

Abstract
Background
Coat colours in canines have many natural phenotypic variants. Some of the genes and alleles involved also cause genetic developmental defects, which are also observed in humans and mice. We studied the genetic bases of the merle phenotype in dogs to shed light on the pigmentation mechanisms and to identify genes involved in these complex pathways. The merle phenotype includes a lack of eumelanic pigmentation and developmental defects, hearing impairments and microphthalmia. It is similar to that observed in microphthalmia mouse mutants.
Results
Taking advantage of the dog as a powerful genetic model and using recently available genomic resources, we investigated the segregation of the merle phenotype in a five-generation pedigree, comprising 96 sampled Australian shepherd dogs. Genetic linkage analysis allowed us to identify a locus for the merle phenotype, spanning 5.5 megabases, at the centromeric tip of canine chromosome 10 (CFA10). This locus was supported by a Lod score of 15.65 at a recombination fraction θ = 0. Linkage analysis in three other breeds revealed that the same region is linked to the merle phenotype. This region, which is orthologous to human chromosome 12 (HSA12 q13-q14), belongs to a conserved ordered segment in the human and mouse genome and comprises several genes potentially involved in pigmentation and development.
Conclusion
This study has identified the locus for the merle coat colour in dogs to be at the centromeric end of CFA10. Genetic studies on other breeds segregating the merle phenotype should allow the locus to be defined more accurately with the aim of identifying the gene. This work shows the power of the canine system to search for the genetic bases of mammalian pigmentation and developmental pathways.
Background
Coat colours in mammals depend on skin and hair pigment synthesis. Melanocytes manufacture two types of melanin: the black/brown photo-protective eumelanin pigment, and the red-yellow cytotoxic phaeomelanin pigment. Several paracrine factors secreted primarily by surrounding keratinocytes are involved in the melanogenic pathway by stimulating the switch between phaeomelanin and eumelanin [1]. In this pathway, microphthalmia transcription factor (MITF) plays a central role by regulating the expression of the TYR (Tyrosinase), TRP-1 (Tyrosine Related Protein) and DCT (Dopachrome Tautomerase) genes that encode enzymes involved in pigment manufacture [2,3].
Coat colour is highly polymorphic in dogs. In 1957, Little described, after observing the possible phenotypes, more than 20 loci affecting coat colours [4,5]. Until recently, only a few genes were recognised as involved in pigmentation. However, more and more genes, alleles and new interactions are being discovered: variants of melanocortine 1 receptor gene (MC1R), (locus previously called extension E) [6-8], variants of Agouti, the antagonist ligand of MC1R [9,10], variants of tyrosinase-related protein 1 (TYRP1) [11] and variants of melanophillin [12]. Three mutations responsible for the brown coat colour versus black coat colour were described in TYRP1 in several dog breeds including the Australian Shepherd dog [11]. Genomic tools are now fully available in canine genetics: dense radiation hybrid maps with 1500 polymorphic microsatellite markers and anchored BAC markers [13,14], a radiation hybrid map comprising 10,000 canine gene-based markers [15], and a whole sequence assembly of the canine genome, build 2.1 [16]. Altogether, the dog appears to be a good model for understanding better the genetics of pigmentation in mammals and for isolating new genes, new variants and interactions between alleles of different loci.
We are interested in the merle phenotype because of its involvement in coat colour and developmental impairments. The merle phenotype is a dominant trait, with heterozygous dogs presenting a coat colour in which eumelanic regions are incompletely and irregularly diluted, leaving intensely pigmented patches. Merle is found throughout the body except on the pheomelanic regions of the black and tan coat colour (Figure 1A, 1B). These dogs often have heterochromia iridis or blue eyes and often have a lack of retinal pigment visible on the fundus. Homozygous merle dogs display a more severe phenotype. The dogs are usually very pale, sometimes completely white and present developmental defects with an incomplete penetrance, microphthalmia and hearing loss (Figure 1C, 1D). In merle European lineages, microphthalmia and/or hearing loss are not frequently observed as breeders avoid mating merle dogs to avoid these developmental defects. However, several veterinary studies on the "merle syndrome", reported retinal defects [17], microphthalmia and coloboma [18]. The non-survival or degeneration of melanocytes in the cochlea have been suggested to explain hearing loss [19].
Figure 1.
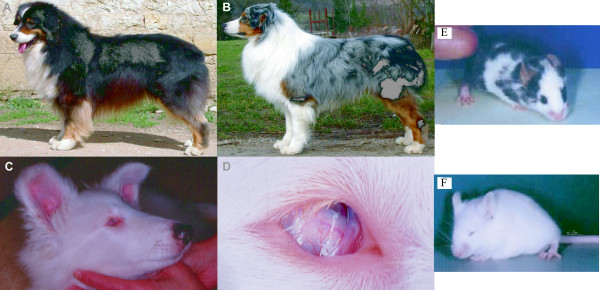
Pictures of none merle and merle dogs and mice microphthalmia mutants. A: Black and tan Australian Shepherd dog. B: Heterozygous merle Australian shepherd dog (pictures from Elevage du Paradis Sauvage de Ménestruel, Poncin, France) [40]. C: Six-month homozygous merle collie: the coat colour is totally white and the dog is blind and deaf. D: The left eye of the same dog, shows a microphthalmia with microcornea and a cataract (pictures from Dr Gilles Chaudieu, DVM, Dip. ECVO, Chamalières, France). E: Mitfmi-sp/Mitfmi-vga9 mouse. F: Mitfmi-vga9/Mitfmi-vga9 mouse with severe microphthalmia (pictures from Steingrímsson [23]).
When analysing the genetic basis of the merle phenotype, Little suggested that a unique locus (called M) was responsible for the merle phenotype in different breeds [4]. It was proposed that the merle coat colour may be due to a transposable element, after the observation of two germinal reversions out of 66 merle offspring of a homozygous merle female [20]. Recently, the Kit Ligand, KITLG, was excluded as a candidate gene for the merle phenotype in dogs [21] and the candidate gene approach has not yet give any conclusive results.
We searched for candidate genes for the merle phenotype in dogs by considering well-described pigment disorders in mice. Mutations in the gene of the Mitf pathway cause specific coat colour phenotypes, some of which are similar to the merle phenotype in dogs. These include dilution of the coat colour in patches and complete or mild microphthalmia (Figure 1E, 1F). Also, the complete abolition of functional Mitf results in loss of the melanocyte lineage, causing a white coat colour due to a lack of pigment cell manufacturer, and additional eye (microphthalmia) and inner ear disorders. Heteroallelic combinations of MITF variants produce animals with normal sized dark eyes and yellowish-brown to grey spotted checker board-like coat colours [22,23]. Mitf is also involved in human Waardenburg syndromes, including pigment cell migration disorders [24] and developmental defects such as deafness. Pax3 (Paired box gene 3) and Sox10 (SRY – Sex determining region Y-box 10), which regulates MITF gene expression, are also associated with this syndrome [25,26]. This genetic evidence suggest that MITF, PAX3 and SOX10 genes may be candidate genes for the merle phenotype.
We collected a pedigree of Australian shepherd dogs and used a genetic linkage approach with microsatellite markers flanking the MITF, PAX3 and SOX10 candidate genes to search for the genetic bases of the merle coat colour in canines. Although these three genes were excluded for the merle phenotype in dogs, we successfully identified the merle locus on canine chromosome 10, close to the centromere, 20 Mb away from Sox10. This locus was restricted to a 5.5 Mb interval and was further confirmed by analysing families of other breeds segregating the merle phenotype.
Results
Pedigrees
A pedigree comprising 96 Australian shepherd dogs (43 brown and 53 black dogs) was collected. This pedigree, called the "complete pedigree", included 42 merle dogs. A sub-pedigree of 38 dogs, including 17 merle dogs, derived from the complete pedigree was used for genotyping (Figure 2). Isolated families from different breeds segregating the merle coat colour were also collected, including three dachshund families (14 dogs); a Beauce shepherd family (five dogs) and a Border collie family (13 dogs).
Figure 2.
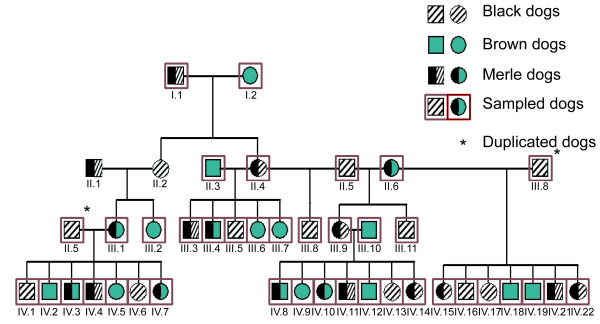
Sub-pedigree of 38 sampled Australian shepherd dogs (19 brown and 19 black dogs).
Genetic linkage analysis of the brown coat colour in the pedigrees
We evaluated the linkage power of the pedigrees by investigating the genetic linkage between the brown phenotype and the TYRP1 gene in the complete pedigree. As the TYRP1 gene was previously associated with the brown coat colour in dogs [11], we genotyped markers linked to TYRP1. These markers, FH2319 and REN105I03, are 1.18 and 5.17 Mb from TYRP1, respectively (see Additional file 1). The Lod scores between the brown phenotype and FH2319 and REN105I03 markers obtained by a two-point analysis on the complete pedigree, were 7.2 and 3.5 respectively, with a recombination fraction of θ = 0. For the sub-pedigree, the Lod scores were 3.6 and 2.4 respectively at θ = 0. The number of informative meiosis for the brown phenotype was 58 in the complete pedigree and 16 in the sub-pedigree. As the number of informative meiosis for the merle phenotype significantly increased to 81 and 33 in the complete pedigree and sub-pedigree, respectively, we expected these present pedigrees to be valuable for linkage analysis of the merle phenotype.
Genetic linkage analysis of the merle trait
As expected, the transmission mode in the collected pedigrees was consistent with an autosomal dominant segregation of the merle phenotype.
Using the sub-pedigree, we carried out genetic linkage analysis on polymorphic markers either flanking or within the intronic part of TYRP1 gene and the candidate genes MITF, PAX3, and SOX 10. Two-point analysis showed no significant linkage between the merle phenotype and markers flanking the MITF and PAX3 genes (Table 1). Therefore, we could exclude MITF and PAX3 being involved in this phenotype. However, we found significant linkages (Lod scores ranging from 3.09 to 3.65) with markers flanking SOX10, with recombination fractions, θ, ranging from 0.08 to 0.14 (Table 2). This suggested that the merle locus was about 10 cM from SOX10. As part of a "chromosome walking strategy", we selected 30 new polymorphic markers spanning a 27 Mb region from the SOX10 region to the centromeric tip of CFA10, as the C10.769 marker telomeric to SOX10 had a decreasing Lod score (Table 2).
Table 1.
Linkage data between the merle phenotype and markers flanking the TYRP1 gene and MITF and PAX3 candidate genes. Two point Lod scores and recombination fractions (θ) between the merle phenotype and the markers flanking TYRP1, MITF and PAX3 genes calculated on the sub-pedigree. a CFA: Canis familiaris chromosome. b Starred markers were selected from the CanFam 1.0 canine sequence draft. Significant exclusions (<-2) are indicated in bold.
| θ at Lod score max | Lod scores at θ | ||||||||||
| CFAa | Gene/Markersb | Gene/Marker Position in bp | θ | Lod score max | 0.001 | 0.01 | 0.05 | 0.1 | 0.15 | 0.2 | 0.3 |
| CFA11 | TYRP1 | 34,664779–34,683148 | |||||||||
| FH2319 | 35,863893–35,864169 | 0.5 | 0.0 | -23.083 | -13.146 | -6.442 | -3.802 | -2.429 | -1.582 | -0.661 | |
| Ren105l03 | 39,857761–39,857999 | 0.5 | 0.0 | -15.888 | -8.939 | -4.278 | -2.471 | -1.549 | -0.995 | -0.417 | |
| Ren96j16 | 22,981531–22,981669 | 0.5 | 0.0 | -13.194 | -8.198 | -4.721 | -3.24 | -2.384 | -1.786 | -0.963 | |
| Ren130E03 | 22,981511–22,981683 | 0.5 | 0.0 | -13.194 | -8.198 | -4.721 | -3.24 | -2.384 | -1.786 | -0.963 | |
| Ren159M20 | 23,456266–23,456464 | 0.5 | 0.0 | -19.789 | -11.82 | -6.37 | -4.141 | -2.915 | -2.1 | -1.061 | |
| CFA20 | MITF | 24,701418–24,735483 | |||||||||
| Ren100j13 | 25,668407–25,668570 | 0.5 | 0.0 | -24.585 | -14.621 | -7.791 | -4.988 | -3.442 | -2.417 | -1.131 | |
| Ren178E07 | 26,911251–26,911383 | 0.5 | 0.0 | -12.593 | -7.605 | -4.164 | -2.729 | -1.923 | -1.377 | -0.671 | |
| Ren105M20 | 19,606338–19,606486 | 0.4 | 0.087 | -8.992 | -5.016 | -2.327 | -1.264 | -0.709 | -0.367 | -0.011 | |
| Ren67C18 | 22,361131–22,361265 | 0.5 | 0.0 | -11.992 | -7.012 | -3.606 | -2.218 | -1.462 | -0.969 | -0.379 | |
| Pax3.2* | 30,185679–30,186031 | 0.27 | 0.147 | -9.89 | -4.957 | -1.763 | -0.644 | -0.159 | 0.063 | 0.137 | |
| CFA37 | PAX3 | 31,332182–31,429995 | |||||||||
| Pax3.1* | 31,368786–31,369020 | 0.5 | 0.0 | -20.085 | -11.159 | -5.204 | -2.924 | -1.779 | -1.101 | -0.418 | |
| Ren75L05 | 31,649572–31,649607 | 0.5 | 0.0 | -9.294 | -5.321 | -2.65 | -1.605 | -1.065 | -0.732 | -0.36 | |
Table 2.
Linkage data between the merle phenotype and CFA10 markers. List of the CFA10 genotyped markers in the sub-pedigree (left), the complete pedigree (middle) and all the studied pedigrees (right), with their two point Lod scores and recombination fractions (θ). All markers were selected from CanFam 1.0 sequence draft except FH2293 and C10769 taken from Guyon et al.[13]. The markers are ordered in the table from the centromere to the telomere. a markers flanking the SOX10 gene. Values in bold correspond to the highest Lod scores obtained in the three conditions.
| Two point Lod scores values | |||||||||||
| θ at Lod max | Lod scores at θ | ||||||||||
| Gene/Marker names | Marker Position (in bp) | θ | Lod max | 0.001 | 0.01 | 0.05 | 0.1 | 0.15 | 0.2 | 0.3 | |
| Lod scores in the sub-pedigree | CFA10.1 | 3,109459–3,109703 | 0.00 | 6.32 | 6.31 | 6.22 | 5.79 | 5.23 | 4.64 | 4.02 | 2.68 |
| CFA10.2 | 3,150322–3,150695 | 0.00 | 6.92 | 6.91 | 6.82 | 6.37 | 5.78 | 5.16 | 4.50 | 3.05 | |
| CFA10.3 | 3,169429–3,169640 | 0.00 | 3.01 | 3.01 | 2.95 | 2.72 | 2.42 | 2.10 | 1.76 | 1.06 | |
| CFA10.4 | 3,452451–3,452757 | 0.04 | 5.94 | 4.82 | 5.71 | 5.90 | 5.55 | 5.04 | 4.44 | 3.04 | |
| CFA10.5 c | 3,533969–3,534312 | 0.00 | 6.92 | 6.91 | 6.81 | 6.35 | 5.73 | 5.09 | 4.40 | 2.90 | |
| CFA10.6 | 4,050670–4,050915 | 0.00 | 3.01 | 3.01 | 2.96 | 2.74 | 2.46 | 2.17 | 1.86 | 1.20 | |
| CFA10.7 | 4,901919–4,902276 | 0.00 | 7.23 | 7.21 | 7.11 | 6.65 | 6.04 | 5.39 | 4.71 | 3.20 | |
| CFA10.8 | 6,938580–6,938928 | 0.00 | 7.23 | 7.21 | 7.11 | 6.65 | 6.04 | 5.39 | 4.71 | 3.20 | |
| CFA10.9 | 8,539383–8,539710 | 0.50 | 0.00 | -20.08 | -11.15 | -5.16 | -2.85 | -1.68 | -0.98 | -0.31 | |
| CFA10.10 | 9,459108–9,459324 | 0.07 | 2.62 | 1.21 | 2.15 | 2.60 | 2.57 | 2.40 | 2.16 | 1.52 | |
| CFA10.11 | 10,599594–10,599989 | 0.04 | 5.08 | 3.91 | 4.82 | 5.07 | 4.79 | 4.35 | 3.84 | 2.64 | |
| CFA10.12 | 12,963916–12,964094 | 0.15 | 2.55 | -4.49 | -0.58 | 1.79 | 2.43 | 2.55 | 2.44 | 1.83 | |
| CFA10.13 | 13,290922–13,291305 | 0.18 | 0.75 | -2.99 | -1.03 | 0.21 | 0.60 | 0.73 | 0.74 | 0.57 | |
| CFA10.14 | 13,687824–13,688059 | 0.11 | 3.44 | -1.49 | 1.42 | 3.09 | 3.43 | 3.36 | 3.10 | 2.25 | |
| CFA10.15 | 15,287513–15,287805 | 0.14 | 2.22 | -2.99 | -0.06 | 1.69 | 2.15 | 2.21 | 2.08 | 1.51 | |
| CFA10.16 | 16,325750–16,325992 | 0.09 | 3.67 | 0.61 | 2.53 | 3.55 | 3.66 | 3.49 | 3.19 | 2.33 | |
| CFA10.17 | 16,883188–16,883575 | 0.12 | 2.95 | -2.09 | 0.83 | 2.53 | 2.92 | 2.91 | 2.72 | 2.02 | |
| CFA10.18 | 17,574178–17,574420 | 0.08 | 3.87 | 0.91 | 2.82 | 3.79 | 3.84 | 3.61 | 3.25 | 2.31 | |
| CFA10.19 | 18,220318–18,220600 | 0.10 | 4.20 | -0.58 | 2.31 | 3.93 | 4.20 | 4.07 | 3.74 | 2.75 | |
| CFA10.20 | 19,075239–19,075433 | 0.12 | 3.19 | -1.79 | 1.12 | 2.81 | 3.17 | 3.13 | 2.90 | 2.10 | |
| CFA10.21 | 19,999011–19,999231 | 0.26 | 0.51 | -11.69 | -5.76 | -1.88 | -0.50 | 0.12 | 0.41 | 0.47 | |
| CFA10.22 | 21,607022–21,607316 | 0.10 | 4.45 | -0.28 | 2.61 | 4.20 | 4.45 | 4.30 | 3.95 | 2.90 | |
| CFA10.23 | 21,956041–21,956337 | 0.10 | 4.45 | -0.28 | 2.61 | 4.20 | 4.45 | 4.30 | 3.95 | 2.90 | |
| CFA10.24 | 23,113457–23,113656 | 0.25 | 0.92 | -13.18 | -6.27 | -1.77 | -0.17 | 0.52 | 0.83 | 0.85 | |
| CFA10.25 | 24,031569–24,031859 | 0.10 | 3.51 | -1.19 | 1.70 | 3.28 | 3.51 | 3.34 | 2.97 | 1.93 | |
| CFA10.26 | 25,504535–25,504764 | 0.14 | 2.73 | -4.19 | -0.29 | 2.05 | 2.65 | 2.73 | 2.57 | 1.89 | |
| CFA10.27 | 25,930447–25,930692 | 0.12 | 1.84 | -1.49 | 0.45 | 1.58 | 1.83 | 1.81 | 1.67 | 1.16 | |
| CFA10.28 | 26,491052–26,491288 | 0.13 | 1.66 | -1.79 | 0.16 | 1.32 | 1.62 | 1.65 | 1.56 | 1.16 | |
| CFA10.29 | 27,391154–27,391817 | 0.18 | 2.36 | -6.88 | -1.98 | 1.09 | 2.04 | 2.33 | 2.33 | 1.87 | |
| CFA10.30 | 28,230039–28,230419 | 0.30 | 0.69 | -18.28 | -9.37 | -3.47 | -1.27 | -0.23 | 0.33 | 0.69 | |
| CFA10.31a | 29,232270–29,232511 | 0.13 | 3.51 | -3.28 | 0.61 | 2.90 | 3.46 | 3.49 | 3.27 | 2.45 | |
| CFA10.32a | 29,355913–29,355989 | 0.14 | 3.09 | -3.88 | 0.02 | 2.37 | 2.99 | 3.08 | 2.94 | 2.24 | |
| CFA10.33a | 29,483066–29,483109 | 0.13 | 2.40 | -2.69 | 0.23 | 1.95 | 2.37 | 2.38 | 2.22 | 1.58 | |
| CFA10.34a | 29,826073–29,826290 | 0.14 | 2.52 | -2.69 | 0.24 | 2.00 | 2.45 | 2.51 | 2.38 | 1.81 | |
| CFA10.35a | 29,834441–29,834690 | 0.16 | 2.74 | -6.28 | -1.39 | 1.63 | 2.51 | 2.73 | 2.67 | 2.08 | |
| SOX10 | 29,856920–29,867728 | ||||||||||
| CFA10.36a | 29,861299–29,861547 | 0.08 | 3.60 | 0.61 | 2.52 | 3.51 | 3.58 | 3.37 | 3.02 | 2.10 | |
| CFA10.37a | 29,868038–29,868267 | 0.14 | 3.38 | -3.58 | 0.32 | 2.67 | 3.29 | 3.37 | 3.21 | 2.47 | |
| CFA10.38a | 30,176995–30,177378 | 0.11 | 3.65 | -1.19 | 1.71 | 3.35 | 3.65 | 3.55 | 3.26 | 2.38 | |
| FH2293a | 31,696028–31,696274 | 0.13 | 3.56 | -3.28 | 0.61 | 2.93 | 3.50 | 3.54 | 3.34 | 2.53 | |
| C10.769a | 36,663349–36,663563 | 0.18 | 1.50 | -5.99 | -2.06 | 0.42 | 1.20 | 1.46 | 1.49 | 1.18 | |
| Lod scores in the complete pedigree | CFA10.1 | 3,109459–3,109703 | 0.00 | 11.74 | 11.72 | 11.54 | 10.70 | 9.61 | 8.46 | 7.27 | 4.71 |
| CFA10.2 | 3,150322–3,150695 | 0.02 | 11.97 | 11.13 | 11.91 | 11.63 | 10.64 | 9.48 | 8.20 | 5.38 | |
| CFA10.3 | 3,169429–3,169640 | 0.00 | 6.62 | 6.61 | 6.50 | 5.98 | 5.30 | 4.60 | 3.86 | 2.33 | |
| CFA10.4 | 3,452451–3,452757 | 0.03 | 13.82 | 11.73 | 13.46 | 13.65 | 12.67 | 11.36 | 9.88 | 6.55 | |
| CFA10.5 c | 3,533969–3,534312 | 0.02 | 8.77 | 7.82 | 8.65 | 8.58 | 7.88 | 7.01 | 6.03 | 3.86 | |
| CFA10.6 | 4,050670–4,050915 | 0.00 | 6.92 | 6.91 | 6.79 | 6.24 | 5.51 | 4.76 | 3.99 | 2.41 | |
| CFA10.7 | 4,901919–4,902276 | 0.00 | 15.65 | 15.63 | 15.39 | 14.30 | 12.88 | 11.40 | 9.84 | 6.54 | |
| CFA10.8 | 6,938580–6,938928 | 0.02 | 14.90 | 14.13 | 14.87 | 14.39 | 13.16 | 11.72 | 10.17 | 6.76 | |
| CFA10.9 | 8,539383–8,539710 | 0.19 | 2.48 | -14.38 | -5.54 | 0.02 | 1.78 | 2.38 | 2.47 | 1.89 | |
|
Lod scores in the complete pedigree plus families of dachshund, Beauce shepherd and Border collie |
CFA10.1 | 3,109459–3,109703 | 0.04 | 10.67 | 8.43 | 10.21 | 10.61 | 9.91 | 8.91 | 7.74 | 5.09 |
| CFA10.2 | 3,150322–3,150695 | 0.04 | 10.97 | 8.73 | 10.51 | 10.91 | 10.21 | 9.21 | 8.04 | 5.39 | |
| CFA10.3 | 3,169429–3,169640 | 0.00 | 6.02 | 6.01 | 5.91 | 5.44 | 4.84 | 4.22 | 3.56 | 2.20 | |
| CFA10.4 | 3,452451–3,452757 | 0.03 | 14.38 | 12.33 | 14.05 | 14.19 | 13.15 | 11.79 | 10.25 | 6.83 | |
| CFA10.5 c | 3,533969–3,534312 | 0.02 | 8.77 | 7.82 | 8.65 | 8.58 | 7.89 | 7.02 | 6.05 | 3.90 | |
| CFA10.6 | 4,050670–4,050915 | 0.03 | 6.99 | 6.01 | 6.86 | 6.85 | 6.23 | 5.46 | 4.61 | 2.82 | |
| CFA10.7 | 4,901919–4,902276 | 0.00 | 19.87 | 19.83 | 19.52 | 18.11 | 16.28 | 14.37 | 12.37 | 8.15 | |
| CFA10.8 | 6,938580–6,938928 | 0.00 | 19.57 | 19.53 | 19.22 | 17.82 | 15.99 | 14.08 | 12.09 | 7.91 | |
| CFA10.9 | 8,539383–8,539710 | 0.16 | 4.23 | -11.97 | -3.16 | 2.25 | 3.83 | 4.22 | 4.10 | 3.02 | |
Linkage analysis allowed us to identify seven markers close to the centromere, CFA10.1 to CFA10.8, which cosegregate with the merle phenotype with significant Lod scores (> 3) (Table 2, see Additional file 2).
We extended the genetic linkage by analysing the nine most centromeric markers in the "complete pedigree" and in three nuclear families of dachshund, Beauce shepherd and Border collie segregating the merle phenotype. We obtained increased Lod scores for markers CFA10.1 to CFA10.8, with maximum Lod scores for CFA10.7 and CFA10.8 (Lod scores at θ = 0 of 15.65 and 14.90 in the complete pedigree and 19.87 and 19.57 in the complete pedigree plus the three other families, respectively). The CFA10.9 marker (telomeric to CFA10.8) is unlinked to the phenotype (Lod scores at θ = 0 of -14.38 in the complete pedigree and -11.97 in the complete pedigree plus the three other families, respectively, Table 2). Haplotype analyses of this region allowed us to detect a recombination event between the merle phenotype and the CFA10.9 microsatellite, thus limiting further the merle locus (Figure 3). These data, as well as the previous results for SOX10 flanking markers (Table 2), allowed us to exclude the SOX10 gene as being involved in the merle phenotype. Our results show that the merle locus is located in a 5.5 Mb region between the end of the centromere, arbitrarily located at 3 Mb, (represented by CFA10.1 located at 3.1 Mb) and the CFA10.9 marker (located at 8.5 Mb) defining the telomeric limit of the critical interval (see Additional file 2).
Figure 3.
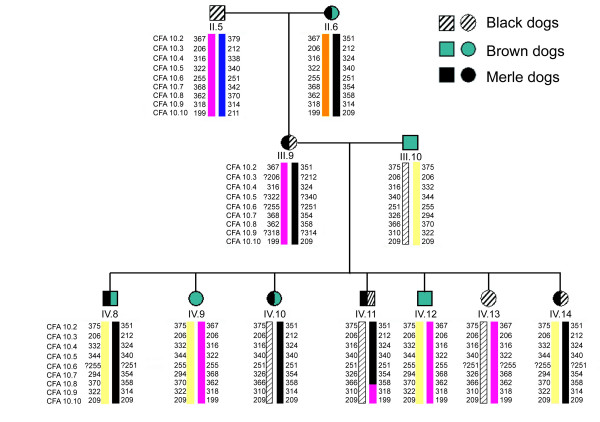
Haplotype analysis of the centromeric end of CFA10. Analysis revealed a crossing-over between CFA10.8 and CFA10.9 markers (dog IV.11), limiting the merle locus in between the centromeric end and the CFA10.9 marker. The merle chromosome is shown in black. Alleles with a question mark indicate that the parental origin of the allele could not be determined.
Discussion
In the present study, we used a genetic linkage approach on a pedigree of Australian shepherd dogs segregating the merle phenotype. We identified with high statistical support a 5.5 Mb locus at the centromeric tip of CFA10 in which the gene responsible for this phenotype should be found. Dog samples were collected from breeders. As phenotyping was easily and immediately detectable after birth by breeders themselves and then officially declared to the breed club, we have been able to obtain an informative family with as many as five generations of Australian shepherd dogs. Moreover, parentage testing ensured good reliability of the pedigrees. Such collected pedigrees from existing families means that housing of dogs is not required, limiting housing costs and ethical issues.
We focused on three candidate genes belonging to the coat colour pathway: MITF, PAX3 and SOX 10. The merle phenotype shares similarities with Mitf mouse mutants in coat colour and ocular and hearing defects, and also with human Waardenburg patients. However, our genetic linkage study ruled out these three genes as being involved in the merle phenotype. Genetic analysis of the SOX10 region in the sub-pedigree, using a "chromosome walking" strategy on CFA10, allowed us to identify the merle locus. It spans a 5.5 Mb region 20 Mb away from SOX10. Genetic linkage analyses on the complete pedigree and on small families from other breeds confirmed that the merle locus was located between the CFA10 centromere (3 Mb) and the CFA10.9 marker (8.5 Mb), with the highest Lod score of 19.87.
The corresponding orthologous human region is HSA12q13-q14 (position from 54.36 Mb to 60.94 Mb) and mouse region is MMU10D3 (position from 122.8 Mb to 128.7 Mb). These orthologous dog, human and mouse regions correspond to a unique conserved ordered segment, which has the same orientation in dogs and humans but is inverted between dogs and mice. In the dog region, 99 genes are predicted and 48 are known (Broad1), in the human region, 134 genes are predicted and 98 are known (NCBI 35), and in the mouse region, 112 genes are predicted and 95 are known (NCBI M34) [27]. These gene numbers may vary due to slight changes in the annotated genes as the versions are updated. In humans, mice and dogs, the conserved segments are totally ordered, making annotation of the dog segment easy, thus helping determination of candidate merle genes. This locus has many candidate genes, with at least a dozen being potential metabolic candidates as they, or their paralogs, belong to the pigmentation pathway. These include proteins involved in neural crest development (such as ERBB3), melanosome motility and transfer to surrounding keratinocytes (such as Silv/Pmel-17 and rab, kinesin, dynactin, myosin proteins).
Although the MITF gene itself has been excluded, the merle mutation should affect a gene interacting directly with the MITF gene in the pigmentation pathway. Alternatively, a more complex mechanism could explain the incomplete penetrance of eye defects observed in homozygous merle dogs. Although hearing loss may be due to an extreme white phenotype, including the absence of melanocytes in the cochlea, as in other white canine breeds [28], less is known about the origins of microphthalmia and other ocular defects. These may be due to another mutation in the same locus.
The merle phenotype occurs in several breeds and is commonly encountered in mongrel dogs. Breeds segregating merle are from the collie lineage (Group 1-FCI – Federation Cynologique Internationale classification): Shetland sheepdog, Border collie, collie, Australian shepherd dog, etc., and from other unrelated breeds belonging to different FCI groups and different clusters, as defined by Parker et al. [29], such as dachshund (Group 4-FCI):, Beauce shepherd (Group 1-FCI), great Dane (Group 2-FCI), Welsh corgi cardigan and Pyrenean shepherd. The merle phenotype is most probably very old, with the merle coat colour being reported in old books [30,31], from which drawings of merle dogs have been selected and reproduced [32].
It is not yet known whether the genetic cause of the merle phenotype is the same in all breeds and mongrels segregating this phenotype. A unique locus has been suggested as responsible for the merle coat colour [4]. In the present study, the increased Lod scores observed for genotyped markers from the merle locus in dachshund, Beauce shepherd and Border collie families is consistent, at least in these breeds, with there being a unique locus for the merle coat colour. If all merle dogs share a common ancestor chromosome, all breeds segregating merle could be used to refine the locus. The sharing of the merle locus by several breeds and also by mongrels may be due either to a common ancestor chromosome region being transmitted throughout canine evolution and/or to backcrosses that introduced a merle haplotype in several breeds at different times.
Conclusion
Using genetic linkage analysis, we excluded the involvement of the MITF, PAX3 and SOX 10 candidates genes in the merle phenotype. However, we identified the merle locus at the centromeric end of CFA10 in pedigrees of Australian shepherd dogs, dachshund, Beauce shepherd dogs and Border collies segregating the merle phenotype. This locus spans 5.5 Mb and is linked to the merle coat colour with a maximum Lod score of 19.87 and a recombination fraction of 0. We are currently analysing this locus in several breeds segregating merle, with a high density of single nucleotide polymorphic markers (SNP). This should help in identifying the merle gene. As well as benefiting breeding practices and canine veterinary medicine, identifying the merle gene will also help in understanding the genetic bases of mammalian pigmentation and developmental pathways.
Methods
Genomic DNA extraction
No dogs were housed for research purposes, and all dogs were privately owned pets.
Blood samples and the accompanying pedigree and coat colour data (with pictures when possible) were collected by DVM veterinarians. All data were entered into a database. Genomic DNA was extracted from 5 ml of blood collected on EDTA, using the nucleon BACC 3 kit (Amersham Biosciences, Piscataway, NJ, USA). For low concentration samples, the extracted DNA was "whole genome amplified" using the genomiphi kit (Amersham Biosciences).
Canine pedigree
Pedigrees were constructed using the Cyrillic software (Cyrillic2.1) [33], which allows haplotypes from the genotyping data to be drawn and the data to be exported in different formats for use in genetic linkage analysis. We carried out genotyping of 20 polymorphic microsatellites from four different chromosomes (CFA10, 11, 20 and 37) to check and validate the parentage compatibility.
Markers selection and Genotyping experiments
Microsatellite markers were selected from RH map data [13,34] or from the CanFam 1.0 draft of the canine genome sequence [35]. Markers were selected from their position and their polymorphism level (see Additional file 1). We used Primer3 software to design PCR primers [36].
Microsatellite markers were labelled using a two-step-PCR fluorescent labelling procedure [37]. The first step was carried out on 50 ng of dog genomic DNA using a classical PCR protocol and a touchdown program of 61°C to 51°C. The second step consisted of a one strand labelling PCR as previously described [37]. The PCR products were purified using Sephadex G50 fine column filtration in a 96 format (Amersham Biosciences). An aliquot of 3 μl fluorescent purified PCR product was mixed with 0.3 μl (0.2 nM) of fluorescent geneScan-500 ROX size standard (Applied Biosystems) and 8.7 μl of formamide, and then loaded onto a 3130 XL genetic analyser (Applied Biosystems). Results were analysed using GeneMapper software v3.7 (Applied Biosystems) and the genotyping data were used by Cyrillic software for the genetic linkage analysis.
Genetic linkage analysis
Haplotypes were constructed using the Cyrillic software. Two-point linkage analysis was carried out between each marker and the merle phenotype using M-LINK software through the GLUE web interface [38] and MultiMap software [39]. We used the 'prepare' option of CRI-MAP to check for Mendelian segregation. The linkage between each pair of markers was carried out with the TWOPOINT option of CRI-MAP. Lod scores were calculated assuming an autosomal dominant transmission with full penetrance and affected individuals were scored as heterozygous at the phenotype locus.
Authors' contributions
BH collected samples, constructed the pedigrees and performed genotyping experiments; BH also interpreted all dataand actively participated in writing the manuscript. SC helped with the genotyping experiments and interpretation of the dataand participated in writing the manuscript. CH carried out the statistical analyses and data interpretation, and critically revised the manuscript. SD helped with the genotyping experiments. TV extracted DNA from blood samples and commented critically on the work and manuscript. TD carried out the synteny analyses. BD contributed with knowledge on canines coat colours and critically revised the manuscript. FG provided intellectual input and critically revised the manuscript. MDG helped conceive and design the work and helped in the writing of the manuscript. CA conceived and designed the work and drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Characteristics of the markers used in the genetic linkage studies. a CFA : Canis familiaris chromosome.b Starred markers were selected from the CanFam 1.0 canine sequence draft, c marker corresponding to marker FH2537 from Guyon et al. [13], d number of alleles as determined from the sub-pedigree. E Markers flanking SOX10 gene.
Scheme of two-point linkage analysis of the merle phenotype in the Australian Shepherd dog pedigrees on CFA10. Two-point linkage analysis of the merle phenotype in the Australian shepherd dog sub-pedigree (in black) and complete pedigree (in brown) (Lod scores at theta = 0) is shown on the right. An ordered list of genotyped markers (right) and genes (left) and their position in Mb are indicated in the middle. An ideogram of the canine chromosome 10 is shown on the left with the corresponding human chromosomal conserved segments. NB: genomic sequence systematically starts at an arbitrary coordinate of 3 Mb to include the non-sequenced centromeric region.
{kind=link}
Acknowledgments
Acknowledgements
We gratefully acknowledge the CNRS, the Conseil Regional de Bretagne for their financial support, as well as Australian shepherd breeders, (especially Mme Boutarfa and Mme Coosemans, Elevage du Paradis Sauvage de Ménestruel, Poncin, France; Mme Bernard, Elevage de l'Orée des Charmois, Bois Le Roi, France), Mme Rougé, Elevage des Corsaires de Feu, Britany, France and Dr. Gilles Chaudieu, DVM and Dr. Philippe Pilorge, DVM for sampling and pictures.
Contributor Information
Benoit Hédan, Email: benoit.hedan@univ-rennes1.fr.
Sébastien Corre, Email: sebastien.corre@univ-rennes1.fr.
Christophe Hitte, Email: christophe.hitte@univ-rennes1.fr.
Stéphane Dréano, Email: stephane.dreano@univ-rennes1.fr.
Thierry Vilboux, Email: thierry.vilboux@univ-rennes1.fr.
Thomas Derrien, Email: thomas.derrien@univ-rennes1.fr.
Bernard Denis, Email: denis.brj@wanadoo.fr.
Francis Galibert, Email: francis.galibert@univ-rennes1.fr.
Marie-Dominique Galibert, Email: marie-dominique.galibert-anne@univ-rennes1.fr.
Catherine André, Email: catherine.andre@univ-rennes1.fr.
References
- Imokawa G. Autocrine and paracrine regulation of melanocytes in human skin and in pigmentary disorders. Pigment Cell Res. 2004;17:96–110. doi: 10.1111/j.1600-0749.2003.00126.x. [DOI] [PubMed] [Google Scholar]
- Busca R, Ballotti R. Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 2000;13:60–69. doi: 10.1034/j.1600-0749.2000.130203.x. [DOI] [PubMed] [Google Scholar]
- Widlund HR, Fisher DE. Microphthalamia-associated transcription factor: a critical regulator of pigment cell development and survival. Oncogene. 2003;22:3035–3041. doi: 10.1038/sj.onc.1206443. [DOI] [PubMed] [Google Scholar]
- Little CC. In: The inheritance of coat color in dogs. Press CU, editor. Ithaca, NY, ; 1957. [Google Scholar]
- Sponenberg DP, Rothschild MF. The Genetics of the Dog. Ruvinsky A , Sampson J. New York, NY, CABI Publishing; 2001. Genetics of coat colour and hair texture; pp. 61 –685. [Google Scholar]
- Everts RE, Rothuizen J, van Oost BA. Identification of a premature stop codon in the melanocyte-stimulating hormone receptor gene (MC1R) in Labrador and Golden retrievers with yellow coat colour. Anim Genet. 2000;31:194–199. doi: 10.1046/j.1365-2052.2000.00639.x. [DOI] [PubMed] [Google Scholar]
- Newton JM, Wilkie AL, He L, Jordan SA, Metallinos DL, Holmes NG, Jackson IJ, Barsh GS. Melanocortin 1 receptor variation in the domestic dog. Mamm Genome. 2000;11:24–30. doi: 10.1007/s003350010005. [DOI] [PubMed] [Google Scholar]
- Schmutz SM, Berryere TG, Ellinwood NM, Kerns JA, Barsh GS. MC1R studies in dogs with melanistic mask or brindle patterns. J Hered. 2003;94:69–73. doi: 10.1093/jhered/esg014. [DOI] [PubMed] [Google Scholar]
- Berryere TG, Kerns JA, Barsh GS, Schmutz SM. Association of an Agouti allele with fawn or sable coat color in domestic dogs. Mamm Genome. 2005;16:262–272. doi: 10.1007/s00335-004-2445-6. [DOI] [PubMed] [Google Scholar]
- Kerns JA, Newton J, Berryere TG, Rubin EM, Cheng JF, Schmutz SM, Barsh GS. Characterization of the dog Agouti gene and a nonagoutimutation in German Shepherd Dogs. Mamm Genome. 2004;15:798–808. doi: 10.1007/s00335-004-2377-1. [DOI] [PubMed] [Google Scholar]
- Schmutz SM, Berryere TG, Goldfinch AD. TYRP1 and MC1R genotypes and their effects on coat color in dogs. Mamm Genome. 2002;13:380–387. doi: 10.1007/s00335-001-2147-2. [DOI] [PubMed] [Google Scholar]
- Philipp U, Hamann H, Mecklenburg L, Nishino S, Mignot E, Gunzel-Apel AR, Schmutz SM, Leeb T. Polymorphisms within the canine MLPH gene are associated with dilute coat color in dogs. BMC Genet. 2005;6:34. doi: 10.1186/1471-2156-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon R, Lorentzen TD, Hitte C, Kim L, Cadieu E, Parker HG, Quignon P, Lowe JK, Renier C, Gelfenbeyn B, Vignaux F, DeFrance HB, Gloux S, Mahairas GG, Andre C, Galibert F, Ostrander EA. A 1-Mb resolution radiation hybrid map of the canine genome. Proc Natl Acad Sci U S A. 2003;100:5296–5301. doi: 10.1073/pnas.0831002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen M, Hitte C, Lorentzen TD, Thomas R, Cadieu E, Sabacan L, Scott A, Evanno G, Parker HG, Kirkness EF, Hudson R, Guyon R, Mahairas GG, Gelfenbeyn B, Fraser CM, Andre C, Galibert F, Ostrander EA. An integrated 4249 marker FISH/RH map of the canine genome. BMC Genomics. 2004;5:65. doi: 10.1186/1471-2164-5-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitte C, Madeoy J, Kirkness EF, Priat C, Lorentzen TD, Senger F, Thomas D, Derrien T, Ramirez C, Scott C, Evanno G, Pullar B, Cadieu E, Oza V, Lourgant K, Jaffe DB, Tacher S, Dreano S, Berkova N, Andre C, Deloukas P, Fraser C, Lindblad-Toh K, Ostrander EA, Galibert F. Facilitating genome navigation: survey sequencing and dense radiation-hybrid gene mapping. Nat Rev Genet. 2005;6:643–648. doi: 10.1038/nrg1658. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulkobas EJ, 3rd, Zody MC, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–19. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Akcan A, Wegner W. Changes in the visual pathways and visual centers in Merle syndrome in the dog. Z Versuchstierkd. 1983;25:91–99. [PubMed] [Google Scholar]
- Gelatt KN, Powell NG, Huston K. Inheritance of microphthalmia with coloboma in the Australian shepherd dog. Am J Vet Res. 1981;42:1686–1690. [PubMed] [Google Scholar]
- Strain GM. Congenital deafness and its recognition. Vet Clin North Am Small Anim Pract. 1999;29:895–907, vi. doi: 10.1016/s0195-5616(99)50079-x. [DOI] [PubMed] [Google Scholar]
- Sponenberg DP. Germinal reversion of the merle allele in Australian shepherd dogs. J Hered. 1984;75:78. doi: 10.1093/oxfordjournals.jhered.a109874. [DOI] [PubMed] [Google Scholar]
- Schmutz SM, Berryere TG, Sharp CA. KITLG maps to canine chromosome 15 and is excluded as a candidate gene for merle in dogs. Anim Genet. 2003;34:75–76. doi: 10.1046/j.1365-2052.2003.00951_5.x. [DOI] [PubMed] [Google Scholar]
- Hansdottir AG, Palsdottir K, Favor J, Neuhauser-Klaus A, Fuchs H, de Angelis MH, Steingrimsson E. The novel mouse microphthalmia mutations Mitfmi-enu5 and Mitfmi-bcc2 produce dominant negative Mitf proteins. Genomics. 2004;83:932–935. doi: 10.1016/j.ygeno.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Arnheiter H, Hallsson JH, Lamoreux ML, Copeland NG, Jenkins NA. Interallelic complementation at the mouse Mitf locus. Genetics. 2003;163:267–276. doi: 10.1093/genetics/163.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251–255. doi: 10.1038/ng1194-251. [DOI] [PubMed] [Google Scholar]
- Potterf SB, Furumura M, Dunn KJ, Arnheiter H, Pavan WJ. Transcription factor hierarchy in Waardenburg syndrome: regulation of MITF expression by SOX10 and PAX3. Hum Genet. 2000;107:1–6. doi: 10.1007/s004390050001. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Takeda K, Ploplis B, Tachibana M. Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat Genet. 1998;18:283–286. doi: 10.1038/ng0398-283. [DOI] [PubMed] [Google Scholar]
- Ensembl http://www.ensembl.org/index.html
- Strain GM. Deafness prevalence and pigmentation and gender associations in dog breeds at risk. Vet J. 2004;167:23–32. doi: 10.1016/S1090-0233(03)00104-7. [DOI] [PubMed] [Google Scholar]
- Parker HG, Kim LV, Sutter NB, Carlson S, Lorentzen TD, Malek TB, Johnson GS, DeFrance HB, Ostrander EA, Kruglyak L. Genetic structure of the purebred domestic dog. Science. 2004;304:1160–1164. doi: 10.1126/science.1097406. [DOI] [PubMed] [Google Scholar]
- Brehm AE. In: La vie des animaux illustrée. Bailleres JB, editor. Paris, ; 1868. [Google Scholar]
- Buffon D. In: Origine des espèces. Bureau-de-la-société-des-Publications-illustrées , editor. Vol. 3. Paris, ; 1839. [Google Scholar]
- Société-Française-de-Cynotechnie . In: Nos chiens d'antan. Maradi , editor. France, ; 1994. [Google Scholar]
- Cyrillic http://www.cyrillicsoftware.com
- Canine-Hybrid-Radiation-project UMR 6061. http://www-recomgen.univ-rennes1.fr/doggy.html
- UCSC http://genome.ucsc.edu
- Primer3 http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
- Jouquand S, Cheron A, Galibert F. Microsatellite analysis using a two-step procedure for fluorescence labeling of PCR products. Biotechniques. 1999;26:902–905. doi: 10.2144/99265st03. [DOI] [PubMed] [Google Scholar]
- Glue http://menu.hgmp.mrc.ac.uk/menu-bin/GLUE/glue.pl
- Matise TC, Perlin M, Chakravarti A. Automated construction of genetic linkage maps using an expert system (MultiMap): a human genome linkage map. Nat Genet. 1994;6:384–390. doi: 10.1038/ng0494-384. [DOI] [PubMed] [Google Scholar]
- Elevage-Menestruel http://menestruel.free.fr
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of the markers used in the genetic linkage studies. a CFA : Canis familiaris chromosome.b Starred markers were selected from the CanFam 1.0 canine sequence draft, c marker corresponding to marker FH2537 from Guyon et al. [13], d number of alleles as determined from the sub-pedigree. E Markers flanking SOX10 gene.
Scheme of two-point linkage analysis of the merle phenotype in the Australian Shepherd dog pedigrees on CFA10. Two-point linkage analysis of the merle phenotype in the Australian shepherd dog sub-pedigree (in black) and complete pedigree (in brown) (Lod scores at theta = 0) is shown on the right. An ordered list of genotyped markers (right) and genes (left) and their position in Mb are indicated in the middle. An ideogram of the canine chromosome 10 is shown on the left with the corresponding human chromosomal conserved segments. NB: genomic sequence systematically starts at an arbitrary coordinate of 3 Mb to include the non-sequenced centromeric region.