

Short abstract
Through understanding the intricacies of host-pathogen interactions, it is now possible to inhibit the growth of microbes, especially viruses, by targeting host-cell proteins and functions.
Abstract
Through understanding the intricacies of host-pathogen interactions, it is now possible to inhibit the growth of microbes, especially viruses, by targeting host-cell proteins and functions. This new antimicrobial strategy has proved effective in the laboratory and in the clinic, and it has great potential for the future.
It is now clear that, despite the optimism of the 1967 US Surgeon General in stating that 'it's time to close the book on infectious diseases', implying that drugs such as antibiotics had made microbial diseases a thing of the past, infectious diseases remain a major cause of continuing human suffering. It is estimated that infectious diseases currently account for one third of global annual deaths. This led the more recent 16th US Surgeon General, David Satcher, to state that 'we are seeing a global resurgence of infectious disease'. Finding ways to treat these diseases is a continuing battle, and any new avenues for discovering therapeutics should be explored.
Current approaches to antimicrobial drug development
The paradigm of antimicrobial drugs, be they against viruses, bacteria, fungi or parasites, has been to target unique processes or enzymes of the pathogen with a specific drug, thus achieving a high antimicrobial effect and low host-cell toxicity because there is no cross-inhibition of host-cell proteins. This paradigm has been used to develop almost all current antimicrobial therapies, with many notable successes, and it will continue to be an important drug-development strategy in the future [1,2]. Even when pressure from a drug results in the selection of drug-resistant variants, the antimicrobial drug pipeline has delivered new therapies that allow us to stay ahead - in some cases just ahead - of a resurgence of old microbial diseases [3].
Despite this success, it is clear that the pipeline of new antimicrobial drugs is not full, and it may never be possible to develop drugs targeted to all the diverse pathogens that cause disease. This is partly because the disease burden of any one pathogen is unlikely to reach sufficient levels for pharmaceutical companies to justify the enormous cost of developing a new drug, which (although hotly debated) is estimated to be between $0.5 billion and $1.7 billion [4-6]. Although legislation over the past 20 years in the USA, especially the 'Orphan Drug Act', is designed to reduce such barriers, the difficulty of developing new antimicrobial drugs remains, and it is compounded by the fact that many infectious diseases requiring treatment occur in developing countries, which cannot cope with the costs of new drugs. In addition, the fact that multiple companies develop drugs against the same 'important' pathogen dilutes an already limited market share; this can be beneficial, however, when drug combination therapy is used to reduce the selection for drug resistance in, for example, human immunodeficiency virus (HIV) in patients with acquired immune deficiency syndrome (AIDS).
Broad-spectrum antibiotics are a way to expand the number of diseases that are treatable with the same medicine, but in the field of antiviral drugs such broad-spectrum classes of drug are less common. Even despite the massive sequencing of microbial genomes, which has identified many new drug targets, the classic antimicrobial paradigm cannot quickly deliver what we need. Given that encounters with existing or new microbial pathogens are unlikely to decrease [7], can we find a new way of combating microbial disease? Evidence suggests that we can, and that by using genome-scale approaches to identifying critical host-cell functions that facilitate microbial replication, we may be able to unlock new potential in both existing and future drugs.
New approaches to antiviral therapy
It is clear that all pathogenic microorganisms interact in some way with host tissues and cells during the disease process. These interactions can now be viewed at the level of whole-genome transcriptional responses [8,9]. In the case of viruses the interaction is obligatory, as viruses require the host-cell environment to replicate. It is the viral world that provides clues to how we could acquire a new range of antimicrobial drugs at relatively little cost and on a short timescale.
The concept of attacking the microbe by altering or augmenting a host-cell function or process is not new. The use of interferon α (IFNα) in combination with ribavirin in the treatment of hepatitis C virus infection is successful in 50% of infected individuals on a long-term basis [10,11], although the exact mode of action, and the reasons why 50% of people do not respond fully, are not understood. Similarly, interleukin-2 (IL-2) has been used to augment 'highly active antiretroviral therapy' (HAART) in HIV infection (HAART is a multiple drug combination therapy with higher antiviral activity than therapy with a single drug). Although this does not directly affect the viral load in the peripheral blood, it accelerates the normalization of CD4+ T-cell counts in infected individuals (HIV-infected individuals have reduced numbers of T cells bearing the CD4 cell-surface molecule because of the effects of HIV replication) [12].
Recently, however, a new anti-infective paradigm has emerged, a logical extension of INFα and IL-2 therapies but more sophisticated. Because pathogens, especially viruses, use host-cell pathways and enzymes for their replicative life cycle, it seems reasonable to expect that inhibiting such cellular processes would have an antiviral effect as a byproduct. There is now ample evidence, both in vitro and in vivo, that this is an effective strategy. Moreover, because all existing licensed drugs that target a human disease process affect the functioning of a cell or organ system in some way, we have in effect a ready-made pharmacy of antimicrobial agents with defined safety-data profiles and clinical-use histories that only require assessment for new or 'off target' second use.
Inhibiting host-cell functions produces an antiviral effect
The evidence that the approach of inhibiting host-cell functions works now extends across many diverse virus types, including poxviruses [13], herpesviruses [14-16], retroviruses [17], hepadnaviruses [18,19] and flaviviruses [20] (Table 1). But how were these novel antiviral targets discovered? In some cases, the insight came from basic knowledge of cellular enzymes and pathways with which the viral proteins interact. In other cases, genome-scale approaches such as gene-expression profiling using DNA microarrays identified genes, upregulated as part of the infected cell response, that were also known targets for drugs.
Table 1.
Host drug targets that have antiviral activity
| Virus family | Virus species | Host-cell target | Drug | Antiviral activity in vitro | Antiviral activity in vivo | References |
| Poxviridae | Vaccinia virus | Abl tyrosine kinase | Gleevec | + | + | [13] |
| Herpesviridae | KSHV | c-Kit | Gleevec | + | + | [14,21] |
| KSHV | Vitamin D receptor | Vitamin D analog EB1089 | + | - | [16] | |
| HSV-1 | EIF-2α | Salubrinal | + | - | [30] | |
| HCMV | COX-2 | BMS-279652 | + | - | [15,23] | |
| BMS-279654 | ||||||
| BMS-279655 | ||||||
| Indomethacin | ||||||
| Aspirin | ||||||
| Unclassified | Hepatitis D virus | Farnesyltransferase | FTI-277 | + | + | [18,19] |
| FTI-2153 | ||||||
| Hepadnaviridae | Hepatitis B virus | Heterogeneous nuclear ribonucleoprotein K (hnRNP K) | siRNA | + | - | [31] |
| Retroviridae | HIV | HMG-CoA reductase | Lovastatin | + | + | [17] |
| HIV | Geranylgeranyltransferase I | GGTI-286 | + | - | [17] | |
| HIV | ATM kinase | KU-55933 | + | - | [28] | |
| HIV | Deoxyhypusine synthase | CNI-1493 | + | - | [29] | |
| HIV | Histone deacetylase | Valporic acid | + | + | [32,33] | |
| Flaviviridae | West Nile disease virus | c-Yes (Src family kinases) | PP2 SU6656 | + | - | [20] |
There are at least three possible functions of these upregulated genes. First, they may be induced specifically by a given virus to facilitate efficient virus replication. Second, they may be induced as part of the cellular response to the pathogen and cause disease pathogenesis. And third, they may be part of the cellular response leading to pathogen clearance. Targeting genes in the first and second categories should lead to reduced virus replication and attenuated disease pathogenesis, whereas inhibiting genes involved in pathogen clearance should be avoided.
The antiviral properties that have been described for Gleevec (Imatinib mesylate), which was originally licensed for the treatment of chronic myelogenous leukemia and inhibits the Abl tyrosine kinase, exemplify the advantages of target discovery that uses both basic knowledge and the genome-scale approach. Knowledge that the protein A36R of the vaccinia virus, a poxvirus, is phosphorylated by both Src and Abl receptor tyrosine kinases led Reeves et al. [13] to examine the effects of inhibiting A36R phosphorylation. It became evident that Gleevec not only decreased virus titer and plaque size in tissue culture, but also prevented the effects of a lethal dose of vaccinia in a mouse nasal challenge assay, showing that Gleevec had the potential to limit the spread of vaccinia in vivo.
For the Kaposi's sarcoma herpesvirus (KSHV), infection of endothelial cells in vitro results in a change in cellular morphology from cobblestone-like to a spindle-cell appearance reminiscent of Kaposi's sarcoma (KS) cells. This was shown by Moses et al. [14] to coincide with increased expression of c-Kit, another target of Gleevec. Gleevec was tested and shown to prevent this change in cellular morphology in vitro [14]. More recently, the results of a small clinical study showed that Gleevec also reduced tumor size in 50% of people with KS [21].
An inhibitor of another KSHV-mediated cellular effect, B-cell lymphoma development, was also identified through changes in gene-expression patterns. My colleagues and I [16] identified the vitamin D receptor as being highly expressed in a subset of B-cell lymphomas, including KSHV primary effusion lymphoma, and showed by using a vitamin D receptor analog that the proliferation of these tumors could be greatly reduced in vitro. Similarly, changes in gene expression following human cytomegalovirus (HCMV) infection in vitro showed increased expression of cyclooxygenase 2 (COX-2) [22]. COX-2, a component of the prostaglandin H2 synthase complex, which is part of the eicosanoid biosynthesis pathway, catalyzes the production of prostaglandin H2 from arachidonic acid. Using COX-2 inhibitors, Zhu et al. [15] went on to show that HCMV titer could be reduced by two orders of magnitude in vitro. This result fits with the fact that inhibitors of the eicosanoid pathway, such as aspirin (a nonsteroidal anti-inflammatory drug that inhibits COX-1 and COX-2), also inhibit HCMV replication [23]. The importance of COX-2 to cytomegalovirus replication is highlighted further by the fact that the related rhesus cytomegalovirus (RhCMV), which infects rhesus macaques, encodes its own COX-2 ortholog [24]. Although the long-term use of COX-2 inhibitors results in an increased risk of cardiovascular toxicity in some individuals, a fact that has resulted in the withdrawal of the drugs for treating arthritis, it is possible that short-term use of the drug would be useful and safe in the treatment of acute HCMV disease.
Two other approved drugs have shown promising effects in the treatment of viral disease; in fact, they converge on a common cellular pathway but affect different components and different viruses (Figure 1). Prenylation, a specific lipid modification of proteins that promotes their association with membranes, is known to be required for the maturation of infectious hepatitis delta virus (HDV). Co-infection of HDV with hepatitis B virus (HBV) causes rapid progression to chronic liver disease. But HDV is a replication-defective virus that requires the HBV surface antigen to become infectious, and the acquisition of this antigen depends on the HDV large delta antigen, a protein that has a prenylation motif recognized by the enzyme farnesyltransferase. Inhibitors of farnesyltransferase have been developed to prevent prenylation of Ras and the subsequent association of Ras with membranes, thereby reducing its transforming properties. Bordier et al. [18,19] have shown that farnesyltransferase inhibitors reduced HDV infectious virion production [18] and also reduced HDV viral load, as assessed by the number of viral RNA copies in the serum of an experimental mouse model for HDV liver infection [19]. This raises the important possibility that prenylation inhibitors, which are known to have low side effects, could be a new class of antiviral agents [25].
Figure 1.
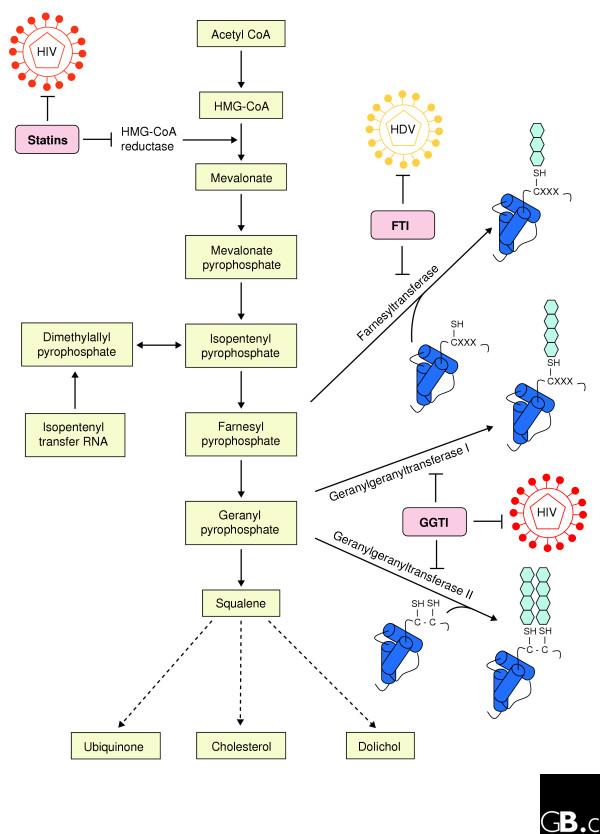
Well characterized drugs that inhibit prenylation can also be used to inhibit viruses. The biosynthetic pathway from acetyl CoA to squalene and then on through multiple other steps (not shown) to ubiquinone, cholesterol and dolichol is shown. Statins inhibit HMG-CoA reductase and thereby prevent the synthesis of mevalonate and subsequent downstream lipids; statins also inhibit the replication of HIV [17]. Farnesyl pyrophosphate and geranyl pyrophosphate are the substrates for farnesyltransferase and geranylgeranyltransferase I and II, respectively. These enzymes catalyze the prenylation of proteins (blue hexagons), with farnesyltransferase catalyzing the addition of the 15-carbon farnesyl prenyl lipids to the cysteine of the tetrapeptide CXXX (where X is one of a possible three amino acids at the carboxyl terminus of the protein) and geranylgeranyltransferase I and II catalyzing the addition of 20-carbon geranyl prenyl lipids to CXXX, CC or CXC motifs. Farnesyltransferase inhibitors (FTI) block the farnesylation of proteins such as Ras and also inhibit the replication of hepatitis delta virus (HDV) [18,19]. Similarly, geranylgeranyltransferase inhibitors (GGTI) block the geranylation of proteins such as Rho and also inhibit the replication of HIV [17]. The prenylation of Ras and Rho proteins promotes their association with membranes and is therefore necessary for the targeting of these proteins to the plasma membrane, where they function.
Inhibiting HIV-1 replication
Applying to HIV infections the approach of looking for known drugs that also affect viruses has resulted in new potential treatment options. The upregulation of genes involved in cholesterol biosynthesis pathways has been observed by gene-expression studies of HIV-infected cells in vitro. The upregulation increases the cellular levels of cholesterol, an effect mediated by the HIV protein Nef [26,27]. Cholesterol-lowering drugs such as statins, which inhibit 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase, the rate-limiting enzyme in the cholesterol biosynthetic pathway, might therefore have an antiviral effect on HIV.
In a ground breaking study, del Real et al. [17] tested the effect of statins on HIV replication both in vitro and in vivo. Amazingly, statins both inhibited HIV replication in culture, an effect that could be reversed by bypassing HMG-CoA reductase by adding the enzyme's product mevalonate (see Figure 1), and reduced HIV viral load by greater than threefold in six patients treated for one month. Interestingly, del Real et al. [17] showed that the anti-HIV effect of the statins was mediated by geranylgeranyl pyrophosphate, the other prenyl lipid involved in prenylating proteins. Inhibition of geranylgeranyltransferase I with a small-molecule inhibitor recapitulated the anti-HIV effect of the statin, whereas far-nesyltransferase inhibitors had no effect. Geranylgeranylation is required for post-translational modification of Rho GTPases, and the HIV envelope protein activates Rho upon binding the cell surface; it is therefore possible that the anti-HIV effects of statins occur through disruption of Rho activation, required for efficient virus entry into the cell, that is induced by the HIV envelope protein gp120 [17].
HIV replication can also be inhibited by targeting other cellular functions. Several studies have shown that the cellular mechanisms responsible for repairing DNA double-strand breaks are required to support retroviral infection and prevent cell death. Lau et al. [28] demonstrated that the ataxia-telangiectasia (ATM) kinase, one of the cellular regulators of responses to DNA damage, is activated by integration of the HIV genome into the host-cell genome. Inhibiting ATM kinase activity efficiently blocked HIV replication in T cells by inducing cell death in the infected cells, most probably as a result of an impaired ability to repair DNA double-stranded breaks arising from retroviral genome integration. Inhibiting ATM kinase also worked synergistically with existing antiretroviral drugs [28].
By a similar rationale, Hauber et al. [29] used knowledge about the HIV Rev protein to identify a novel antiviral agent: Rev uses the eukaryotic initiation factor 5A (eIF-5a) as a cofactor for the nuclear export of unspliced viral RNAs containing the Rev-responsive element. Activity is conferred on eIF-5a by the modification of the amino acid lysine to hypusine. The catalysis of the hypusine modification is achieved by two enzymes, deoxyhypusine synthase (DHS) and deoxyhypusine hydrolase. A small-molecule inhibitor of DHS, CNI-1493, is known to have antiproliferative effects on human cells in vitro and is currently being assessed clinically for the treatment of Crohn's disease. Hauber et al. [29] showed that CNI-1493 could inhibit HIV replication in vitro at drug concentrations below those that are being used in the clinical trials, suggesting that CNI-1493 is also a potential antiviral.
Inhibition of basic host-cell functions
Targeting aspects of the host mRNA transport and translation initiation mechanisms are also ways of inhibiting herpes simplex virus (HSV) and HBV replication. Research aimed at protecting cells from endoplasmic reticulum (ER) stress showed that the small molecule salubrinal could block dephosphorylation of the eukaryotic initiation factor eIF-2α, and that this inhibited HSV replication. All eukaryotic cells respond to ER stress, including the stress induced by viral infection, by inducing a set of pathways known collectively as the unfolded protein response. The unfolded protein response leads in part to the phosphorylation of eIF-2α and subsequent transient translational arrest, a cytoprotective response. Virus infection can also induce eIF-2α phosphorylation through the double-stranded RNA-activated protein kinase (PKR). To counteract this activity HSV encodes a protein (ICP34.5) that interacts with cellular proteins to mediate eIF-2α dephosphorylation.
Testing of salubrinal in an HSV-infection assay showed that the small molecule could block HSV-mediated eIF-2α dephosphorylation and prevent HSV replication [30]. Similarly, knowledge that the HBV genome enhancer region binds heterogeneous nuclear ribonucleoprotein K (hnRNP K), a pre-mRNA-binding protein that shuttles between the nucleus and cytoplasm, led Ng et al. [31] to remove hnRNP K using RNA interference (RNAi) gene silencing and to demonstrate that this reduces HBV DNA replication by up to 50%.
The ability to target host-cell proteins as antivirals is not limited to DNA viruses (poxviruses and herpesviruses) or viruses that use DNA intermediates as part of their replicative life cycle (hepadnaviruses and retroviruses). Studies of gene-expression changes caused by infection with West Nile virus (WNV), a flavivirus the genome of which consists of a single-stranded positive-sense RNA molecule, showed that the gene encoding the Src-family kinase (SFK) c-Yes was upregulated fivefold [20]. Specifically inhibiting SFKs with small molecules and removing c-Yes by RNAi significantly inhibited WNV replication in vitro [20].
Treating persistent virus infections
Many viruses, especially herpesviruses and retroviruses, persist for the lifetime of the host after the initial infection. These 'latent' infections are the source of continued virus production, but they are generally not affected by the range of drugs that target the lytic side of the viral life cycle. Recently, the prospect of using licensed drugs to deplete latently HIV-infected cells in patients has shown promising results.
Histone deacetylase 1 (HDAC1), which mediates chromatin remodeling, has been implicated in the repression of HIV gene expression in infected cells, and inhibition of HDAC1 by the anticonvulsant valporic acid (VPA) results in reactivation of HIV-1 replication in latently infected T cells [32]. This may seem like a bad effect, but it raises the possibility of using VPA to induce a 'flushing out' of latently infected cells in combination with antiretroviral drugs to prevent infection of new cells. The idea was tested in a small group of patients, and the combination of VPA and the antiretroviral enfuvirtide resulted in a significant decline in latently infected resting T cells [33]. It may therefore be possible in the future to reactivate latent virus infections in a controlled way and thereby perhaps eliminate the virus from the individual completely.
Systematic hunting for new antiviral targets
Together, the approaches described above provide sufficient evidence that targeting host-cell processes is a good approach to finding new antiviral drugs. More of the same kind of research will clearly uncover other cell processes that can be similarly inhibited. Fewer than half of the studies discussed so far, however, used DNA microarray technology to provide the first clue to which drug to use [14-16,20], suggesting that greater use of unbiased approaches will uncover a greater range of novel drug targets.
The use of genomics and bioinformatics in drug discovery is now part of the core business of many pharmaceutical companies [34,35]. This has resulted in a rationalization of the sort of data required and how they are integrated to facilitate discovery of new drugs. The simplest unbiased approach to finding new antivirals would be to assess systematically the gene-expression changes induced in appropriate target cells by a range of viruses, and then link the induced genes to known drugs that target the protein or pathway encoded by the induced genes. This can be undertaken in vitro, as the data generated from appropriate cells seem to be robust and can be translated directly into therapies for patients [17,21,33].
As it is unlikely that one laboratory will generate all possible virus-induced host gene-expression data, it is clear that data sharing and integration will be required, an idea championed by the Katze laboratory through their view of a 'virus compendium' to capture such information [36]. If such a compendium were integrated with data on existing drugs and their protein or pathway targets, we would have an easily accessible means of finding potential antivirals. Even in the absence of a unified compendium, meta-analysis of host gene-expression programs altered by infection has revealed over 500 genes that can now be explored [9]. If these are not direct targets themselves, the clever use of 'pathway expansion' methods should reveal alternative proteins that could be used to achieve an antiviral effect. Pathway expansion allows a given protein (which may or may not be the target of an existing drug) to be placed in a functional pathway, thereby identifying additional pathway proteins as potential drug targets. When unraveling the anti-HIV effects of statins, knowledge that HMG-CoA reductase is part of the pathway producing farnesyl and geranyl pyrophosphate (see Figure 1) allowed the identification of farnesyl- and geranyltransferase inhibitors as additional drugs in the expanded pathway [17].
To find more general targets, this strategy could be integrated with high-throughput loss-of-function assays, in which the function being assessed is the ability of the cells to support viral replication. This could be undertaken either in model systems or in cell types that can actually be infected by the viruses of interest. The idea of using model systems stems from the work of two groups [37-39], who assessed yeast single-gene-deletion libraries for their ability to support the replication of RNA virus genomes. Each study identified a range of yeast genes that were required for viral-genome replication or recombination. A concern must be raised about the low level of concordance between genes identified by the two groups, however. In fact, comparing genes required for tomato bushy stunt virus (TBSV) replication with those for brome mosaic virus (BMV) replication identified only four genes needed for both viruses. This small overlap can be explained in part by different experimental systems and the fact that TBSV and BMV represent different supergroups of viruses. Nevertheless, the real proof that these surrogate systems are meaningful will require testing the effect of inhibiting some of the genes in real viral replication systems.
A more authentic method, and one supported by some of the examples described above, would be the large-scale loss-of-function screening of human cells with RNAi libraries [40]. When combined with expression vectors to produce short interfering RNAs (siRNAs), such screens have been successful in identifying new modulators of the proliferation arrest that is dependent on the transcription factor p53 [41].
A related method of using genetic suppressor elements has been used in the context of HIV infection. Genetic suppressor elements work by blocking target-gene function through an inhibitory RNA or the production of trans-dominant peptides. When used to screen for genes important for HIV infection, this method revealed known proteins, such as the chemokine receptor CXCR4 and cyclin T1, together with additional proteins that were subsequently confirmed to affect HIV replication [42]. Such approaches could identify many important host-cell factors, but care will be needed to exclude the possibility that they do not induce an apparent antiviral response by simply activating classical cellular sensors of viral infection, such as PKR.
Combined with existing mechanistic insights into host-pathogen interactions, the coordinated use of unbiased, large-scale genomic methods will reveal a richer picture of the integrated host-cell response to infection and could well unlock the largely untapped potential of inhibiting virus replication through targeting the host. Whether using existing drugs in an infectious disease setting will result in additional side effects is not known, but the safety profiles of drugs generated from their current clinical use suggest that this will not be a large concern. The use of existing drugs for a new antiviral purpose, although in some cases not as effective as traditional antiviral drugs in preventing virus replication, will also enable combination therapies and may be less likely to cause selection for drug-resistant viruses.
In conclusion, the constraints on developing drugs to treat diseases caused by the wide variety of microbes has left us with a gap in healthcare that existing and emerging pathogens can all too easily colonize. The potential to target host-cell functions that prevent or reduce pathogen replication may be a quick and cost-effective way to plug this gap.
References
- Schmid MB. Seeing is believing: the impact of structural genomics on antimicrobial drug discovery. Nat Rev Microbiol. 2004;2:739–746. doi: 10.1038/nrmicro978. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Strategies in the design of antiviral drugs. Nat Rev Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- Weber J, Courvalin P. An emptying quiver: antimicrobial drugs and resistance. Emerg Infect Dis. 2005;11:791–793. doi: 10.3201/eid1106.050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development. J Health Econ. 2003;22:151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- Frank RG. New estimates of drug development costs. J Health Econ. 2003;22:325–330. doi: 10.1016/S0167-6296(03)00002-X. [DOI] [PubMed] [Google Scholar]
- Mullin R. Drug development costs about $1.7 billion. Chem Eng News. 2003;81:8. [Google Scholar]
- Weiss RA, McMichael AJ. Social and environmental risk factors in the emergence of infectious diseases. Nat Med. 2004;10:S70–S76. doi: 10.1038/nm1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellam P. Host-pathogen studies in the post-genomic era. Genome Biol. 2000;1:reviews1009–1009.4. doi: 10.1186/gb-2000-1-2-reviews1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner RG, Young RA. Insights into host responses against pathogens from transcriptional profiling. Nat Rev Microbiol. 2005;3:281–294. doi: 10.1038/nrmicro1126. [DOI] [PubMed] [Google Scholar]
- Davis GL, Esteban-Mur R, Rustgi V, Hoefs J, Gordon SC, Trepo C, Shiffman ML, Zeuzem S, Craxi A, Ling M-H, Albrecht J. Interferon alfa-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. N Engl J Med. 1998;339:1493–1499. doi: 10.1056/NEJM199811193392102. [DOI] [PubMed] [Google Scholar]
- Schalm SW, Hansen BE, Chemello L, Bellobuono A, Brouwer JT, Weiland O, Cavalletto L, Schvarcz R, Ideo G, Alberti A. Ribavirin enhances the efficacy but not the adverse effects of interferon in chronic hepatitis C. Meta-analysis of individual patient data from European centers. J Hepatol. 1997;26:961–966. doi: 10.1016/S0168-8278(97)80103-1. [DOI] [PubMed] [Google Scholar]
- Stellbrink HJ, van Lunzen J, Westby M, O'Sullivan E, Schneider C, Adam A, Weitner L, Kuhlmann B, Hoffmann C, Fenske S, et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial). AIDS. 2002;16:1479–1487. doi: 10.1097/00002030-200207260-00004. [DOI] [PubMed] [Google Scholar]
- Reeves PM, Bommarius B, Lebeis S, McNulty S, Christensen J, Swimm A, Chahroudi A, Chavan R, Feinberg MB, Veach D, et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med. 2005;11:731–739. doi: 10.1038/nm1265. [DOI] [PubMed] [Google Scholar]
- Moses AV, Jarvis MA, Raggo C, Bell YC, Ruhl R, Luukkonen BG, Griffith DJ, Wait CL, Druker BJ, Heinrich MC, et al. Kaposi's sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J Virol. 2002;76:8383–8399. doi: 10.1128/JVI.76.16.8383-8399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Cong JP, Yu D, Bresnahan WA, Shenk TE. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc Natl Acad Sci USA. 2002;99:3932–3937. doi: 10.1073/pnas.052713799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner RG, Maillard K, Cattini N, Weiss RA, Boshoff C, Wooster R, Kellam P. Kaposi's sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc Natl Acad Sci USA. 2003;100:10399–10404. doi: 10.1073/pnas.1630810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Real G, Jimenez-Baranda S, Mira E, Lacalle RA, Lucas P, Gomez-Mouton C, Alegret M, Pena JM, Rodriguez-Zapata M, Alvarez-Mon M, et al. Statins inhibit HIV-1 infection by down-regulating rho activity. J Exp Med. 2004;200:541–547. doi: 10.1084/jem.20040061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordier BB, Marion PL, Ohashi K, Kay MA, Greenberg HB, Casey JL, Glenn JS. A prenylation inhibitor prevents production of infectious hepatitis delta virus particles. J Virol. 2002;76:10465–10472. doi: 10.1128/JVI.76.20.10465-10472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordier BB, Ohkanda J, Liu P, Lee SY, Salazar FH, Marion PL, Ohashi K, Meuse L, Kay MA, Casey JL, et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J Clin Invest. 2003;112:407–414. doi: 10.1172/JCI200317704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch AJ, Medigeshi GR, Meyers HL, DeFilippis V, Fruh K, Briese T, Lipkin WI, Nelson JA. The Src family kinase c-Yes is required for maturation of West Nile virus particles. J Virol. 2005;79:11943–11951. doi: 10.1128/JVI.79.18.11943-11951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koon HB, Bubley GJ, Pantanowitz L, Masiello D, Smith B, Crosby K, Proper J, Weeden W, Miller TE, Chatis P, et al. Imatinib-induced regression of AIDS-related Kaposi's sarcoma. J Clin Oncol. 2005;23:982–989. doi: 10.1200/JCO.2005.06.079. [DOI] [PubMed] [Google Scholar]
- Zhu H, Cong JP, Mamtora G, Gingeras T, Shenk T. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:14470–14475. doi: 10.1073/pnas.95.24.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir E, Yu ZX, Ferrans VJ, Huang ES, Epstein SE. Aspirin attenuates cytomegalovirus infectivity and gene expression mediated by cyclooxygenase-2 in coronary artery smooth muscle cells. Circ Res. 1998;83:210–216. doi: 10.1161/01.res.83.2.210. [DOI] [PubMed] [Google Scholar]
- Rue CA, Jarvis MA, Knoche AJ, Meyers HL, DeFilippis VR, Hansen SG, Wagner M, Fruh K, Anders DG, Wong SW, et al. A cyclooxygenase-2 homologue encoded by rhesus cytomegalovirus is a determinant for endothelial cell tropism. J Virol. 2004;78:12529–12536. doi: 10.1128/JVI.78.22.12529-12536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einav S, Glenn JS. Prenylation inhibitors: a novel class of antiviral agents. J Antimicrob Chemother. 2003;52:883–886. doi: 10.1093/jac/dkg490. [DOI] [PubMed] [Google Scholar]
- van 't Wout AB, Swain JV, Schindler M, Rao U, Pathmajeyan MS, Mullins JI, Kirchhoff F. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J Virol. 2005;79:10053–10058. doi: 10.1128/JVI.79.15.10053-10058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van 't Wout AB, Lehrman GK, Mikheeva SA, O'Keeffe GC, Katze MG, Bumgarner RE, Geiss GK, Mullins JI. Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4(+)-T-cell lines. J Virol. 2003;77:1392–1402. doi: 10.1128/JVI.77.2.1392-1402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Swinbank KM, Ahmed PS, Taylor DL, Jackson SP, Smith GC, O'Connor MJ. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat Cell Biol. 2005;7:493–500. doi: 10.1038/ncb1250. [DOI] [PubMed] [Google Scholar]
- Hauber I, Bevec D, Heukeshoven J, Kratzer F, Horn F, Choidas A, Harrer T, Hauber J. Identification of cellular deoxyhypusine synthase as a novel target for antiretroviral therapy. J Clin Invest. 2005;115:76–85. doi: 10.1172/JCI200521949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Ng LF, Chan M, Chan SH, Cheng PC, Leung EH, Chen WN, Ren EC. Host heterogeneous ribonucleoprotein K (hnRNP K) as a potential target to suppress hepatitis B virus replication. PLoS Med. 2005;2:e163. doi: 10.1371/journal.pmed.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS. 2004;18:1101–1108. doi: 10.1097/00002030-200405210-00003. [DOI] [PubMed] [Google Scholar]
- Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005;366:549–555. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searls DB. Data integration: challenges for drug discovery. Nat Rev Drug Discov. 2005;4:45–58. doi: 10.1038/nrd1608. [DOI] [PubMed] [Google Scholar]
- Kramer R, Cohen D. Functional genomics to new drug targets. Nat Rev Drug Discov. 2004;3:965–972. doi: 10.1038/nrd1552. [DOI] [PubMed] [Google Scholar]
- Katze MG, He Y, Gale M., Jr Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Kushner DB, Lindenbach BD, Grdzelishvili VZ, Noueiry AO, Paul SM, Ahlquist P. Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus. Proc Natl Acad Sci USA. 2003;100:15764–15769. doi: 10.1073/pnas.2536857100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panavas T, Serviene E, Brasher J, Nagy PD. Yeast genome-wide screen reveals dissimilar sets of host genes affecting replication of RNA viruses. Proc Natl Acad Sci USA. 2005;102:7326–7331. doi: 10.1073/pnas.0502604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serviene E, Shapka N, Cheng CP, Panavas T, Phuangrat B, Baker J, Nagy PD. Genome-wide screen identifies host genes affecting viral RNA recombination. Proc Natl Acad Sci USA. 2005;102:10545–10550. doi: 10.1073/pnas.0504844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JM, Mizuno H, Brady A, Lucito R, Hannon GJ. RNA interference microarrays: high-throughput loss-of-function genetics in mammalian cells. Proc Natl Acad Sci USA. 2004;101:6548–6552. doi: 10.1073/pnas.0400165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Dunn SJ, Khan IH, Chan UA, Scearce RL, Melara CL, Paul AM, Sharma V, Bih F-Y, Holzmayer TA, Luciw PA, Abo A. Identification of cell surface targets for HIV-1 therapeutics using genetic screens. Virology. 2004;321:260–273. doi: 10.1016/j.virol.2004.01.010. [DOI] [PubMed] [Google Scholar]