM-1–deficient livers to activate fat oxidation.
When an external source of glucose is absent, such as during fasting, glucose homeostasis is maintained by breakdown of glycogen and through gluconeogenesis in the liver from precursors supplied by liver, muscle, and adipose tissues. It is reasonable to expect that impaired fat oxidation during fasting would result in faster depletion of liver glycogen. We observed much lower liver glycogen content after fasting in ICAM-1–deficient mice. These results suggest that liver gluconeogenesis is impaired, causing more rapid glycogen depletion in ICAM-1–deficient mice compared with WT mice.
Changes in Leukocyte Distribution Induced by Fasting Are Modified in ICAM-1– or CD18-Deficient Mice
In malnutrition, one of the earliest changes in immune function is leukopenia due to the simultaneous decrease in granulocyte, lymphocyte, and monocyte counts (16). The mechanism of leukopenia during fasting is unknown. Both ICAM-1 and CD18 molecules play an essential role in recruitment of leukocytes to the sites of tissue damage or inflammation (5,17). Therefore, we hypothesized that deficiency of these molecules may interfere with recruitment of leukocytes to organs subjected to the highest metabolic stress during fasting. We measured differential leukocyte counts in fed and fasted mice. In 24-h fasted WT mice, peripheral neutrophil, lymphocyte, and monocyte counts were significantly decreased compared with fed mice (Figure 2A). In ICAM-1–deficient mice, fasting also produced leukopenia. However, this leukopenia was without significant decrease in neutrophil counts (see Figure 2B). In fasted CD18-deficient mice both peripheral neutrophil and monocyte counts did not change (see Figure 2C). The results indicate that these adhesion molecules affect peripheral neutrophils and monocytes counts during fasting.
Figure 2.
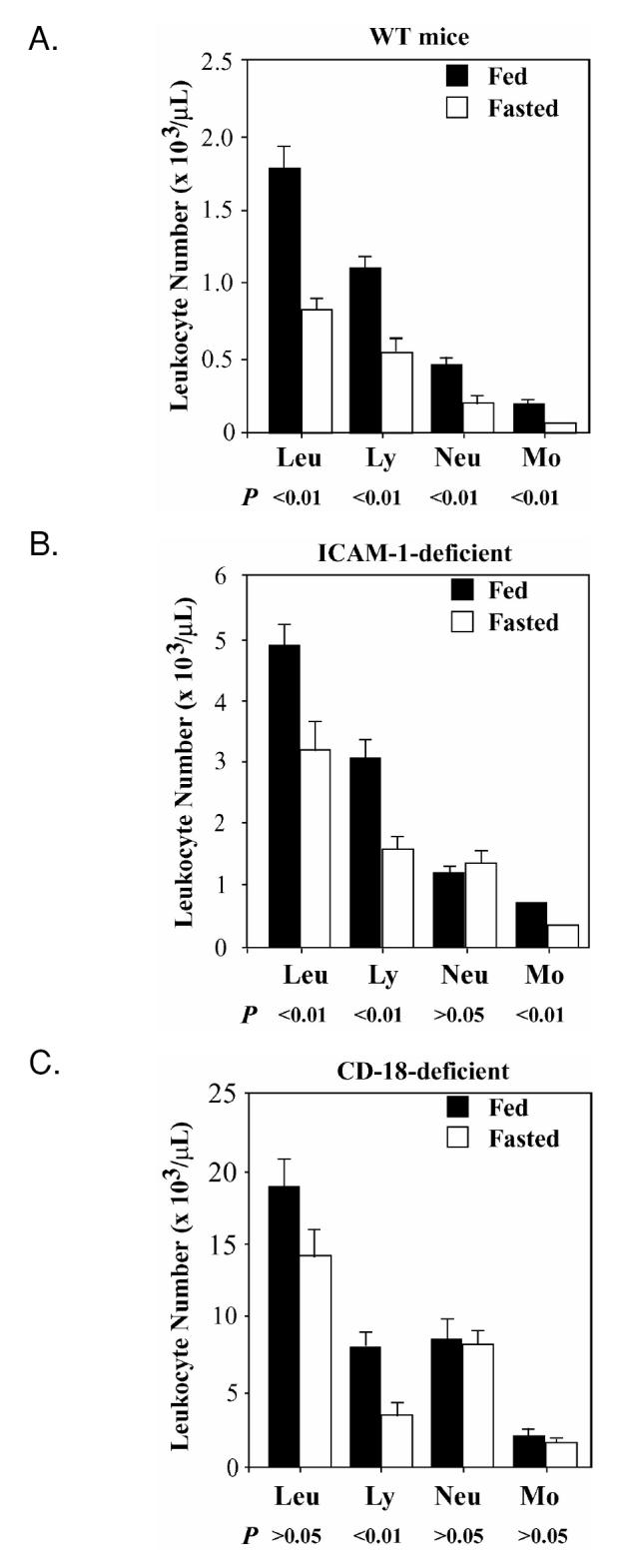
Changes in leukocyte distribution during fasting differ in ICAM-1– and CD18-deficient mice compared with WT mice. A. Peripheral blood leukocyte differential count in WT mice in fed state and after 24-h of fasting. Each experimental group had 7 mice. Data were analyzed using Mann-Whitney nonparametric test. B: Peripheral blood leukocyte differential count in ICAM-1–deficient mice that were either fed or after 24 h of fasting. Each experimental group had 7 mice. Data were analyzed using Mann-Whitney nonparametric test. C: Peripheral blood leukocyte differential count in CD18-deficient mice that were fed or after 24 h of fasting. Each experimental group had from 8 to 9 mice. Data were analyzed using nonparametric Mann-Whitney test.
The liver holds the central position coordinating the metabolic response to fasting. Previous studies showed that increased adhesion of leukocytes to endothelium of liver sinusoids is one of the earliest steps in the liver’s response to stress (18,19). Could fasting, as a form of stress, cause up-regulation of ICAM-1 expression in the liver? ICAM-1 protein level was indeed increased (P < 0.01, n = 7 to 8, paired t test) in livers of 24-h fasted (593.4 ± 37.1 ng/mg of liver protein) compared with fed WT mice (352.6 ± 24.6 ng/mg of liver protein). To measure recruitment of granulocytes and monocytes to the liver, we measured liver myeloperoxidase (MPO) content. Fasted WT mice had much higher levels of MPO compared with fed WT (Figure 3A). However, liver MPO level did not differ significantly between fed and fasted ICAM-1–deficient mice (see Figure 3A). To confirm this finding we measured the number of CD11b/CD18 + cells in frozen sections from livers of fed and fasted WT and ICAM-1–deficient mice (see Figure 3B). We obtained results that were similar to the results from the previous experiment. Thus, during fasting liver increased its expression of ICAM-1, which coincided with increased recruitment of MPO+ and CD11b/CD18+ cells.
Figure 3.
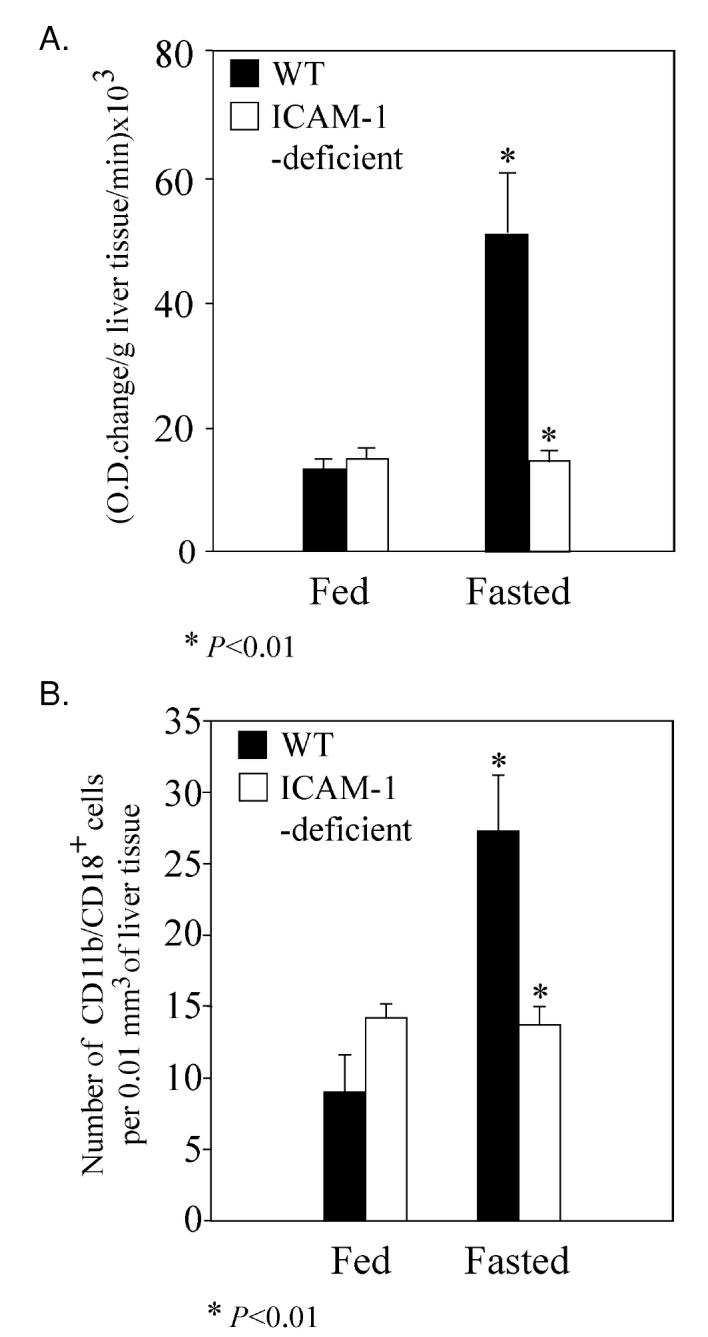
Recruitment of neutrophils and/or monocytes into the liver is impaired in ICAM-1–deficient mice compared with WT mice after 24 h of fasting. A: MPO content of mouse livers was estimated by measuring enzymatic activity. Each experimental group had from 6 to 7 mice. Statistical analysis was performed using unpaired t test followed by normality posttest. B: Numbers of CD11b/CD18 + cells in histological sections of mouse livers. Each experimental group had 6 mice. Data were analyzed using nonparametric Mann-Whitney test.
Serum Insulin and Glucose Concentrations in ICAM-1– and CD18-Deficient Mice during Fasting
Insulin plays a dominant regulatory role in the metabolic flux of glucose and lipids. In peripheral tissues, fasting low blood insulin reduces glucose uptake and utilization and stimulates lipolysis and proteolysis. Failure of insulin levels to decrease during fasting would inhibit these metabolic changes. We measured serum insulin levels in the fed and fasted mice (Figure 4A). In WT mice, serum insulin level during fasting was predictably lower compared with serum insulin level in the fed state (see Figure 4A). In contrast, serum insulin levels failed to decrease during fasting in both ICAM-1–deficient mice (P > 0.05, n = 7, see Figure 4A) and CD18-deficient mice (P > 0.05, n = 7). The fed state insulin concentrations in ICAM-1–deficient and CD18-deficient mice were similar to serum insulin concentrations in corresponding fed WT mice. In addition, fasting serum glucose levels were significantly lower in both ICAM-1–deficient and CD18-deficient mice compared with fasted WT mice, as it would be expected in hyperinsulinemic state (see Figure 4B). These results document the inability of ICAM-1– and CD18-deficient mice to appropriately respond to fasting by lowering their circulating insulin concentration.
Figure 4.
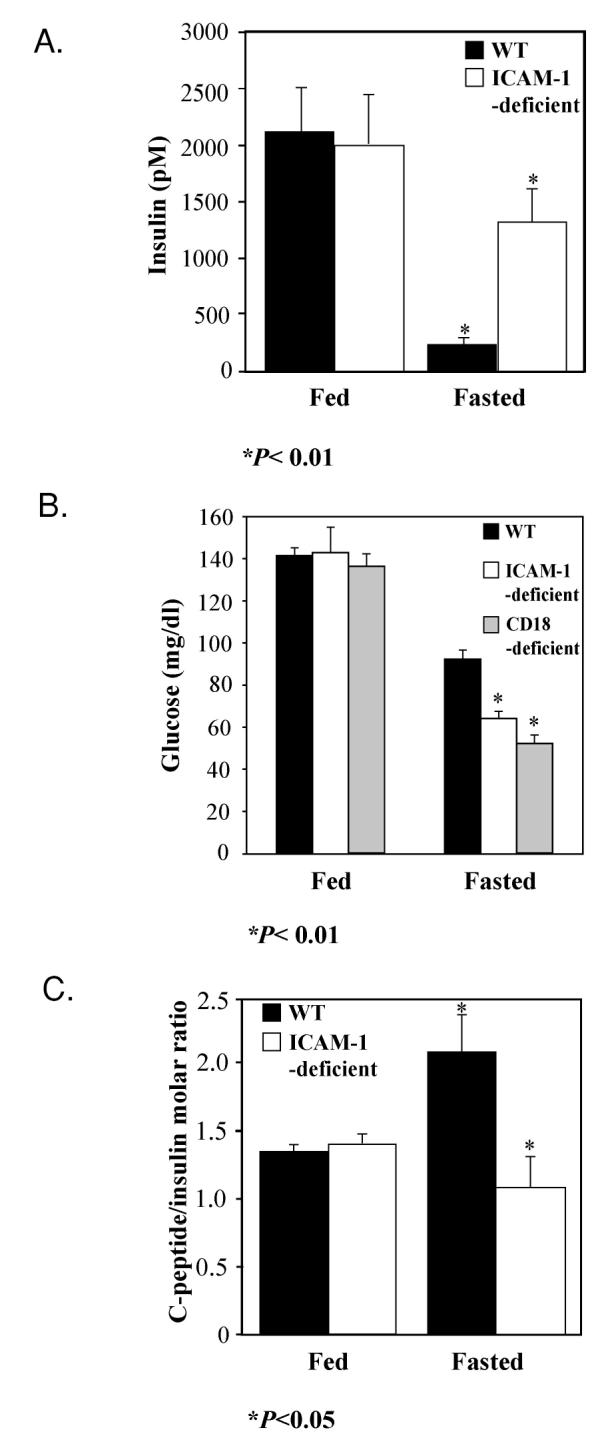
Serum glucose, insulin levels, and C-peptide/insulin ratio in fasting WT and adhesion receptor–deficient mice. A: Serum insulin concentration did not decrease significantly in ICAM-1–deficient mice after 24 h of fasting (P > 0.05), whereas it did in WT mice (P < 0.01). Each experimental group had 7 to 10 mice. Statistical analysis was performed using Kruskal-Wallis test (nonparametric ANOVA) followed by Dunn’s multiple comparisons test. B: Blood glucose concentration decreased more profoundly in ICAM-1–deficient (P < 0.01) and CD18-deficient mice (P < 0.01) than in WT mice. Each group had 6 to 10 mice. Statistical analysis was performed using Tukey-Kramer multiple comparisons test followed by Bartlett’s standard deviation normality test and Kolmogorov and Smirnov gaussian normality test. C: Serum C-peptide/insulin molar ratio after 24 h of fasting is significantly higher in WT mice than in ICAM-1–deficient mice (P < 0.05). Each experimental group had 5 to 6 mice. Statistical analysis was performed using unpaired t test followed by normality post-test.
Leukocyte Adhesion Affects Clearance of Insulin on Fasting
Blood insulin level is regulated by pancreatic insulin secretion and insulin clearance by peripheral tissues. Liver is the predominant organ clearing insulin during fasting (20). In a rat model, pancreatic insulin secretion and, consequently, portal insulin concentration shows very little diurnal variation while at the same time large differences exist in peripheral arterial and venous insulin levels (21). We hypothesized that fasting hyperinsulinemia in ICAM-1– and CD18-deficient mice may be due to impaired insulin clearance by the liver. We measured concentration of C-peptide, a pro-peptide released during biosynthesis of insulin, and insulin concentrations and calculated C-peptide/insulin ratio. Because C-peptide is cleared independently of insulin in the kidney (22), this ratio is used to estimate the liver’s ability to remove insulin (23). We observed a markedly lower C-peptide/insulin ratio in 24-h fasted ICAM-1–deficient compared with 24-h fasted WT mice (see Figure 4C). This was due to an increase in circulating insulin concentration because C-peptide levels between fasted WT (864.2 ± 127.6 pmol/L) and ICAM-1–deficient mice (1098.2 ± 326.4 pmol/L) were not different (P > 0.05, n = 9). The fed state C-peptide/insulin ratio in ICAM-1–deficient mice was similar to C-peptide/insulin ratio in corresponding fed WT mice (see Figure 4C). Our results indicate that the most likely mechanism of impaired insulin decrease during fasting is a defect in insulin clearance.
Consequences of High-Fasting Insulin Level on Key Enzymes Regulating Fat Metabolism
In WT animals, fasting causes increased ACC phosphorylation by AMP-activated protein kinase (AMPK) (24) and consequently a decreased production of malonyl-CoA, an inhibitor of fatty acid import into mitochondria. Conversely, fasting hyperinsulinemia would cause inhibition of long-chain fatty acid import into mitochondria at the level of carnitine palmitoyl transferase I (CPT I), which would result in increased ratio of free carnitine to the sum of palmitoyl-carnitine and stearoylcarnitine (CR) (25). In ICAM-1–deficient mice, CR was higher (CR = 13.27, pooled plasma from 4 mice) compared with WT mice (CR = 6.15, pooled plasma from 4 mice). To further study the effect of ICAM-1 deficiency on CPT I regulation, we evaluated the levels of phosphorylation of AMPK (active enzyme is phosphorylated) in the liver and muscle and phosphorylation of ACC (phosphorylated enzyme is inactive and the inhibition of CPT I is removed) in the muscle in ICAM-1–deficient mice, using phosphorylation-specific antibodies to these proteins. Fasted ICAM-1–deficient mice had significantly lower levels of phosphorylation of soleus muscle AMPK and ACC and liver AMPK compared with WT mice (Figure 5). At the same time, there was no significant difference between fasted WT and ICAM-1–deficient mice in their protein level of AMPK in muscle and liver tissues. We have also compared AMPK in muscle in fed state of WT and ICAM-1–deficient mice. Both genotypes showed similar weak phosphorylation of the enzyme. Because concentration of FFAs in the circulation was not a limiting factor for mitochondrial fatty acid oxidation, these results suggest that insulin inhibited fatty acid oxidation in ICAM-1–deficient mice through inhibition of AMPK activity (26) and consequently prevented the import of fatty acids into mitochondria.
Figure 5.
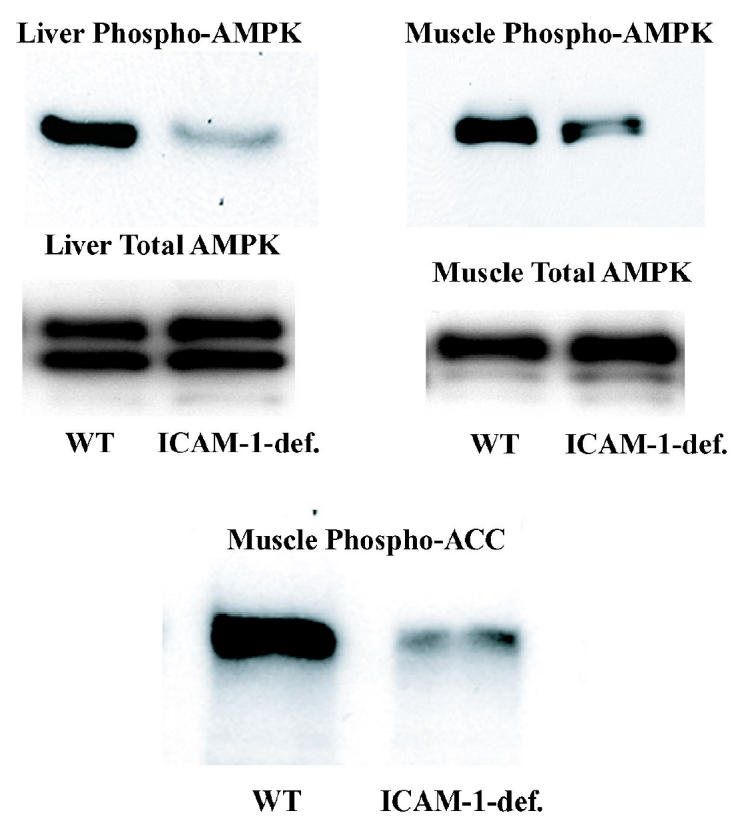
Measurement of the phosphorylation status of AMPK and ACC in fasted WT and ICAM-1–deficient mice. Western blot analysis of total protein from soleus muscles and from livers of WT or ICAM-1–deficient mice fasted for 24 h. Protein extract was made from soleus muscles and livers of WT or ICAM-1–deficient mice sacrificed at the end of the dark cycle. Antibodies used were either anti-phosphoAMPK, anti-AMPK-α, or anti-phosphoACC as indicated. Each lane contains combined protein extract from 2 mice. Each experiment was repeated 2 times.
DISCUSSION
Our results demonstrate that deficiency of either ICAM-1 or CD18 impairs physiologic response to fasting. Fasted ICAM-1–deficient and CD18-deficient mice show impaired ability to oxidize fat. This is reflected in the development of liver steatosis and inappropriately low blood levels of ketone bodies and glucose (hypoketotic hypoglycemia). We have also seen excessive weight gain in mice lacking CD11b (αM integrin) (7) and, more recently, in mice overexpressing soluble ICAM-1, which acts as an inhibitor of leukocyte recruitment (Hong-Wei Wang, manuscript submitted for publication).
One of the earliest changes in immune response during fasting is neutropenia. However, fasted ICAM-1–deficient and CD18-deficient mice do not become neutropenic. Failure to develop fasting neutropenia coincides with the failure of fasted ICAM-1–deficient mice to recruit neutrophils to the liver. ICAM-1–deficient and CD18-deficient mice exhibit fasting hyperinsulinemia, which is most likely caused by impaired liver insulin clearance during fasting (see Figure 4C). Indeed, we established for ICAM-1–deficient mice that hyperinsulinemia caused decreased AMPK phosphorylation in muscle and liver resulting in decreased import of long chain fatty acids into mitochondria and impaired fat oxidation. Thus, ICAM-1 and CD18 leukocyte adhesion receptors are necessary for the regulation of whole body fat oxidation and overall energy balance (see Figure 1A) during fasting.
The 2 main causes of fasting hypoketotic hypoglycemia in people are (1) inborn errors of fatty acid oxidation and ketone body production and (2) hyperinsulinemia. The defect in fat oxidation observed in ICAM-1– and CD18-deficient mice is in many ways similar to the fat oxidation defect observed in PPARα-deficient mice. Fasted PPARα-deficient mice show accumulation of lipid in their livers. At the same time they display hypoglycemia, hypoketonemia, and elevated plasma FFA levels, indicating inhibition of fatty acid uptake and oxidation in the liver (12). Several lines of evidence argue against errors affecting expression of enzymes of the PPARα-regulated pathway of fatty oxidation being the cause of metabolic defect in ICAM-1–deficient and CD18-deficient mice. We did not detect significant differences in fasting mRNA expression levels of either PPARα or acetyl-CoA oxidase and liver form of fatty acid binding protein, genes known to be regulated by PPARα (27), between WT and ICAM-1–deficient mice. Additionally, the in vitro ability of liver mitochondria to oxidize exogenously supplied short and long chain fatty acids to β-hydroxybutyrate was not significantly different between WT and ICAM-1–deficient mice (not shown). Similarly, we did not detect any differences in PPAR-γ levels, the nuclear receptor involved in adipogenesis (unpublished observation).
An alternative mechanism by which fat oxidation could be inhibited in fasting mice is hyperinsulinemia. Upon stimulation by nutrient secretagogues, insulin is co-secreted with C-peptide by the pancreatic β cells in equimolar amounts into portal circulation (28). Liver is the principal site where insulin is enzymatically degraded and cleared from the circulation. In contrast to insulin, C-peptide is not significantly extracted by the liver and its metabolic clearance rate is fairly constant (22,23). Therefore, clinical studies of insulin clearance use circulating C-peptide/insulin molar ratios as an index of hepatic insulin uptake (22). Several lines of evidence suggest that insulin clearance by the liver is a regulated process markedly stimulated by fasting. In humans, oral glucose administration decreases hepatic extraction of insulin as indicated by a decrease of C-peptide insulin ratio (29). Furthermore, experiments using a perfused rat pancreas-liver preparation showed that the activity of liver in degradation of insulin changes from close to 0% in the fed state to 45% in the fasted state (30). After 24-h fasting, both ICAM-1–deficient and CD18-deficient mice had serum insulin concentrations that did not differ significantly from their fed state. At the same time, fasted WT mice decreased their insulin concentration several fold compared with their fed serum insulin concentration (see Figure 4A). C-peptide concentration was not significantly different between WT and ICAM-1–deficient mice. In contrast, C-peptide/insulin molar ratio is significantly lower in ICAM-1–deficient mice compared with WT (see Figure 4C). Together, these findings suggest failure of insulin clearance as the possible cause of fasting hyperinsulinemia in ICAM-1–deficient mice and likely in CD18-deficient mice as well. In addition, the high levels of insulin in these mice explain why their body is not responding to fasting in the expected way. Insulin is indeed a very powerful regulator of metabolism. We injected insulin into fasting wild-type mice while in the metabolic chamber and their RQ transiently increased to fed-state levels (unpublished observation).
Because both ICAM-1–deficient and CD18-deficient mice have a similar defect in fat oxidation and insulin metabolism, it is reasonable to suspect that interaction between ICAM-1 and CD18, such as occurs during leukocyte adhesion, is required for physiologic regulation of insulin metabolism and fat oxidation. The fact that CD18 (β2 integrin) is a leukocyte-specific integrin implicates leukocytes and, with them, the immune system in metabolic response to fasting. Recent reports of macrophage accumulation in adipose tissue from obese people and mice (8,9) together with this report may show a new paradigm in the role of immune system in regulation of fat metabolism.
Hyperinsulinemia can inhibit fat oxidation directly through inhibition of intracellular fat oxidation pathways and indirectly through regulation of FFA availability (31). In ICAM-1–deficient mice FFA availability is not impaired (Table 1). Alternatively, examination of the free carnitine and acylcarnitine concentrations in plasma of fasted WT and ICAM-1–deficient mice suggested decreased CPT I activity. To confirm this mechanism, we measured phosphorylation level of AMPK and ACC that together control fatty acid import into mitochondria (32). Insulin antagonizes activation of AMPK and in that way inhibits phosphorylation of downstream targets of AMPK (26,31). One of the main targets of AMPK is ACC that produces malonyl-CoA, which allosterically inhibits CPT I, the main enzyme for transport of long-chain fatty acids into mitochondria. Decreased phosphorylation of ACC by AMPK sustains malonyl-CoA production and ultimately decreases fat oxidation. Our results suggest that AMPK is less active in both fasting muscle and liver cells of ICAM-1–deficient mice (see Figure 5).
Metabolic abnormalities in ICAM-1–deficient and CD18-deficient mice can be directly related to recently described hyperinsulinemia and hypoglycemia in a family with LAD I (6). LAD I is an autosomal-recessive hereditary disorder characterized by CD18 deficiency resulting in delayed umbilical cord separation, persistent granulocytosis, recurrent cutaneous abscesses, and periodontal infections, and bacterial sepsis (33). The finding of a similar metabolic phenotype in both mice and people with deficiency of the same leukocyte adhesion molecule suggests evolutionary conservation of a mechanism for leukocyte-mediated regulation of fat oxidation during fasting.
The mainstream view of regulation of metabolism during fasting holds that lack of food leads to decreased pancreatic secretion of insulin due to falling blood glucose concentration which then represents the critical signal for increase of fat oxidation. However, several reports published in the late 1970s and early 1980s suggested that, in addition, the liver had an important role in decreasing peripheral insulin concentration in the early stages of fasting, thereby helping the body to adjust to fasting stress (21,30). Our work supports the importance of insulin degradation in the normal response to fasting and furthermore suggests that this is a highly regulated process in which innate immune response plays an important role. Recently, the focus has shifted from the view of hyperinsulinemia as a compensatory mechanism for peripheral insulin resistance to an alternative view in which hyperinsulinemia in some cases could be the primary metabolic abnormality causing obesity (34), possibly through inhibition of lipolysis (35). Furthermore, emerging evidence suggests that hyperinsulinemia can cause accumulation of β-amyloid in the brain and in doing so significantly increases the risk of Alzheimer’s disease (36). With a growing interest in developing therapeutic options for treatment of obesity and Alzheimer’s disease, a deeper understanding of this new mechanism of insulin homeostasis regulated by leukocytes and their adhesion receptors will have future clinical significance.
Acknowledgments
We thank Barbara Corkey, Jeffrey Friedman, Ronald Kahn, and Harvey Lodish for helpful discussions and the many scientists who patiently listened to our story and gave us advice. We thank Leonardo Ganem for PPAR analysis. The work was supported by National Heart, Lung, and Blood Institute/NIH grant RO1-HL53756 to DDW. TWF was supported by a research stipend from the Deutsche Forschungsgemeinschaft (FE 537/1-1).
Note: Thomas Daniels and Alain Stricker-Krongrad were employees of Millennium Pharmaceuticals.
Footnotes
Online address: http://www.molmed.org>
REFERENCES
- 1.Staunton DE, Dustin ML, Erickson HP, Springer TA. The arrangement of the immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinovirus. Cell. 1990;61:243–54. doi: 10.1016/0092-8674(90)90805-o. [DOI] [PubMed] [Google Scholar]
- 2.Ho MK, Springer TA. Mac-1 antigen: quantitative expression in macrophage populations and tissues, and immunofluorescent localization in spleen. J Immunol. 1982;128:2281–6. [PubMed] [Google Scholar]
- 3.Kurzinger K, Reynolds T, Germain RN, Davignon D, Martz E, Springer TA. A novel lymphocyte function-associated antigen (LFA-1): cellular distribution, quantitative expression, and structure. J Immunol. 1981;127:596–602. [PubMed] [Google Scholar]
- 4.Xu H, et al. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J Exp Med. 1994;180:95–109. doi: 10.1084/jem.180.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scharffetter-Kochanek K, Lu H, Norman K, et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998;188:119–31. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suliaman F, Jabbar MA. The association of leukocyte adhesion defect type I and persistent hyperinsulinemic hypoglycemia of infancy in a Saudi Arabian family. Pediatr Hematol Oncol. 2002;19:429–32. doi: 10.1080/08880010290097206. [DOI] [PubMed] [Google Scholar]
- 7.Dong ZM, Gutierrez-Ramos JC, Coxon A, Mayadas TN, Wagner DD. A new class of obesity genes encodes leukocyte adhesion receptors. Proc Natl Acad Sci USA. 1997;94:7526–30. doi: 10.1073/pnas.94.14.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehrke M, Lazar MA. Inflamed about obesity. Nat Med. 2004;10:126–7. doi: 10.1038/nm0204-126. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz MW, Seeley RJ. Seminars in medicine of the Beth Israel Deaconess Medical Center. Neuroendocrine responses to starvation and weight loss. New Engl J Med. 1997;336:1802–11. doi: 10.1056/NEJM199706193362507. [DOI] [PubMed] [Google Scholar]
- 12.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–98. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roe JH, Dailey RE. Determination of glycogen with the anthrone reagent. Anal Biochem. 1966;15:245–50. doi: 10.1016/0003-2697(66)90028-5. [DOI] [PubMed] [Google Scholar]
- 14.Schutz Y. The basis of direct and indirect calorimetry and their potentials. Diabetes Metab Rev. 1995;11:383–408. doi: 10.1002/dmr.5610110406. [DOI] [PubMed] [Google Scholar]
- 15.Yasuhara M, et al. Induction of fatty liver by fasting in suncus. J Lipid Res. 1991;32:887–91. [PubMed] [Google Scholar]
- 16.Drenick EJ, Alvarez LC. Neutropenia in prolonged fasting. Am J Clin Nutr. 1971;24:859–63. doi: 10.1093/ajcn/24.7.859. [DOI] [PubMed] [Google Scholar]
- 17.Lloyd CM, Gonzalo JA, Salant DJ, Just J, Gutierrez-Ramos JC. Intercellular adhesion molecule-1 deficiency prolongs survival and protects against the development of pulmonary inflammation during murine lupus. J Clin Invest. 1997;100:963–71. doi: 10.1172/JCI119647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhabhar FS. Acute stress enhances while chronic stress suppresses skin immunity. The role of stress hormones and leukocyte trafficking. Ann NY Acad Sci. 2000;917:876–93. doi: 10.1111/j.1749-6632.2000.tb05454.x. [DOI] [PubMed] [Google Scholar]
- 19.Volpes R, van den Oord JJ, Desmet VJ. Immunohistochemical study of adhesion molecules in liver inflammation. Hepatology. 1990;12:59–65. doi: 10.1002/hep.1840120110. [DOI] [PubMed] [Google Scholar]
- 20.Duckworth WC, Hamel FG, Peavy DE. Hepatic metabolism of insulin. Am J Med. 1988;85:71–6. doi: 10.1016/0002-9343(88)90399-3. [DOI] [PubMed] [Google Scholar]
- 21.Balks HJ, Jungermann K. Regulation of peripheral insulin/glucagon levels by rat liver. Eur J Biochem. 1984;141:645–50. doi: 10.1111/j.1432-1033.1984.tb08240.x. [DOI] [PubMed] [Google Scholar]
- 22.Polonsky K, et al. Metabolism of C-peptide in the dog. In vivo demonstration of the absence of hepatic extraction. J Clin Invest. 1983;72:1114–23. doi: 10.1172/JCI111036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castillo MJ, Scheen AJ, Letiexhe MR, Lefebvre PJ. How to measure insulin clearance. Diabetes Metab Rev. 1994;10:119–50. doi: 10.1002/dmr.5610100205. [DOI] [PubMed] [Google Scholar]
- 24.Munday MR, Milic MR, Takhar S, Holness MJ, Sugden MC. The short-term regulation of hepatic acetyl-CoA carboxylase during starvation and re-feeding in the rat. Biochem J. 1991;280 (Pt 3):733–7. doi: 10.1042/bj2800733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fingerhut R, et al. Hepatic carnitine palmitoyltransferase I deficien-cy:acylcarnitine profiles in blood spots are highly specific. Clin Chem. 2001;47:1763–8. [PubMed] [Google Scholar]
- 26.Witters LA, Kemp BE. Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5′-AMP-activated protein kinase. J Biol Chem. 1992;267:2864–7. [PubMed] [Google Scholar]
- 27.Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res. 1996;37:907–25. [PubMed] [Google Scholar]
- 28.Zavaroni I, Deferrari G, Lugari R, et al. Renal metabolism of C-peptide in man. J Clin Endocrinol Metab. 1987;65:494–8. doi: 10.1210/jcem-65-3-494. [DOI] [PubMed] [Google Scholar]
- 29.Tranberg KG, Thorell J. Variation in the disappearance of unlabeled insulin from plasma: studies with portal and peripheral infusions. Diabetes. 1979;28:846–51. doi: 10.2337/diab.28.9.846. [DOI] [PubMed] [Google Scholar]
- 30.Striffler JS, Curry DL. Effect of fasting on insulin removal by liver of perfused liver-pancreas. Am J Physiol. 1979;237:E349–355. doi: 10.1152/ajpendo.1979.237.4.E349. [DOI] [PubMed] [Google Scholar]
- 31.Beauloye C, et al. Insulin antagonizes AMP-activated protein kinase activation by ischemia or anoxia in rat hearts, without affecting total adenine nucleotides. FEBS Lett. 2001;505:348–52. doi: 10.1016/s0014-5793(01)02788-0. [DOI] [PubMed] [Google Scholar]
- 32.Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol. 1999;277:E1–10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- 33.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–94. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 34.Sigal RJ, El-Hashimy M, Martin BC, Soeldner JS, Krolewski AS, Warram JH. Acute postchallenge hyperinsulinemia predicts weight gain: a prospective study. Diabetes. 1997;46:1025–9. doi: 10.2337/diab.46.6.1025. [DOI] [PubMed] [Google Scholar]
- 35.Arner P. Control of lipolysis and its relevance to development of obesity in man. Diabetes Metab Rev. 1988;4:507–15. [PubMed] [Google Scholar]
- 36.Taubes G. Neuroscience. Insulin insults may spur Alzheimer’s disease. Science. 2003;301:40–1. doi: 10.1126/science.301.5629.40. [DOI] [PubMed] [Google Scholar]