

Abstract
Prion diseases such as Creutzfeldt-Jakob disease are believed to result from the misfolding of a widely expressed normal cellular prion protein, PrPc. The resulting disease-associated isoforms, PrPSc, have much higher β-sheet content, are insoluble in detergents, and acquire relative resistance to proteases. Although known to be highly aggregated and to form amyloid fibrils, the molecular architecture of PrPSc is poorly understood. To date, it has been impossible to elicit antibodies to native PrPSc that are capable of recognizing PrPSc without denaturation, even in Prn-Pº/º mice that are intolerant of it. Here we demonstrate that antibodies for native PrPc and PrPSc can be produced by immunization of Prn-Pº/º mice with partially purified PrPc and PrPSc adsorbed to immunomagnetic particles using high-affinity anti-PrP monoclonal antibodies (mAbs). Interestingly, the polyclonal response to PrPSc was predominantly of the immunoglobulin M (IgM) isotype, unlike the immunoglobulin G (IgG) responses elicited by PrPc or by recombinant PrP adsorbed or not to immunomagnetic particles, presumably reflecting the polymeric structure of disease-associated prion protein. Although heat-denatured PrPSc elicited more diverse antibodies with the revelation of C-terminal epitopes, remarkably, these were also predominantly IgM suggesting that the increasing immunogenicity, acquisition of protease sensitivity, and reduction in infectivity induced by heat are not associated with dissociation of the PrP molecules in the diseased-associated protein. Adsorbing native proteins to immunomagnetic particles may have general applicability for raising polyclonal or monoclonal antibodies to any native protein, without attempting laborious purification steps that might affect protein conformation.
INTRODUCTION
The prion diseases are a closely related group of invariably fatal neurodegenerative disorders that affect both humans and animals (1). The human diseases comprise kuru, Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), and sporadic and familial fatal insomnia (FFI). GSS, FFI, and 10% to 15% of CJD are dominantly inherited disorders associated with specific mutations in the prion protein gene (PRNP) (2), whereas 85% to 90% of CJD occurs sporadically. In animals, they include scrapie, affecting sheep and goats, transmissible mink encephalopathy, chronic wasting disease of mule deer and elk (3), and bovine spongiform encephalopathy (BSE) (4). The appearance in UK cattle of BSE, which rapidly evolved into a major epidemic (5), is widely attributed to the transmission of sheep scrapie to cattle via contaminated feed prepared from rendered carcasses (6). A specific prion strain causing BSE in cattle is believed to be the aetiological agent responsible for the novel human prion disease, variant Creutzfeldt-Jakob disease (vCJD) (7–9).
Prion diseases are believed to result from the posttranslational conversion of a normal cell membrane sialoglycoprotein PrPc, composed primarily of α-helical structure, into a disease specific isoform, PrPSc, that is rich in β-sheet and partially proteinase-resistant (10). Experimental models of scrapie have demonstrated that PrPc plays a central role in the pathogenesis of prion disease (11). Generation of Prn-Pº/º mice (12,13) has established the fact that PrPc is necessary for generating infectious particles as well as for developing neurodegenerative disease (12,14).
PrPSc accumulates rapidly in lymphoid tissues following experimental peripheral infection of rodents and sheep (15) well before it is detected in the central nervous system (CNS). Secondary lymphoid tissues of patients with vCJD contain readily detectable amounts of PrPSc allowing ante-mortem diagnosis (16), or in sheep affected with scrapie by 3rd eyelid immunostaining (17). Tonsil biopsy has also successfully been used to detect PrPSc in sheep (18) and in humans with vCJD (19).
Attempts have been made by several groups to develop reagents that specifically bind PrPSc, including anti-PrP antibodies (20–24), human plasminogen (25), RNA aptamers (26), β-sheet breaker peptides (27), and sodium phosphotungstate (28). Most recently monoclonal antibodies have been generated to the prion Tyr-Tyr-Arg (YYR) repeat motif that shows selectivity for disease-associated prion protein (24). In PrP-sufficient animals (Prn-P+/+), both humoral and cellular immune responses to prion protein are not induced during the course of natural prion disease (29). In fact the immune system appears to propagate the disease (30). No antibody responses can be elicited in experimentally infected mice (29) partly because of the widespread expression of PrPc in the immune system (31). However PrPSc has not been considered immunogenic (32), possibly because antigen-presenting cells (APC) are incapable of processing the highly aggregated, protease-resistant protein. Classical immunization techniques applied to raise specific antibodies have generally been unsuccessful, and until recently, only a handful of monoclonal antibodies (mAbs) had been produced that recognized any form of PrP (33). Moreover, when purified PrPSc has been used to elicit responses, the mAbs subsequently characterized for example, 3F4, have little or no affinity for native PrPSc (33).
In this report, we show that Prn-Pº/º mice immunized with partially purified native PrPSc immunoadsorbed to Dynabeads using anti-PrP monoclonal antibodies raised in our laboratory (34) produce an immunoglobulin M (IgM) antibody response unlike the immunoglobulin G (IgG) responses elicited by normal cellular prion protein isoforms. Thus polymeric proteins induce IgM responses in much the same way as do other polymeric immunogens, such as the polysaccharides associated with bacterial cell membranes (35). Given the scarcity of reagents that recognize native PrPSc and the urgent need to develop diagnostic tests for prion diseases such as CJD, antibodies generated using this approach should have wide applicability.
MATERIALS AND METHODS
Mice
Mice with ablation of both alleles of the single copy Prnp gene (Prn-Pº/º [36]) backcrossed onto an FVB/N strain background (Harlan-Olac UK) were used for these experiments. All animals were housed in the Prion Immunology facility at Imperial College, Charing Cross campus. Procedures involving experimental animals were carried out under project and personal license authority issued in accordance with the UK Animals (Scientific Procedures) Act 1986. FVB/N Prn-Pº/º mice were subcutaneously (S/C) immunized with emulsified antigen at day 0 and at day 21 (in incomplete Freund’s adjuvant [IFA] (Sigma, Dorset, UK).
Preparation of Native PrP Immunogens
Brains from Rocky Mountain Laboratory (RML) prion-infected terminally sick mice were homogenized in phosphate-buffered saline (PBS) (10% w/v) using a Ribolyser (Hybaid, Ashford, UK). The homogenate was split into aliquots and frozen at −70 °C until use. The infectious titer of the homogenate was determined as 8.1 log LD50/g brain by infectivity bioassay in tga20 mice as described previously (37).
Monoclonal Antibodies
ICSM anti-PrP mAbs were produced as described (34). ICSM 35 binds both normal and disease-associated prion protein isoforms from all species tested, whether the protein is in a native conformation or denatured. Its epitope lies between residues 91–110 (data not shown). ICSM 18 binds PrPc strongly in its native state, weakly to native PrPSc, and to both isoforms in Western blots. Its epitope lies between codons 144–156.
Recombinant α and β PrP Production
The production and purification of recombinant prion protein (rPrP) was performed as described (38).
Murine Synthetic Peptides
Synthetic peptides were made by automated solid phase stepwise synthesis using Fmoc N-terminal protection chemistry (ABC Laboratories, Charing Cross campus, Imperial College, London, UK). They were purified to greater than 98% purity by reverse phase high-performance liquid chromatography and were analyzed by MALDI mass spectrometry. Purified fractions were freeze-dried and then reconstituted in either water or PBS prior to use. The murine peptides found in Table 1 were synthesized.
Table 1.
Peptide sequences
| 91 | GGGTHNQWNKPSKPKTNLKH | 110 |
| 101 | PSKPKTNLKHVAGAAAAGAV | 120 |
| 111 | VAGAAAAGAVVGGLGGYMLG | 130 |
| 121 | VGGLGGYMLGSAMSRPMIHF | 140 |
| 131 | SAMSRPMIHFGNDWEDRYYR | 150 |
| 141 | GNDWEDRYYRENMYRYPNQV | 160 |
| 151 | ENMYRYPNQVYYRPVDQYSN | 170 |
| 161 | YYRPVDQYSNQNNFVHDCVN | 180 |
| 171 | QNNFVHDCVNITIKQHTVTT | 190 |
| 181 | ITIKQHTVTTTTKGENFTET | 200 |
| 191 | TTKGENFTETDVKMMERVVE | 210 |
| 201 | DVKMMERVVEQMCVTQYQKE | 220 |
| 211 | QMCVTQYQKESQAYYDGRRS | 230 |
Peptide and Recombinant PrP Enzyme-Linked Immunosorbent Assay (ELISA)
Synthetic peptides (50 μL of a 10 μg/mL peptide solution dissolved in 35 mM NaHCO3, 15 mM Na2CO3, pH 9.6) and 50 μL of a 10 μg/mL solution of recombinant murine PrP91–231 similarly dissolved were absorbed respectively to high and medium binding, 96 well plates (Greiner) for 1 h at 37 °C. The plates were then washed 3 times with PBS-0.05% Tween and blocked with RPMI (Gibco, Paisley, UK)/10% fetal calf serum (RF10) for 1 h at room temperature. After decanting RF10, 50 μL of the relevant diluted serum sample and positive control mAb was added and incubated for 1 h at 37°C. The plates were then washed 3 times with PBS-0.05% Tween and a 1/1000 dilution of horseradish-peroxidase–conjugated anti-mouse Ig (Serotec, Oxford, UK) added for 25 min at 37°C. The plates were again washed 4 times with PBS-0.05% Tween, and finally, the substrate reaction was induced with o-phenylenediamine dihydrochloride (OPD) buffer and stopped with 3 M sulphuric acid prior to spectrophotometric analysis at 490 nm.
Ig Isotype ELISA
Isotype ELISA was performed using a kit purchased from Becton Dickinson (Oxford, UK) with slight variation of the protocol. Briefly, medium binding, 96 well plates (Greiner) were coated with 50 μL of appropriate isotype-specific rat anti-mouse purified mAb solution in PBS. The plates were incubated for 1 h at 37 °C, washed 3 times with PBS-0.05% Tween, and then blocked with 1.0% bovine serum albumin in PBS for 1 h at room temperature. After decanting the blocking buffer, 100 μL of the relevant sera (1 in 100 dilution) was added and incubated for 1 h at 37°C. The plates were then washed 3 times with PBS-0.05% Tween, and a 1/100 dilution of horseradish-peroxidase–labeled anti-mouse Ig solution (Becton Dickinson) was added for 25 min at 37 °C, followed by 4 washes with PBS-0.05% Tween. Finally, the substrate reaction was induced and then stopped with 2 N NaOH prior to spectrophotometric analysis at 405 nm.
Immunoprecipitation
Immunoprecipitation of PrP from murine brain tissues was performed as described (34). Briefly, brain tissues from mice were homogenized (10% w/v in PBS) using a Ribolyser (Hybaid) and centrifuged at 1000g. The supernatants were stored at −70 °C until use. For the purpose of immunoprecipitation, brain homogenates were diluted to 0.5% in lysis buffer with protease inhibitors (Roche Biochemicals). The mixture was then incubated (1:1 dilution) with mAb antibody supernatant for 2 h, rotated continuously at 4 °C. The immune complexes were then adsorbed overnight to protein G Dynabeads (Dynal) rotated continuously at 4 °C. The Dynabeads were then washed. The PrP adsorbed mAb-coated Dynabeads were resuspended in Laemmli buffer (39) and heated to 100 °C for 5 min. The beads were finally pelleted, and the supernatants used for subsequent Western blotting.
Western Blot Analysis
Heat-denatured samples were separated by electrophoresis through 12% polyacrylamide gels, electrotransferred onto polyvinylidene difluoride membranes, (Millipore, Watford, UK) and immunoblotted with 0.2 μg/mL of biotinylated ICSM 18. The membranes were blocked for 1 h using 5% nonfat dried milk in PBS-Tween 20 (0.05%) (PBST). After 4 washes in PBST, membranes were incubated (1 h at RT) with a 1/5000 dilution of streptavidin-horseradish peroxidase (Sigma). Immunoreactivity was visualized with an enhanced chemiluminescence kit on autoradiographic films (ECL+; Amersham Biosciences, Chalfont St Giles, UK).
Adsorption of PrPSc to Dynabeads for Immunization
Sheep anti-mouse IgG coupled 4.5 μm immunomagnetic particles (Dynabeads M450) were washed 4 times in sterile PBS to remove preservatives, resuspended in varying concentrations of ICSM anti-PrP mAb, and rotated continuously overnight at 4 °C. After 7 washes to remove unbound mAb, the Dynabeads were resus-pended at 107/mL in sterile PBS, then stored at 4 °C until use. To adsorb PrP, 1 × 107 mAb-coated Dynabeads were incubated with 100 μL of varying concentrations of scrapie-infected brain homogenate for at least 2 h at room temperature. The final immunoisolate was washed 7 times and resuspended in PBS. This immunocomplex, hereafter called PrP-Dynabeads, was subsequently used for immunizations. To emulsify the antigen, an equal volume (1:1 v/v) of both complete Freund’s adjuvant (CFA) and the PrP solution adsorbed or not to Dynabeads was aspirated into 2 separate 5-mL glass syringes with a Luer lock attachment, and the mixture was passed rapidly back and forth between the syringes a few dozen times.
To determine conditions for maximal specific adsorption, we optimized the concentration of the anti-PrP mAb ICSM 35 required to saturate the sheep anti-mouse Dynabeads (107 beads/mL) by varying the binding concentration and then using the washed mAb-coated beads to immunoprecipitate the same quantity of PrPSc derived from proteinase K (PK)–digested murine scrapie brain homogenates (Figure 1). These experiments indicated that mAb coupling concentrations equaling 5 × 10−2 mg/mL saturated the beads. We also determined that 2.5 × 106 PrP-Dynabeads loaded onto each lane gave a readily detectable signal in standard Western blots using biotinylated ICSM 18 for detection.
Figure 1.
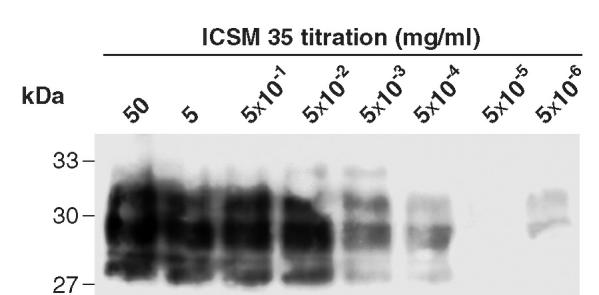
Adsorption of PrPSc is proportional to ICSM 35 concentration. Decreasing 10-fold dilutions of ICSM 35 (starting at 50 mg/mL) were adsorbed to Dynabeads as described in Materials and Methods. Next 107 of these beads were added to 50 μL of a 10% RML brain homogenate spiked into an equal volume of 10% Prn-Pº/º brain homogenate. Representative of 3 experiments.
We next titrated in increasing amounts of PrPSc to achieve maximal specific antigen loading onto Dynabeads saturated with ICSM 35, keeping the concentration of other brain constituents constant by spiking increasing quantities of scrapie brain homogenate into decreasing volumes of homogenates (at equivalent w/v) derived from Prn-Pº/º mice (Figure 2). Again PrPSc binding to the bead surface was highly dependent on the presence of the capturing mAb, although at the highest concentrations used, some non-specific adsorption to the surface of the Dynabeads was evident (see Figure 2). Generally 100 μL of a relevant 1% brain homogenate was used to adsorb native PrPC and PrPSc for immunization. When the supernatant from which PrP had been adsorbed to the Dynabeads was assayed for residual PrPSc by Western blotting, only partial depletion of the protein was observed under adsorption conditions used. In other experiments (not shown), increasing the bead numbers depleted the homogenate of PrPSc.
Figure 2.
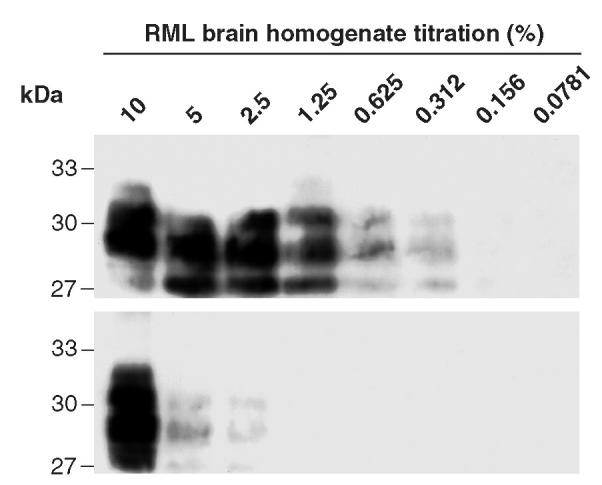
Optimal detection and depletion of PrPSc from homogenized brain. RML brain homogenate spiked into brain homogenate derived from Prn-Pº/º mice was proteinase K (PK)-treated then incubated with 107 ICSM 35 saturated Dynabeads (upper panel). In the lower panel, ICSM 35 was omitted from the immunocomplex, keeping the other conditions identical. Representative of 3 experiments.
RESULTS
Polyclonal Responses
In preliminary experiments, PrPSc-Dynabeads elicited readily detectable polyclonal (pAb) anti-PrP responses in Prn-Pº/º mice. We also found that PrPc and rPrP were highly immunogenic when adsorbed to Dynabeads. After the mice were immunized with bead-adsorbed antigens (PrPc, PrPSc, and rPrP) in CFA then IFA (Sigma) on 2 occasions 3 wk apart, specific antibody was assayed using recombinant PrP and synthetic peptide PrP, thereby confirming the specificity of the pAb for prion protein (Figure 3 and 5).
Figure 3.
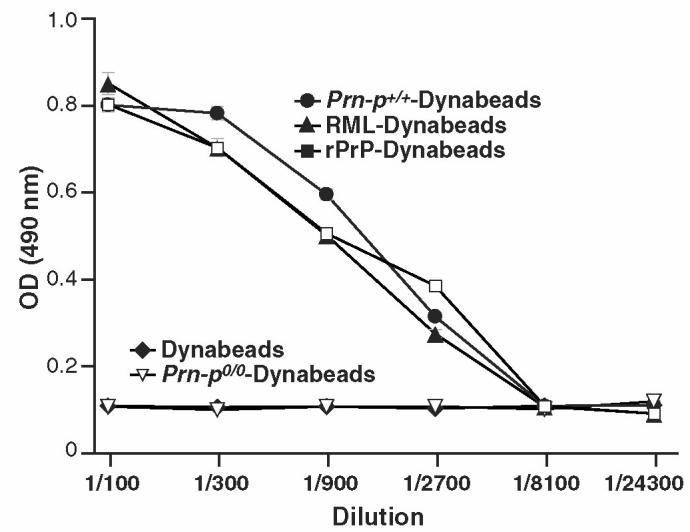
Polyclonal antibody responses to bead-adsorbed prion proteins including normal (•) and RML (▴) brain homogenates, and rPrP (▪) in FVB-N Prn-Pº/º mice (n = 3). Several specificity controls were used including omitting the homogenate/antigen (♦), adsorbing antigens from Prn-Pº/º mice (▿), or using an irrelevant isotype-matched mAb control for adsorption.
Figure 5.
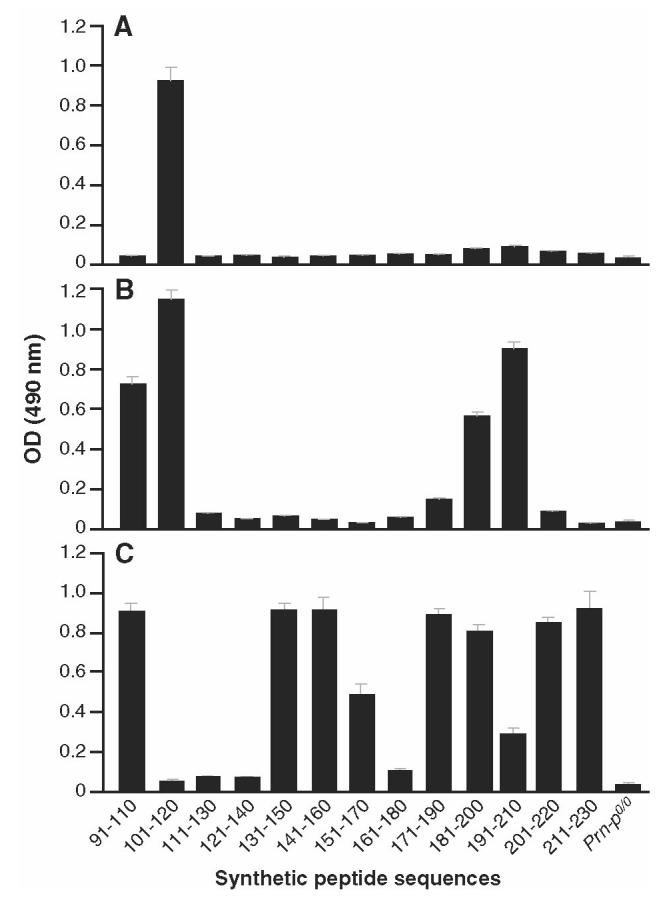
Pep-scan of sera generated with bead-bound prion proteins. A: PrPSc-Dynabeads sera from Prn-Pº/º mice were assayed against 20-mer peptides spanning the 91–231 region of the mouse PrP protein. B: PrPSc-Dynabeads (heat-treated prior to adsorption) sera from Prn-Pº/º mice were assayed against 20-mer peptides spanning the 91–231 region of the mouse PrP protein. C: PrPc-Dynabeads sera from Prn-Pº/º mice were assayed against 20-mer peptides spanning the 91–231 region of the mouse PrP protein.
We then determined whether responses to PrPSc were dependent on using Dynabead-adsorbed antigen, because in other experiments (not shown), we found that as little as 100 μL of a 1% normal brain homogenate elicited readily detectable anti-PrP responses in Prn-Pº/º mice. A 1/100 dilution of the sera was used to screen rPrP in ELISA. Although very weak responses to PrPSc emulsified in adjuvants were evident (Figure 4), robust responses were induced only when the antigen was adsorbed to the ICSM 35-Dynabeads (see Figure 4). As expected from the above experiments, responses were undetectable when the ICSM mAbs were either omitted or substituted with an irrelevant isotype matched control mAb (BRIC 126; see Figure 4) (40). These results highlight the need for solid-phase immobilization/concentration of the antigen when raising antibodies to native PrPSc.
Figure 4.
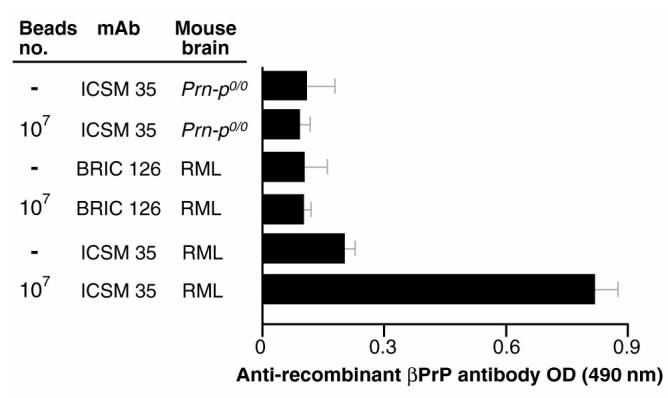
Polyclonal antibodies from mice immunized with PrPSc-Dynabeads recognize mouse rβPrP in ELISA. Anti-PrP responses were assayed in mice (n = 5 for each arm) immunized with RML (PrPSc) or Prn-Pº/º brain homogenates adsorbed or not to antibody (ICSM or an isotype control mAb BRIC 126)-coated sheep anti-mouse Dynabeads-M450. PrP was adsorbed from 100 μL of a 1% PK-digested homogenate, or for arms without Dynabeads, 100 μL of a 1% homogenate emulsified in Freunds was used for immunization. Sheep anti-mouse Dynabeads-M450 coated with anti-PrP (ICSM 35) or an isotype-match control mAb (BRIC 126) were used to adsorb after proteinase K digestion. After omission of the Dynabeads, anti-PrP (ICSM 35) or isotype-match control mAb (BRIC 126) were used to bind RML (PrPSc) or Prn-Pº/º brain homogenates. Serum were assayed at 1:100 dilution. The assay negative control (Prn-Pº/º mouse serum) gave optical density (OD) values of 0.1 ± 0.03.
Given that PrPSc has previously been thought not to be immunogenic and that we had found that responses to soluble PrPc were inducible, it was possible that the immune response induced by PrPSc-Dynabeads was focused on small quantities of PrPc that had not been adequately digested by proteinase K. We therefore looked for qualitative differences in the pAb response to normal and disease-associated PrP isoforms using peptide ELISA and also by determining the Ig-subclass of the responding antibodies. The peptide ELISA, which used overlapping 20-mer spanning the mouse PrP sequence starting from codon 91, demonstrated that the response to PrPSc-Dynabeads was highly focused compared with responses against PrPc-Dynabeads (Figure 5c) and to the α isoform of rPrP (data not shown), with most of the antibody recognizing the PrP 101–120 region (see Figure 5a). However, it should be noted that many conformational epitopes are not mapped using peptide ELISA. In fact, pre-binding of PrPSc-Dynabead serum to peptide 101–120 did not appreciably reduce pAb binding to recombinant PrP91–231 (data not shown). Interestingly, responses to Dynabead-adsorbed PrPSc that had 1st been heat treated to partially denature it (95 °C for 15 min) were more heterogeneous with more C-terminal PrP regions recognized (see Figure 5b), similar to responses to PrPc-Dynabeads (see Figure 5c) and to monomeric recombinant α-PrP (data not shown).
Further evidence of the specificity of the antibody responses to PrPSc was sought by determining the isotype of the induced anti-PrP pAb. Extensive experience of using recombinant α and β PrP to produce monoclonal antibodies indicates that IgG responses are predominantly induced by these soluble immunogens (34). We therefore measured the isotype of polyclonal antibodies induced by native and recombinant bead-bound PrP. Interestingly, the pAbs raised against α rPrP were of the IgG1 isotype and mainly IgG1/2a/b when mice were immunized with β rPrP, whether or not the immunogen was adsorbed to ICSM 35-Dynabeads (Figure 6a). IgG responses were similarly induced by PrPc-Dynabeads (see Figure 6c). However in contrast, responses to PrPSc-Dynabeads were predominantly of the IgM isotype (see Figure 6b), indicating the profound effect of protein polymerization and/or conformation on the quality of the immune response. The predominantly IgM anti-PrPSc responses were clearly not related to targeting solid-phase antigen to APCs, as the subclass of antibodies induced by soluble and bead-bound recombinant α and β PrP were identical (see Figure 6a). Repeated immunization with PrPSc-Dynabeads failed to induce class switching from IgM to IgG anti-PrP (data not shown).
Figure 6.
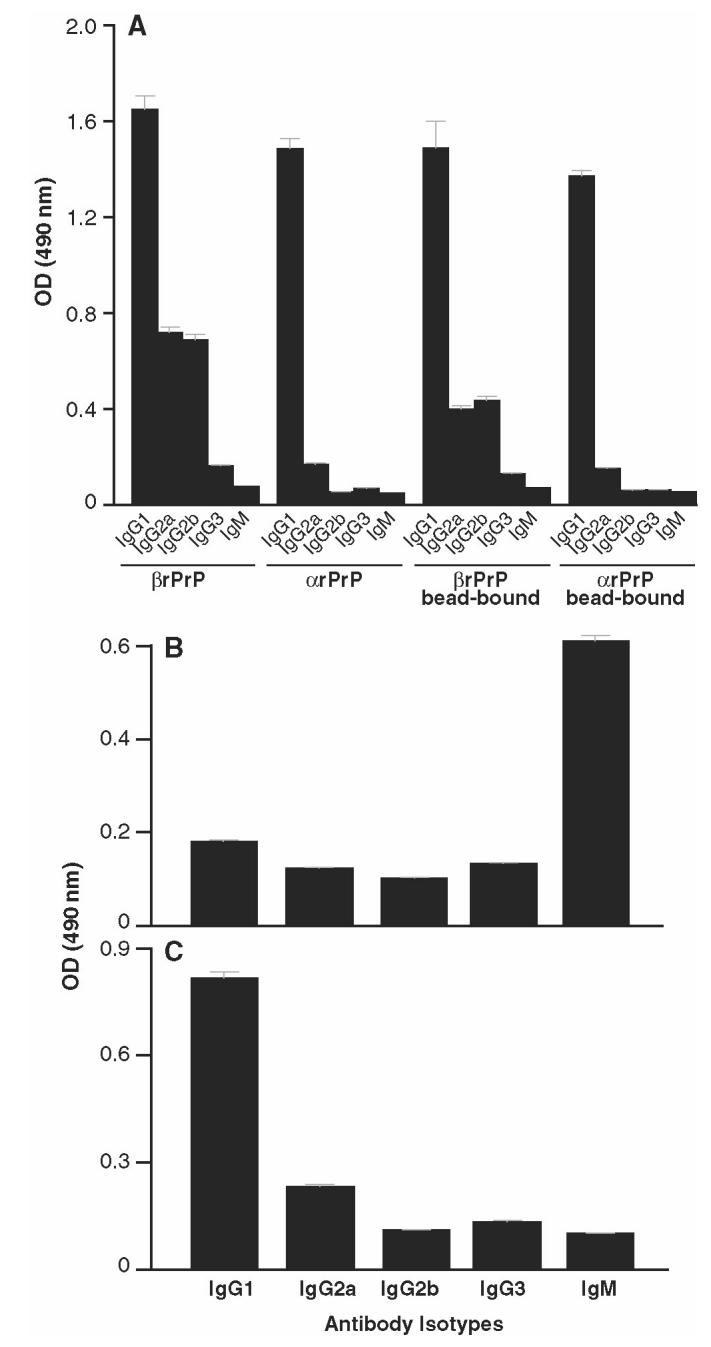
Isotype profile of anti-PrP polyclonal responses. FVB-N Prn-Pº/º mice were immunized with either soluble or bead-bound recombinant α or β PrP91–231 (panel A), PrPSc-Dynabeads (panel B), or PrPc-Dynabeads (panel C), and the isotype of anti-PrP produced was determined using Ig subclass–specific secondary antibodies in the standard recombinant PrP ELISA (see Materials and Methods). Data points represent the mean serum OD values of 1.24 ± 0.41, derived from 5 mice immunized with each antigen. Sera from unimmunized mice or mice similarly immunized with CFA/IFA emulsified with PBS gave OD values < 0.05. Representative of 2 experiments.
Perhaps most interesting was the finding that heat-denatured PrPSc also induced a near exclusively anti-PrP IgM response (data not shown). In other work we have shown that mouse PrPSc is not recognized in two-site immunoassays using ICSM 18 and ICSM 35 unless the protein is similarly heated for 5 min to 95 °C. This led us to conclude that heating dispersed or disaggregated the protein, allowing the antibodies to bind. Heating is well recognized, of course, to substantially reduce prion infectivity (41). However, given that the heat-denatured protein still elicits an IgM response, it seems likely that the protein retains its polymeric structure in a partially unfolded state.
Polyclonal Antibodies Induced by Dynabead Adsorbed PrPSc Bind to Native PrPSc
It was important to show that the antibodies raised against native PrPSc have the capacity to recognize the immunogen. We therefore immunoprecipitated native PrP from normal and PK-treated scrapie brain homogenates and found that the pAb recognized both native PrPc and PrPSc (Figure 7). The results were similar to those obtained with ICSM 35 and/or ICSM 18 (not shown). Further analysis of the selectivity of the anti-PrP responses for disease-associated prion protein awaits the production of monoclonal antibodies from these immunized mice.
Figure 7.

Immunoprecipitation of mouse PrP using polyclonal sera derived from Prn-Pº/º mice immunized (serum 1 or 2) or not (Prn-Pº/º) with PrPSc-Dynabeads. These sera were incubated with brain homogenates prepared from scrapie-affected and normal animals. Bound antibody was immuno-precipitated with protein G agarose. For comparison, ICSM 35 was incubated with brain homogenates prepared from scrapie and normal mice.
Finally, we were interested to determine whether antibody responses to other brain constituents had been induced by the Dynabead-adsorbed antigen, given that a large number of genes are thought to be expressed exclusively in the brain, and their protein products might have adsorbed nonspecifically to the particles. We therefore probed denatured brain homogenates from wild-type and Prn-Pº/º mice with the polyclonal antibodies and found surprisingly few non–PrP bands in the wild-type homogenate and little binding to homogenates derived from Prn-Pº/º, indicating that antibodies induced were highly focused on the specifically adsorbed immunogen (Figure 8).
Figure 8.
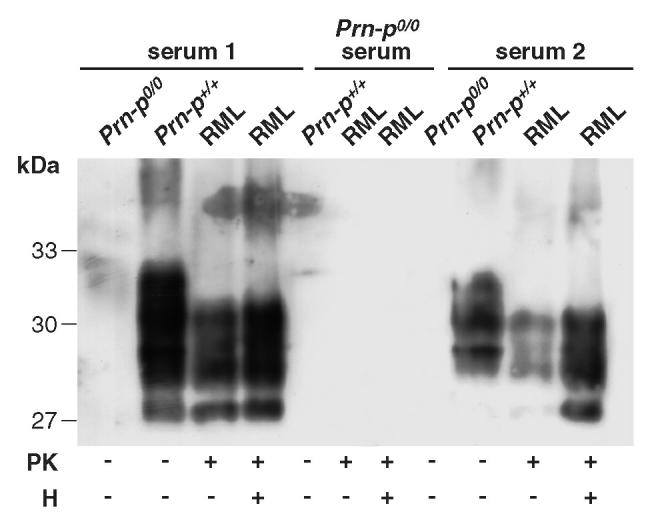
Western blotting of mouse PrP from proteinase K (PK) and/or heat-treated (H) Prn-Pº/º, Prn-P+/+, and RML brain homogenates with polyclonal sera from Prn-Pº/º (serum 1 and 2). Prn-Pº/º serum was used as negative control.
DISCUSSION
Attempts to generate immune responses to disease-associated prion protein have generally been unsuccessful. In fact, the majority of the available anti-PrP antibodies bind weakly to native PrPSc or not at all unless the protein is 1st denatured in strong ionic detergents, chaotropic reagents (32), or by heat-denaturing the protein (20,33,42,43). Recently a panel of monoclonal antibodies with PrPSc selectivity was produced by immunizing Prn-Pº/º mice with synthetic peptides containing a YYR epitope found to be exposed on β-PrP and not in α-isoforms (24). This is in line with our current strategy of raising monoclonal antibodies against full-length and truncated versions of recombinant human β-PrP (44). While these approaches hold promise, artificially produced proteins are structurally dissimilar to disease-associated isoforms as they do not contain posttranslational modifications present in native forms of the protein. Differences in the ratio of PrP glycoforms are readily apparent biochemically in different prion strains (7), although consistent differences in the glycoform composition between normal and disease states have not been identified. Using mAbs specific for different glycoforms and truncations of PrP, we have shown that the normal protein is expressed in alternative states within different brain regions (34), thereby potentially explaining the biochemical differences seen with different prion strains. The availability of a panel of conformation-dependent, strain-specific mAbs would greatly increase our understanding of the fundamental processes involved in prion replication.
Here we show for the 1st time that native normal and disease-associated prion protein can be used to generate anti-PrP responses. The antibodies produced are highly specific, allowing the immunoprecipitation of native PrP both in its normal and disease-associated states. It is remarkable that robust responses were generated to what were seemingly very low numbers of PrP-Dynabeads. Although we did not quantitate the amount of antigen loaded onto the beads, this is reminiscent of the highly efficient targeting of bead-bound antigen to APCs that was evident with the stimulation of a thymus-derived acetylcholine receptor-specific T-cell line from a patient with myasthenia gravis (45). On the other hand, in that situation, targeting antigen to APCs, such as macrophages, that have the capacity to internalize the beads and then strip the antigen to allow its processing provides a ready explanation for T-cell stimulation. However for generating antibody responses, it seems unlikely that specific B cells could have been stimulated unless there was dissociation of the antigen/bead complex, at least for any IgG responses that were induced. Given that B cells do not generally phagocytose large particulate antigens, perhaps they possess cell surface proteases that cleave off the antigen from the beads.
The ability to manipulate the specificity of this response by changing the antibody suggests that this method will prove extremely useful for raising antibodies to native proteins. Although it was conceivable that other brain immunogens might have adsorbed nonspecifically to the beads, we found surprisingly little recognition of brain homogenates derived from Prn-Pº/º mice; although it is likely that very few protein antigens survive digestion by proteinase K. In our view, the specificity of responses to proteins purified from tissue homogenates needs additional confirmation such as we obtained here by demonstrating reactivity to independently synthesized recombinant proteins (46). Dissection of the fine specificity of the response requires producing a panel of monoclonal antibodies and this is in progress.
Of note, antibodies generated in this system appear to bind the same linear epitope regardless of the host mouse strain (data not shown), and our results were similar to the antibody epitopes that have been defined in the N-terminal region of the PrP protein (22,47) except when PrPSc was 1st heat-denatured, in which case C-terminal region epitopes were revealed. These results suggested that heat denaturation might break up the protein polymer, conferring on it a more PrPc-like conformation, possibly even completely dissociating the aggregates into individual molecules.
We then studied the Ig subclass of the responding anti-PrP antibodies and found that whereas rPrP and PrPc induced predominantly IgG responses, PrPSc induced predominantly IgM. Therefore, a higher IgG/IgM ratio reflected PrPc and rPrP monomeric protein conformation whether the latter was folded into the α or β isoform, and polymeric, aggregated PrPSc induced IgM. Given that heat-denatured PrPSc also induced specific IgM responses, one can argue that heat does not dissociate the polymeric structure of disease-associated PrP. IgM responses to bacterial capsular polysaccharides are well-recognized components of the early defenses to infection and are generally T-cell independent (48). We are currently determining if responses to PrPSc are similarly independent of T-cell help and, if so, whether they are also elicitable in wild-type mice. If this was the case, one could conceive that such responses might play a role in resistance to prion infection in the periphery as has been shown for early resistance to other infections (48).
Interestingly, IgM responses to other polymeric proteins have recently been identified. The IgM antibodies produced by the amyloid fibril state of Aβ 1–40 (49), not only bound the immunogen, but remarkably also to other disease-related amyloid fibrils and amyloid-like aggregates derived from proteins of unrelated sequence (49). These data suggest a common repetitive immunogenic unit, though the relevance of these observations to disease resistance or susceptibility is unknown. Intriguingly, antibodies with PrPSc selectivity raised against repetitive YYR motif synthetic peptides were also of the IgM sub-class (24), and we are currently producing panels of monoclonal antibodies, hoping to identify mAbs with similar PrPSc selectivity. Finally, having the ability to induce antibodies to native PrPSc now offers the possibility of generating panels of strain specific antibodies, i.e., antibodies focused on strain-specific posttranslational modifications of the protein including conformation, glycosylation, and truncation.
Acknowledgments
We thank Ray Young for preparation of figures and the animal team at the Charing Cross prion facility. This work is supported by grants from the Medical Research Council (UK).
Footnotes
Online address: http://www.molmed.org>
REFERENCES
- 1.Prusiner SB. Molecular biology and genetics of prion diseases. Philos Trans R Soc Lond B Biol Sci. 1994;343:447–63. doi: 10.1098/rstb.1994.0043. [DOI] [PubMed] [Google Scholar]
- 2.Gray F, et al. Creutzfeldt-Jakob disease and cerebral amyloid angiopathy. Acta Neuropathol (Berl) 1994;88:106–11. doi: 10.1007/BF00294366. [DOI] [PubMed] [Google Scholar]
- 3.Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis. 1980;16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 4.Wells GAH, et al. A novel progressive spongiform encephalopathy in cattle. Vet Record Oct. 1987;31:419–20. doi: 10.1136/vr.121.18.419. [DOI] [PubMed] [Google Scholar]
- 5.Wilesmith JW, Wells GA, Cranwell MP, Ryan JB. Bovine spongiform encephalopathy: epidemiological studies. Vet Record. 1988;123:638–44. [PubMed] [Google Scholar]
- 6.Simmons MM, et al. Scrapie surveillance in Great Britain: results of an abattoir survey, 1997/98. Vet Record. 2000;146:391–5. doi: 10.1136/vr.146.14.391. [DOI] [PubMed] [Google Scholar]
- 7.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the etiology of ‘new variant’ CJD. Nature. 1996;383:685–90. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 8.Hill AF, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–50. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 9.Bruce ME, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 10.Prusiner SB. The prion diseases. Brain Pathol. 1998;8:499–513. doi: 10.1111/j.1750-3639.1998.tb00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajdusek DC. Hypothesis: interference with axonal transport of neurofilament as a common pathogenetic mechanism in certain diseases of the central nervous system. N Engl J Med. 1985;312:714–9. doi: 10.1056/NEJM198503143121110. [DOI] [PubMed] [Google Scholar]
- 12.Prusiner SB, et al. Immunologic and molecular biologic studies of prion proteins in bovine spongiform encephalopathy. J Infect Dis. 1993;167:602–13. doi: 10.1093/infdis/167.3.602. [DOI] [PubMed] [Google Scholar]
- 13.Bueler H, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–47. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 14.Weissmann C, et al. The use of transgenic mice in the investigation of transmissible spongiform encephalopathies. Rev Sci Tech. 1998;17:278–90. doi: 10.20506/rst.17.1.1079. [DOI] [PubMed] [Google Scholar]
- 15.Mabbott NA, Mackay F, Minns F, Bruce ME. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat Med. 2000;6:719–20. doi: 10.1038/77401. [DOI] [PubMed] [Google Scholar]
- 16.Hill AF, et al. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet. 1999;353:183–9. doi: 10.1016/s0140-6736(98)12075-5. [DOI] [PubMed] [Google Scholar]
- 17.O’Rourke KI, et al. Preclinical diagnosis of scrapie by immunohistochemistry of third eyelid lymphoid tissue. J Clin Microbiol. 2000;38:3254–9. doi: 10.1128/jcm.38.9.3254-3259.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreuder BE, Van Keulen LJ, Vromans ME, Langeveld JP, Smits MA. Tonsillar biopsy and PrPSc detection in the preclinical diagnosis of scrapie. Vet Record. 1998;142:564–8. doi: 10.1136/vr.142.21.564. [DOI] [PubMed] [Google Scholar]
- 19.Hill AF, Zeidler M, Ironside J, Collinge J. Diagnosis of new variant Creutzfeldt-Jakob disease by tonsil biopsy. Lancet. 1997;349:99–100. doi: 10.1016/S0140-6736(97)24002-X. [DOI] [PubMed] [Google Scholar]
- 20.Prusiner SB, et al. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci USA. 1993;90:10608–12. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prusiner SB, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673–86. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 22.Krasemann S, Groschup M, Hunsmann G, Bodemer W. Induction of antibodies against human prion proteins (PrP) by DNA-mediated immunization of PrPº/º mice. J Immunol Methods. 1996;199:109–18. doi: 10.1016/s0022-1759(96)00165-2. [DOI] [PubMed] [Google Scholar]
- 23.Korth C, et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature. 1997;390:74–7. doi: 10.1038/36337. [DOI] [PubMed] [Google Scholar]
- 24.Paramithiotis E, et al. A prion protein epitope selective for the pathologically misfolded conformation. Nat Med. 2003;9:893–9. doi: 10.1038/nm883. [DOI] [PubMed] [Google Scholar]
- 25.Maissen M, Roeckl C, Glatzel M, Goldmann W, Aguzzi A. Plasminogen binds to disease-associated prion protein of multiple species. Lancet. 2001;357:2026–8. doi: 10.1016/S0140-6736(00)05110-2. [DOI] [PubMed] [Google Scholar]
- 26.Weiss S, et al. RNA aptamers specifically interact with the prion protein PrP. J Virol. 1997;71:8790–7. doi: 10.1128/jvi.71.11.8790-8797.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soto C, et al. Reversion of prion protein conformational changes by synthetic β-sheet breaker peptides. Lancet. 2000;355:192–7. doi: 10.1016/s0140-6736(99)11419-3. [DOI] [PubMed] [Google Scholar]
- 28.Safar J, et al. Eight prion strains PrPSc molecules with different conformations. Nat Med. 1998;4:1157–65. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 29.Porter DD, Porter HG, Cox NA. Failure to demonstrate a humoral immune response to scrapie infection in mice. J Immunol. 1973;111:1407–10. [PubMed] [Google Scholar]
- 30.Aguzzi A. Protein conformation dictates prion strain. Nat Med. 1998;4:1125–6. doi: 10.1038/2621. [DOI] [PubMed] [Google Scholar]
- 31.McBride PA, Eikelenboom P, Kraal G, Fraser H, Bruce ME. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J Pathol. 1992;168:413–8. doi: 10.1002/path.1711680412. [DOI] [PubMed] [Google Scholar]
- 32.Peretz D, et al. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol. 1997;273:614–22. doi: 10.1006/jmbi.1997.1328. [DOI] [PubMed] [Google Scholar]
- 33.Kascsak RJ, et al. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–93. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beringue V, et al. Regional heterogeneity of cellular prion protein isoforms in the mouse brain. Brain. 2003;126:2065–73. doi: 10.1093/brain/awg205. [DOI] [PubMed] [Google Scholar]
- 35.Mond JJ, Vos Q, Lees A, Snapper CM. T cell independent antigens. Curr Opin Immunol. 1995;7:349–54. doi: 10.1016/0952-7915(95)80109-x. [DOI] [PubMed] [Google Scholar]
- 36.Bueler H, et al. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 37.Brandner S, et al. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci USA. 1996;93:13148–51. doi: 10.1073/pnas.93.23.13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson GS, et al. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science. 1999;283:1935–7. doi: 10.1126/science.283.5409.1935. [DOI] [PubMed] [Google Scholar]
- 39.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Anstee DJ, et al. New monoclonal antibodies in CD44 and CD58: their use to quantify CD44 and CD58 on normal human erythrocytes and to compare the distribution of CD44 and CD58 in human tissues. Immunology. 1991;74:197–205. [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor DM, Fernie K, Steele PJ, McConnell I, Somerville RA. Thermostability of mouse-passaged BSE and scrapie is independent of host PrP genotype: implications for the nature of the causal agents. J Gen Virol. 2002;83:3199–204. doi: 10.1099/0022-1317-83-12-3199. [DOI] [PubMed] [Google Scholar]
- 42.Serban D, Taraboulos A, DeArmond SJ, Prusiner SB. Rapid detection of Creutzfeldt-Jakob disease and scrapie prion proteins. Neurology. 1990;40:110–7. doi: 10.1212/wnl.40.1.110. [DOI] [PubMed] [Google Scholar]
- 43.Krasemann S, Jürgens T, Bodemer W. Generation of monoclonal antibodies against prion proteins with an unconventional nucleic acid-based immunization strategy. J Biotechnol. 1999;73:119–29. doi: 10.1016/s0168-1656(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 44.Jackson GS, et al. Multiple folding pathways for heterologously expressed human prion protein. Biochim. Biophys. Acta. 1999;1431:1–13. doi: 10.1016/s0167-4838(99)00038-2. [DOI] [PubMed] [Google Scholar]
- 45.Hawke S, Willcox N, Harcourt G, Vincent A, Newsom-Davis J. Stimulation of human T cells by sparse antigens captured on immunomagnetic particles. J Immunol Methods. 1992;155:41–8. doi: 10.1016/0022-1759(92)90269-y. [DOI] [PubMed] [Google Scholar]
- 46.Hawke S, et al. Autoimmune T cells in myasthenia gravis: heterogeneity and potential for specific immunotargeting. Immunol Today. 1996;17:307–11. doi: 10.1016/0167-5699(96)10022-0. [DOI] [PubMed] [Google Scholar]
- 47.Harmeyer S, Pfaff E, Groschup MH. Synthetic peptide vaccines yield monoclonal antibodies to cellular and pathological prion proteins of ruminants. J Gen Virol. 1998;79:937–45. doi: 10.1099/0022-1317-79-4-937. [DOI] [PubMed] [Google Scholar]
- 48.Mond JJ, Vos Q, Lees A, Snapper CM. T cell independent antigens. Curr Opin Immunol. 1995;7:349–54. doi: 10.1016/0952-7915(95)80109-x. [DOI] [PubMed] [Google Scholar]
- 49.O’Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci USA. 2002;99:1485–90. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]