

Abstract
B cells play a variety of immunoregulatory roles through their antigen-presentation ability and through cytokine and chemokine production. Innate immune activation of B cells may play a beneficial role through the generation of natural cross-reactive antibodies, by maintaining B cell memory and by exercising immunomodulatory functions that may provide protection against autoimmunity. In this article, we review human B cell populations and their functional properties, with a particular focus on a population of inherently autoreactive B cells, which seem to play an important physiological role in innate immunity, but which, if selected into adaptive immune responses, appear to become pathogenic agents in systemic lupus erythematosus.
Keywords: B cells/B lymphocytes, Autoimmunity, Innate immunity, Immunoregulation, Human
Introduction
B cells have been considered to be part of the adaptive immune system, specializing in the generation of protective high-affinity antibodies. While this indeed represents an important function, at least some B cell subsets [in particular B-1 B cells and marginal zone (MZ) B cells] also participate in innate immune responses and may represent a bridge between the innate and the adaptive immune responses [1]. Moreover, it appears that even memory B cells belonging to the adaptive arm of the immune response can also be regulated through innate immune receptors in an antigen-independent fashion [2]. Current views of B cell biology are also enhanced by the realization that in addition to antibody production, B cells play a variety of immunoregulatory roles through their antigen-presentation ability and through cytokine and chemokine production [3, 4, 5, 6]. Innate immune activation of B cells may play a beneficial role through the generation of natural cross-reactive antibodies, by maintaining B cell memory and by exercising immunomodulatory functions that may provide protection against autoimmunity [2, 7, 8, 9, 10, 11, 12]. On the other hand, it is also apparent that innate immune activation of autoreactive B cells, a frequent component of a healthy immune system, has the potential to break tolerance and trigger autoimmunity [13]. This danger is also illustrated by the observation that expansion of MZ B cells may be associated, at least in some models, with autoimmunity [14].
While many of these issues have been substantially explored in the mouse, our understanding of the biology of human B cell subsets and the division of labor among them is more rudimentary. In this article, we shall review our current understanding of mature human B cell populations and their putative functions. We shall also review the immunobiology of a subset of human B cells (which we designate, 9G4 B cells) whose function appears to be at the interface between the innate and adaptive immune responses [15]. The study of 9G4 B cells offers important insight into the delicate balance between beneficial and pathological immune responses.
Human mature B cell subsets
Three major subsets of peripheral mature B cells are typically identified in the mouse: B-1 cells (divided into B-1a, which express surface CD5, and B-1b, which do not); B2 cells; and MZ B cells [16, 17]. B2 cells, also known as follicular B cells, are the archetypical B cell of the adaptive immune system. They participate in T-dependent germinal center (GC) reactions and give rise to isotype-switched, high-affinity memory cells and long-lived plasma cells. In contrast, B-1 and MZ B cells are located in specific anatomical compartments (peritoneal/pleural cavities and splenic MZ, respectively) and are responsible for early antibody responses (typically T-independent, low-affinity and frequently polyreactive). MZ B cells, like B-1 cells, are important not only to combat infections but also in the maintenance of host homeostasis [18]. Both populations are thought to play important roles in microbial surveillance, at least in part due to their anatomical location. In particular, MZ B cells are ideally suited to protect against blood-borne pathogens and have been proposed to participate in T-independent responses against encapsulated bacteria [18]. Both populations share the property of being T cell independent and of producing natural antibodies, which may serve protective functions in early response to infections and autoimmunity. It has been postulated that in the mouse B-1 cells are the mobile arm of the innate B cell response and MZ B cells are a sessile component, restricted to the spleen [19].
In comparison to the mouse, the definition of human B-1, B2, and MZ B cell subsets is less well defined. The existence in humans of an equivalent to mouse B-1 cells is controversial, and there is little consensus as to how to define human B-1 B cells. The expression of CD5, which identifies B-1a B cells in the mouse, does not seem to be a good marker for human B-1 cells [20]. Further, the very presence of B cells in serosal cavities, a B-1 B cell hallmark in the mouse, has been questioned in humans [20]. However, the existence of B-1 B cells in humans has been gleaned from early studies showing that some CD5 B cells have the ability to produce polyreactive antibodies, and more recent studies suggesting that a population of IgM memory cells responsible for providing protection against Streptococcus pneumoniae infections may represent the human equivalent to murine B-1a B cells [20, 21]. At least from a functional standpoint, human MZ B cells also remain to be fully defined. Anatomically, however, a well-defined MZ compartment can be observed in infant spleens by 2 years of age. Interestingly, while in adults the vast majority of MZ B cells express the memory cell marker CD27 and express at least some level of somatic hypermutation, the infant MZ appears to be devoid of CD27+ B cells [22]. These observations need to be reconciled with the presence in the peripheral blood of children under 2 years of age of somatically mutated B cells with an IgM/IgD MZ phenotype [23].
In humans, mature B cells corresponding to naïve, pre-GC, GC and memory B cell subsets have been delineated using a variety of surface markers including CD19, CD20, IgD, CD38, CD21, CD23 and CD27 [24, 25]. Classifications based on the expression of CD22 and CD27 have also been proposed [17, 20]. Examples of these subsets are shown in Fig. 1.
Fig. 1.
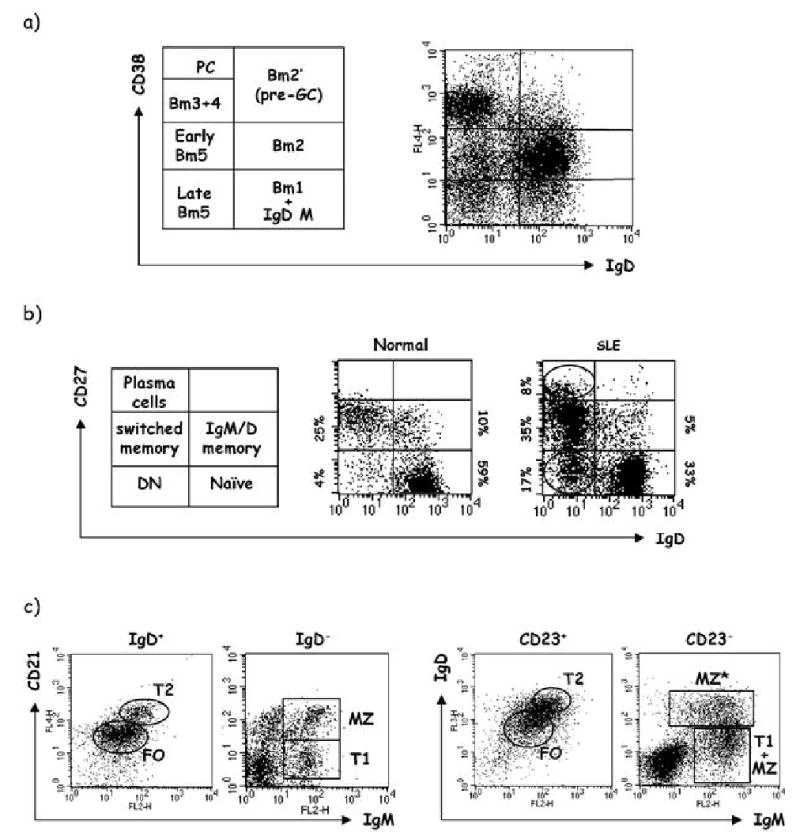
Human B cell subsets. Representative examples of human B cell populations are shown. a Analysis of normal tonsils by flow cytometry using antibodies against IgD and CD38 reveals the following subsets: Bm1 (virgin naïve, IgD++CD38−); Bm2 (antigen-experienced naïve, IgD++CD38+); Bm2’ (pre-GC, IgD+CD38++); Bm3 (centroblast, IgD−CD38++); Bm4 (centrocyte, IgD−CD38++); Bm5 (memory, IgD−CD38+/−). b Similar analysis of peripheral blood using IgD and CD27 markers. Healthy subjects demonstrate two dominant populations, naïve (IgD+, CD27−) and memory (identified as CD27+). In turn memory cells can be classified into either IgD+/IgM+, IgM-only (not shown) and isotype switched. This type of analysis also reveals a very minor fraction of IgD/CD27 double negative B cells (DN). Our preliminary studies suggest that DN B cells contain CD27− memory cells and can be induced to acquire CD27 expression under some culture conditions that include stimulation with CpG DNA (Cappione et al., in preparation). DN B cells, which usually go undetected in normal subjects due to their low frequency, are frequently greatly expanded in patients with active SLE in whom they sometimes represent the dominant peripheral blood subset [132]. Similarly, circulating CD27High plasmablasts which are quite scarce in healthy subjects are frequently increased in active SLE. c Characterization of B cell subsets in normal human spleen using staining for CD21/CD23, IgM/IgD. This type of staining generates a pattern very similar to the one obtained with mouse spleen and permits separation into follicular (FO), marginal zone (MZ) and transitional B cell populations (T1/T2). An additional subset can be seen in human spleen that appears to represent an IgD+ marginal zone population (MZ*) (SLE systemic lupus erythematosus)
One important difference between humans and mice is the frequency of peripheral blood (PBL) memory B cells, which in humans may represent 40–60% of all PBL B cells and are recognized by their expression of CD27 [26]. This difference has been attributed to the accumulation of long-lived memory cells during the longer human life-span [26]. Also of significant interest is that only about half of all human memory B cells have undergone isotype switch, while the rest express surface IgM. Of importance, our preliminary studies indicate that memory B cells can also be found among CD27− cells (Fig. 1b).
Whether IgM memory cells represent a homogeneous subset or not remains controversial. Initially, it was reported that populations of IgM-only and IgM/IgD memory cells could be clearly differentiated [26]. However, other reports and our own experience indicate that the vast majority of IgM memory cells also express at least low levels of surface IgD [20, 23]. The actual developmental origin of these subsets of memory cells and their specific role in functional immune responses remain to be elucidated. Although the phenotypic difference may be difficult to discriminate, evidence indicates that these represent two distinct memory populations. Thus, it has been found that in the absence of AID (e.g., in patients with AID mutations), memory B cells preferentially express IgM but not IgD [23]. Because the block in these patients is in the isotype-switch mechanism, it is postulated that the accumulated IgM-only memory cells are precursors to isotype-switched memory cells. In this model, IgM-only memory cells would belong to the B2 subpopulation. It should, however, be pointed out that there is no direct evidence that these cells persist in the presence of functional AID.
More data are available pertaining to memory B cells that express both IgM and IgD. Thus, patients with X-linked hyper-IgM syndrome, which is characterized by defective CD40L function and absence of GC, have significant levels of IgM/IgD memory cells, indicating that the development of this population may proceed independently of GC reactions [27]. In addition, two studies have linked IgM/IgD memory cells with both the B-1 and the MZ B cell populations. As cited above, Kruetzmann et al. [20] showed that the presence of IgM/IgD memory cells correlated with serum levels of anti-pneumococcal polysaccharides IgM antibodies and with protection against pneumococcal infections. Interestingly, these authors also found that these memory cells were absent in patients without spleen and were not detected in infants. Accumulation was first observed in children at 2 years of age. Based on their observation that B-1a cells are lacking in asplenic mice, the investigators postulated that IgM/IgD memory cells may have similar survival requirements and function as murine B-1a B cells [20].
In contrast, Weller et al. [23] have proposed that these memory cells represent a recirculating fraction of MZ B cells which are already present in early life. Consistent with Kruetzmann’s study, IgM/IgD memory cells were shown to participate in the immune response against encapsulated bacteria. Interestingly these investigators showed that these cells develop and mutate early in ontogeny, prior to their differentiation into T-independent antigen-responsive cells. Although their phenotypic identifiers have yet to be fully and satisfactorily described, some degree of consensus appears to be emerging regarding the existence of human B-1 and MZ equivalents. That these B cells participate in the innate immune system response is also gaining acceptance.
The two faces of innate immune B cells: a link between innate immunity and autoimmunity
Innate immune B cells are T cell-independent B cells present in the primary, pre-antigen-driven repertoire. These innate immune B cells participate in the host response against microbial antigens through the generation of natural IgM antibodies that do not require affinity maturation to provide early protection. Innate immune B cells are also likely to play beneficial roles through the production of natural IgM autoantibodies, which may contribute to the physiological clearance of apoptotic cells and help suppress pathogenic IgG autoantibody responses [11, 28]. These cells, while beneficial for the host, are also enriched in autoreactivity and, therefore, present a challenge for a healthy immune system which, to avoid pathogenic autoimmunity, must strike a difficult regulatory balance. Consequently, it can be envisioned that an important function of B cell tolerance must be to prevent the incorporation of autoreactive innate B cells into the T cell-dependent long-lived IgG memory compartment.
Here, we analyze a population of human B cells (designated 9G4 B cells) to gain insight into such regulatory processes. This population of B cells can be readily identified and isolated using a rat monoclonal antibody, 9G4 [29]. The 9G4 monoclonal antibody binds specifically to a unique epitope encoded in framework 1 of the human heavy chain variable region gene V 4–34. Thus, by definition, all 9G4 B cells express the V 4–34 VH gene. Most light chains do not affect expression of the 9G4 epitope and, so far as is known, light chain utilization by 9G4 B cells is not restricted. Interestingly, the same framework 1 sequence that encodes the 9G4 epitope, is responsible for the superantigen binding property, and autoreactivity, of 9G4 B cells, as described below [30]. The 9G4 epitope can be lost consequent to the accumulation of somatic mutations in the region of framework 1 that encodes the 9G4 epitope. Thus, it cannot be said that all V 4–34-expressing B cells are 9G4 B cells. Furthermore, mutational analysis indicates that the loss of the 9G4 epitope also results in loss of autoreactivity [31]. Therefore, the detection of the 9G4 epitope can be considered synonymous with autoreactivity, and the analysis of 9G4 expression affords the opportunity to determine the fate of autoreactive B cells and to investigate the mechanisms involved in the regulation of these cells.
9G4 B cells can be considered innate B cells because they do represent a large fraction of the primary pre-antigenic repertoire (approximately 5–10% of mature naïve B cells), and are expanded in the context of at least some infections (such as Infectious mononucleosis and Mycoplasma pneumoniae) in which they presumably play a protective role [32, 33]. Importantly for their classification as innate B cells, the antigenic reactivities of 9G4 B cells (both for autoantigens and for pathogens) are determined by the germline sequence of VH4–34, and do not appear to require somatic hypermutation or undergo antigen-selected affinity maturation (although admittedly, the latter issue needs formal exploration) [30]. In addition, as detailed below, 9G4 B cells are also represented in the IgM/IgD memory compartment and in the MZ B cell population but are excluded from the T cell-dependent IgG memory repertoire and therefore, do not normally participate in adaptive B cell responses.
This functional sequestration of 9G4 B cells in the innate immune system is subject to abrogation in certain situations. In systemic lupus erythematosus (SLE) patients, 9G4 B cells are highly expanded in the IgG memory population and may contribute to the pathogenesis of this autoimmune disease [15, 34]. As will be discussed below, these observations suggest the hypothesis that SLE represents a situation where the inappropriate positive selection of innate B cells into adaptive immune responses results in chronic autoimmunity.
9G4 antibodies in health and disease
9G4 antibodies are intrinsically autoreactive with the poly-N-acetyllactosamine (NAL) determinants of the I/i blood group antigens targeted by pathogenic cold agglutinins (CA) [35]. Strikingly, all 9G4 monoclonal antibodies thus far characterized possess anti-I/i reactivity. In addition, all anti-I/i antibodies thus far studied were encoded by VH4-34, thereby indicating that the expression of this gene segment is mandatory for anti-I/i CA activity [36, 37, 38]. The ability of 9G4 antibodies to bind NAL reflects their ability to recognize this antigen in a superantigen-like fashion through residues encoded in the germline sequence of the first framework region and largely irrespective of the somatically generated third hypervariable region and of the associated light chain [33, 39, 40, 41]. Indeed, it appears that most light chains (either κ or λ) are permissive of anti-I/i binding [40].
The fact that the infection-triggered immune response to I/i antigens selectively targets 9G4 B cells suggests that the VH4-34 gene segment may have been selected during evolution for its ability to encode protective antibodies. This speculation is supported by the observation that a close VH4-34 homolog is not present in the mouse genome and is also absent from the germline of rhesus monkeys even though these lower primates possess a VH4 gene family closely related to its human counterpart [42]. Chimpanzees, on the other hand, possess a close homologue of VH4–34, having only two nucleotide differences. Interestingly, both nucleotide substitutions result in amino acid replacements, and both occur in framework 1 within the site that mediates NAL binding (E.C.B. Milner, unpublished data). Thus, given that humans are the only natural host for both M. pneumoniae and EBV infections, it is tempting to speculate that these infectious organisms may constitute the evolutionary pressure responsible for the selection and highly conserved maintenance of the VH4-34 gene segment in the human germline (remarkably VH4-34 has been shown to be non-polymorphic and present in every subject thus far studied regardless of ethnic origin) [43, 44, 45, 46].
The closest sequence homology with VH4–34 among mouse VH genes is found with VH12 (I. Sanz, unpublished observations). Interestingly, VH12 B cells offer some striking parallels with 9G4 cells in that they represent a substantial fraction of the physiological B-1 repertoire (5–8%), are autoreactive against red blood cells, are positively selected, and appear to play a protective antimicrobial role [47].
It is important to realize that NAL is expressed in a variety of glycoproteins including CD45 and other differentiation antigens, gangliosides, gastrointestinal mucins and glycolipids as well as by apoptotic cells [48, 49, 50, 51, 52, 53]. Consequently, NAL-bearing self-antigens are widely available to engage 9G4 B cells both in soluble form and in the plasma membrane of RBC, lymphocytes, epithelial cells, tumor cells and apoptotic cells and indeed 9G4 antibodies have been shown to react with many of the antigens or cells described above ([33, 37, 38, 41, 54, 55, 56, 57, 58, 59, 60] and our own observations that will be discussed below in more detail).
Even though the vast majority of 9G4 antibodies recognize the I/i antigen, cross-reactivity with other relevant antigens (devoid of I/i epitopes) can be demonstrated in many cases depending on the expression of specific HCDR3 regions, associated light chains and charged residues present on the conventional antigen binding site [59, 60, 61, 62]. Cross-reactive antigens prominently include dsDNA and the lipid A (endotoxin) anchor moiety of bacterial lipopolysaccharides (LPS) [61, 63, 64, 65]. These important reactivities of 9G4 antibodies have been used to propose a physiological role for 9G4 B cells in the innate immune response and a pathogenic one in SLE, an autoimmune disease where a link between infection and anti-DNA responses has been frequently invoked [64, 66, 67, 68].
The central observation concerning 9G4 antibodies is that, despite the abundance of 9G4 B cells in the primary normal repertoire, they are scarcely represented in healthy sera (whether IgM or IgG) [34, 35, 69, 70, 71]. Yet, IgM 9G4 antibodies are substantially expanded in the serum of otherwise healthy individuals in response to acute infections with herpes viruses such as EBV and CMV and in acute M. pneumoniae infections [32, 36]. In these infections, 9G4 antibodies are responsible for the presence of pathogenic CA that mediate autoimmune hemolytic anemia [72]. Interestingly, while the fetal RBC linear NAL i-antigen is the usual target of CA in EBV infections, the branched NAL I-antigen expressed by adult RBC is overwhelmingly preferred by CA antibodies induced by M. pneumoniae [73]. We are thus presented with a fascinating scenario in which different infectious agents break, at least temporarily, B cell tolerance preferentially targeting autoreactive 9G4 B cells, yet resulting in the production of either anti-i or anti-I autoantibodies in a mutually exclusive fashion. A detailed discussion of this phenomenon is outside the scope of this review and has been recently published elsewhere [74].
In these infections, chronic autoimmunity appears to be prevented by the generation of autoantibodies largely of the IgM class, which tend to be of lower affinity, mostly limited to the intravascular space and relatively short-lived. Furthermore, healthy individuals without genetic predisposition to autoimmunity appear to possess the appropriate regulatory mechanisms necessary to prevent these autoreactive B cells from entering the long-lived IgG compartment. It is likely that these mechanisms would act in concert to prevent long-term systemic autoimmunity by avoiding chronic expansion of autoreactive B cells, isotype switch to more pathogenic IgG and epitope spreading.
According to this model, the chronic production of either IgM or IgG 9G4 antibodies would be expected to result in sustained autoimmunity. Indeed, this scenario is illustrated by two clinical situations: HIV infection and SLE (for 9G4 IgM and IgG antibodies, respectively). Thus, chronic CA disease can be demonstrated in 20–60% of HIV-infected patients with the large majority of cases being due to IgM anti-i/I autoantibodies [75]. While systematic studies of serum 9G4 antibodies remain to be done in HIV infections, the almost absolute restriction of anti-i/I responses to VH4-34 antibodies and the identification of VH4-34 in monoclonal CA derived from HIV patients strongly suggest that IgM 9G4 antibodies are chronically expanded in a substantial fraction of HIV patients and account for chronic CA disease [76].
In contrast to the previously discussed infections, sustained increases of serum IgG 9G4 antibodies are restricted to patients with SLE and the presence of high levels of these autoantibodies is very specific for this autoimmune disease [34, 69, 70, 71, 77]. While most reports only detected total 9G4 Ig levels two recent studies, including our own, also determined IgG levels [34, 77]. Our data demonstrate that IgG 9G4 antibodies are quite elevated in up to 75% of patients with active SLE where they represent from 10% to a stunning 40% of total serum IgG. Overall, serum 9G4 antibodies have been shown to correlate positively with global disease activity and with the presence of lupus nephritis and central nervous system involvement [34, 69, 71]. While a direct pathogenic effect of these antibodies in SLE remains to be formally established, several lines of evidence (in addition to their sheer abundance and disease association) support this contention. First, 9G4 antibodies may contribute to the anti-dsDNA and anti-Smith response in SLE and can be found in kidney eluates [69, 70, 78]. Secondly, 9G4 antibodies bind to the vast majority of naïve B cells by recognizing a glycoform of CD45/B220 both in vivo and in vitro [34]. It is tempting to speculate that this property could contribute to the naïve lymphopenia typically associated with active disease by inducing killing of naïve lymphocytes and/or promoting their differentiation into class-switched B cells [56, 79]. Moreover, the ability of 9G4 antibodies to bind B220 may explain our observation that these antibodies recognize apoptotic lymphocytes (Fig. 2) [34, 80]. Therefore, it is plausible that 9G4 antibodies could contribute to the ability of SLE sera to induce the sustained production of IFN-α, a cytokine currently considered central to the pathogenesis of this disease [81, 82]. In addition, by binding to CD45, 9G4 antibodies could contribute to the breakdown of B cell tolerance by modulating the activation threshold of B cells [83, 84]. Finally, 9G4 antibodies could contribute to the pathogenesis of neuro-psychiatric SLE through their ability to recognize gangliosides, which are abundantly expressed in neuronal cells [59]. Indeed, this antigenic reactivity could help explain the described association between anti-lymphocyte antibodies and central nervous system disease in SLE [85]. Whatever the pathogenic role(s) of 9G4 antibodies in SLE may be, the large contribution of these antibodies to the IgG repertoire in this disease strongly indicate that the tolerance mechanisms that normally act upon 9G4 cells to prevent their expansion into the memory and plasma cell repertoire are faulty in SLE. These aspects will be discussed in the following sections.
Fig. 2.
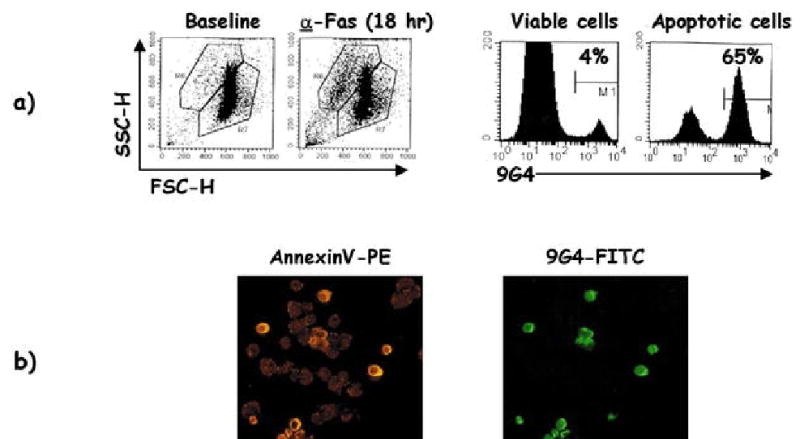
Recognition of apoptotic cells by 9G4 antibodies. a Dot plots in the left panels demonstrate induction of apoptosis in a CD45− Jurkat cell line treated with anti-Fas antibodies. The histograms shown in the right panels demonstrate that 9G4 antibodies (derived in this example from lupus serum) to apoptotic cells but not to viable cells after anti-Fas treatment. b The binding of 9G4 antibodies to apoptotic cells was corroborated by immunofluorescence. In this example, cells were incubated with either AnnexinV-PE or 9G4-FITC after 18 h of anti-Fas treatment. Only AnnexinV+ cells were also stained with 9G4, indicating specific binding to apoptotic cells
Autoreactive 9G4 B cells are censored in a healthy immune system
The need for strict censoring of 9G4 B cells is predicated on their abundance in the pre-immune repertoire and on the pathogenic potential of 9G4 antibodies. Yet, as previously discussed, 9G4 B cells can be activated under some conditions even in healthy individuals. This begs the question as to how this dynamic form of B cell tolerance is achieved. The answer seems to reside on the ability of healthy subjects to censor the progression of 9G4 cells through productive GC reactions. Indeed, we have previously shown that, while 9G4 B cells represent a substantial fraction of the follicular naïve repertoire (5–10%), they decrease by 80–90% in the IgM memory and plasma cell compartment and are rarely found in the IgG memory and plasma cell subsets, where they represent less than 0.5% of these two compartments ([15] and Cappione et al., Defective germinal censoring of autoreactive B-cells in human SLE, submitted). This regulation appears to be accomplished by strict censoring in the GC, the critical anatomical and functional environment responsible for the generation and expansion of high-affinity memory B cells and long-lived plasma cells in a T cell-dependent fashion. Strikingly, our published and unpublished studies have failed to identify productive GC reactions in any of more than 350 secondary follicles from 15 independent tonsils obtained from healthy donors (Fig. 3a). While 9G4-positive GCs have been recently reported in healthy tonsils, the frequency of such events was not provided and, therefore, it is hard to understand the significance of these findings [31]. In fact, in the absence of counterselection, the expected frequency of 9G4+ GC in healthy individuals would be 12–20%. This calculation is based on the observed frequency in our studies of mature GC formed by expansions of other VH4 B cells (40%, as identified by the Lc1 idiotype) and the relative contribution of 9G4 B cells to the total VH4 naïve B cell repertoire (30–50%) [31, 86, 87]. In contrast, our extensive tonsil studies as well as similar studies of healthy spleens show that the actual frequency of 9G4+ GCs is less than 1% of all mature GCs analyzed, a finding indicative of strong counterselection and well in keeping with the absence of 9G4+ GCs observed by other groups (J. Spencer and D. Dunn-Walters, Guy’s, King’s and St Thomas Medical School, London, UK; personal communication). Our unpublished observations also indicate that the censoring of naïve 9G4 B cells occurs at the transition between the pre-GC and GC stages of B cell differentiation and more specifically, at the GC founder cell level (Cappione et al., submitted).
Fig. 3.
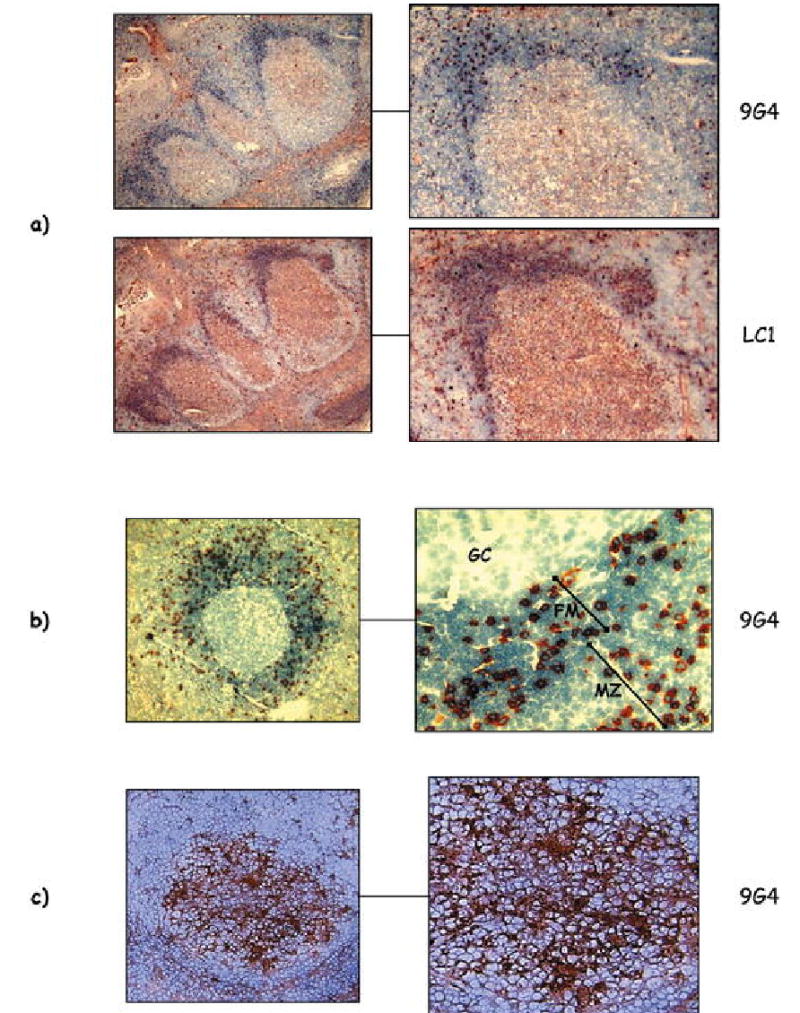
Histological distribution of 9G4 B cells. a Representative examples of GC from normal tonsils are shown. While 40% of all GC were positive for expansions of Lc1+ B cells (representing control VH4 B cells), autoreactive 9G4 do not form productive GC. b In normal spleen, 9G4 B cells accumulate in the follicular mantle (FM) and can also be seen within the anatomical marginal zone (MZ) but are absent from the GC proper. c Staining of tonsil biopsy specimens obtained from patients with SLE demonstrates mature GC reactions formed by expansions of 9G4 B cells (GC germinal center)
We believe that, at least in part, such censoring is the result of anergy induced by chronic exposure to self antigens. Thus, phenotypic and signaling studies indicate that naïve 9G4 B cells express a partially activated phenotype characterized by down-regulation of surface IgM and reduced Ca2+ flux in response to BCR stimulation similar to mouse anergic B cells [88, 89]. This observation would be consistent with the absence of intracellular calcium oscillations observed in B cell activation induced by T cell-independent type 2 antigens [90]. Furthermore, preliminary gene expression experiments using DNA microarrays indicate that as compared to other naïve B cells that regularly participate in productive GC reactions, 9G4 naïve cells express decreased levels of NF-κB and JNK kinase, a profile characteristic of anergic autoreactive transgenic B cells [91, 92]. Several mechanisms could be postulated to explain the failure of pre-GC 9G4 B cells to successfully participate in a mature GC reaction including defective responses to stimulation through the BAFF receptor [93, 94]. In addition, when stimulated in the presence of surrogate T cell help, healthy naïve 9G4 B cells readily differentiate into antibody-producing plasma cells (PC), thereby suggesting that their inability to do so in vivo may be due, at least in part, to the absence of T cell help, a mechanism known to contribute to the maintenance of tolerance in transgenic anti-DNA B cells [15, 95].
Interestingly, in addition to the follicular mantle, 9G4 B cells are also abundant in the MZ of the spleen (Fig. 3b). This finding points to the possibility that, as suggested in murine models, both negative selection (from the GC) and positive selection (into the MZ resulting in sequestration from self-antigens and/or T cell help) could both contribute to the censoring of 9G4 B cells.
Despite the obvious efficacy of these mechanisms, 9G4 B cells still make up approximately 1% of all IgM memory cells, a non-negligible amount whose presence needs be explained and which creates the need for additional censoring mechanisms. Interestingly, virtually all 9G4 IgM memory cells belong to the IgM/IgD memory population recently proposed to develop in a GC-independent fashion and which may represent a recirculating fraction of MZ B cells. Our observations with 9G4 cells strongly support these concepts and provide further evidence for the concept that IgM/IgD CD27+ memory cells may develop and accumulate in the MZ without participating in GC reactions. As for autoreactive memory B cells in general, the regulatory mechanisms acting upon memory 9G4 cells remain to be further explored. Nonetheless, our preliminary studies indicate that 9G4 cells almost universally express surface Fas and may therefore be destined to die by apoptosis should they encounter activated T cells which might otherwise induce them to differentiate into PC [96].
The physiological regulation of 9G4 B cells is faulty in SLE
The high levels of 9G4 IgG antibodies present in the serum of patients with active SLE strongly suggests that the censoring imposed in the GC by a healthy immune system is faulty in this autoimmune disease. This suggestion is strongly supported by our observation that 9G4 IgG memory cells and plasma cells are greatly expanded (10- to 25-fold) in SLE patients and represent 10–33% and 10–40% of all IgG memory B cells and plasma cells, respectively ([15, 34] and Cappione et al., submitted). To directly address this question, we have investigated the distribution of 9G4 B cells in tonsil biopsies obtained from SLE patients (Cappione et al., submitted). This unique approach provides access to secondary lymphoid tissue in patients with active disease and minimal treatment thereby circumventing the limitations created by the study of peripheral blood or of tissue obtained from patients treated with cytotoxic/immunosuppressive agents. Flow cytometric studies of tonsil biopsies demonstrate that the block observed in healthy subjects in the progression of 9G4 pre-GC cells is absent in SLE. Instead, these patients display a significantly increased frequency of 9G4 B cells with a GC surface phenotype (average expansion 8- to 10-fold) and with a memory phenotype (10- to 20-fold). Moreover, histological studies of the biopsy samples conclusively show that 9G4+ GC represent 15–25% of all productive GC reactions analyzed (Fig. 3c). Interestingly, however, active GC reactions formed by proliferating 9G4 B cells can also be observed in SLE patients without significant disease activity. Of significant importance, tonsil biopsy specimens of patients with other autoimmune diseases such as rheumatoid arthritis and vasculitis resembled normal samples and did not share the abnormalities described for SLE patients. This finding is consistent with the specificity of elevated titers of serum 9G4 antibodies for SLE. Together, these observations strongly suggest that the autoreactivity encoded by the 9G4 idiotype is positively selected by the autoimmune process underlying SLE, and is highly likely to play a pathogenic role in this disease. Outstanding issues that need to be understood include the actual antigen(s) responsible for the triggering and/or selection of 9G4 B cells in SLE and whether or not affinity maturation plays a role in this process. The fact that the expression of the germline-encoded 9G4 idiotype is favored even in the presence of enhanced activity of the mutational machinery in SLE, strongly indicates that NAL epitopes (possibly expressed by apoptotic cells) may be the selecting antigen. It is still possible, however, that these cells might also recognize additional antigens through the conventional antigen-binding site and that this reactivity might be created or enhanced by somatic hypermutation. The final elucidation of this important issue will require systematic analysis of the binding specificity of mutated and unmutated 9G4 B cells in normal subjects and in SLE patients.
Our current interpretation of our data is that abnormal censoring of autoreactive B cells in the GC, leading to B cell tolerance breakdown, is an intrinsic defect in SLE, which may be necessary but not sufficient for full clinical expression of the disease process. This interpretation would be consistent with the prevailing view that primary B cell defects, presumably of a genetic basis, are central to the pathogenesis of SLE but would require (environmental) triggers to induce disease expression [97]. Unfortunately, at this point, the actual triggering factors remain unknown. Popular candidates include infectious agents such as EBV and possibly other infections for which indirect, albeit in some cases quite strong, evidence has been reported [98]. Whatever the ultimate nature of the precipitating antigen may turn out to be, our unpublished results indicate that isolated stimulation of SLE 9G4 B cells through the BCR is not enough to induce full activation of these cells as demonstrated by low Ca2+ oscillations comparable to those observed with healthy 9G4 cells [91]. We believe that these results point to the need for co-stimulation through other receptors as a necessary factor to overcome anergic responses in 9G4 cells. This could be accomplished in SLE by the provision of T cell help, by defective T cell regulatory activity or by the contribution of an overactive innate immune system in a T cell-independent fashion. Examples of such mechanisms could include unabated IFN-α activity, direct dendritic cell/B cell interactions, excessive BLyS-mediated stimulation and co-stimulation of Toll-like receptors (TLR) [99, 100, 101, 102]. A tantalizing example of the latter mechanisms has indeed been described in rheumatoid factor (RF) transgenic mice in which tolerant RF B cells can be activated by co-stimulation of the BCR and TLR9 induced by IgG-chromatin immune complexes [13]. As previously discussed in this review, a similar mechanism has been invoked in human SLE to explain the production of IFN-α by dendritic cells (in this case by co-stimulation of Fcγ receptors and TLR9 by immune complexes of IgG-apoptotic cells) [81]. Along these lines, it is important to note that 9G4 B cells have the potential to recognize antigens such as LPS and apoptotic cells both through their BCR for antigen as well as through TLR4, and, therefore, it is plausible that these antigens might be responsible for disrupting tolerance in genetically predisposed individuals [64, 103, 104].
Concluding remarks
We propose that 9G4 autoantibodies play a beneficial role by participating in important physiological functions. Although the ultimate reason for the selection of autoreactive 9G4 B cells into the follicular and MZ compartments is still unknown, important clues are available. Thus, while virtually all 9G4 mAbs recognize RBC carbohydrate antigens, many such antibodies cross-react with other non-protein antigens such as bacterial LPS, DNA, and tumor gangliosides. In our working model, one could envision surface 9G4 antibodies as a type of innate immune receptor that could recognize repetitive patterns present in self- and microbial antigens [16]. Under conditions that provide strong stimulation these cells could differentiate into MZ cells and play a protective role either by clearing self antigens or by swiftly reacting with bacterial antigens. The required stimulation could be provided by several factors including the nature of the antigen and the engagement of appropriate coreceptors and signaling pathways. For example, the highest expression of the B cell-specific Toll-like receptor RP105, which is required for responsiveness to LPS via TLR4, is found in humans in the mantle zones where 9G4 cells are concentrated, suggesting that innate receptors may play a role in selecting these cells [105, 106].
Alternatively, 9G4 B cells could function as regulatory B cells interacting with innate T cells. Indeed, our unpublished results indicate that 9G4 B cells express substantial levels of CD1d (typical of MZ B cells) and CD1b (an antigen not previously recognized on human B cells) (Fig. 4). Both the expression of these MHC class I-like molecules and the nature of the antigens recognized by 9G4 B cells strongly suggest that they may play a role in the presentation of “unconventional” glycolipid antigens to CD1-restricted T cells. The exact nature of the antigens/epitopes presented by 9G4 B cells remains to be determined. Yet it seems reasonable that they could be derived from the same antigens recognized by 9G4 antibodies (NAL-containing glycosphingolipids/gangliosides, bacterial glycolipids) [59, 63, 107].
Fig. 4.
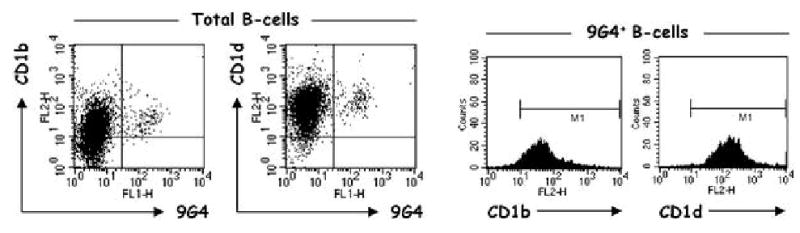
Expression of CD1 antigens in human B cells. Total CD19+ human tonsil B cells were stained with antibodies against CD1b and CD1d as well as 9G4. Both CD1b and CD1d are expressed in virtually 100% of 9G4 B cells with higher relative levels of CD1d. The universal expression of CD1b by 9G4 cells is in contrast to total B cells, which can be divided into CD1b-positive and -negative fractions
While the actual antigens presented by CD1 molecules in general remain fairly obscure and represent an evolving and controversial field, the available information is of significant help when thinking about 9G4 B cells. Thus, while some evidence indicates that all CD1-restricted T cells may recognize endogenous ligands [108], it is apparent that human CD1b may also present microbial lipid antigens such as mycobacterial glycolipids and bacterial lipoarabinomannan (LAM) in addition to self glycosphingolipids such as GM1 gangliosides [109, 110, 111]. Of significant interest, the GM1 epitope recognized by the autoreactive T cells is also present in bacterial carbohydrates, suggesting a possible cross-reactivity between self- and microbial glycolipid antigens [112]. In contrast to CD1b, no physiological ligands have been yet identified for CD1d since a compound that efficiently activates all human and mouse CD1d-restricted T cells (α-galactosylceramide; αGalCer) is merely a useful surrogate antigen derived from marine sponges but not present in mammalian cells [113]. However, the current consensus of opinion is that endogenous, self-glycolipid antigens represent the physiological ligand presented by CD1d to the main subset of human regulatory NKT cells (Vα24J αQ/V β11 invariant NKT cells) [108, 114, 115, 116].
Subsequently, 9G4 B cells could establish cognate interactions with antigen-specific CD1-restricted T cells belonging to different subsets since human CD1d antigens engage mostly TCR invariant NKT cells [either single-positive (SP) CD4+ or double-negative (DN) CD4−CD8− NKT cells], while CD1b molecules preferentially interact with a more diverse TCR repertoire expressed on either SP (CD4+ or CD8+) or DN αβ T cells [117, 118].
Finally, the functional consequences of these interactions could also vary substantially. Thus, it has been shown that CD4+ and CD8+ (CD1d-restricted) NKT cells derived from non-autoimmune BALB/c mice are capable of stimulating syngeneic B cells to secrete antibodies, and can also induce lupus-like autoimmune disease in vivo. In contrast, CD4/CD8 DN NKT cells (producing large amounts of IL-4 but no IL-10 or IFN-γ) suppressed the generation of autoimmunity [119]. On that basis, it can be postulated that in healthy individuals 9G4 B cells would present self glycolipids to CD1d-restricted DN NKT cells, and that this interaction would result in the survival of autoreactive 9G4 B cells, while suppressing autoantibody production. This scenario is certainly consistent with the immunoregulatory role proposed for DN NKT cells which, upon activation with αGalCer, have been shown to suppress autoimmune disease and to have anti-tumor effects [120, 121, 122]. On the other hand, 9G4 B cells could fulfill their role in microbial surveillance by presenting microbial glycolipids to CD1b-restricted αβ T cells.
In patients with SLE, the balance between these interactions would be shifted in the direction of autoimmunity either due to intrinsic B cell defects, abnormal T cell responses or both [123, 124] and MZ B cells stimulated by T cells could enter the GC reaction [125]. Of particular interest in this respect is the expansion of αβ DN T cells observed in murine and human lupus and in patients with the autoimmune lymphoproliferative syndrome (ALPS), who have a very high incidence of autoantibody-mediated cytopenias [119, 126, 127, 128, 129, 130, 131]. It is therefore possible that in SLE, 9G4 B cells could receive abnormal stimulation by αβ DN T cells and/or by the absence of regulatory NKT cells.
In summary, 9G4 B cells represent a fascinating population of autoreactive B cells, which, when restricted to innate immune responses, appear to play an important physiological role. In contrast, if selected into adaptive immune responses, 9G4 B cells become pathogenic agents and are likely to play an important role in SLE (Fig. 5). Understanding the behavior of these cells should contribute important insights into the regulation of human B cell tolerance and its breakdown in SLE.
Fig. 5.
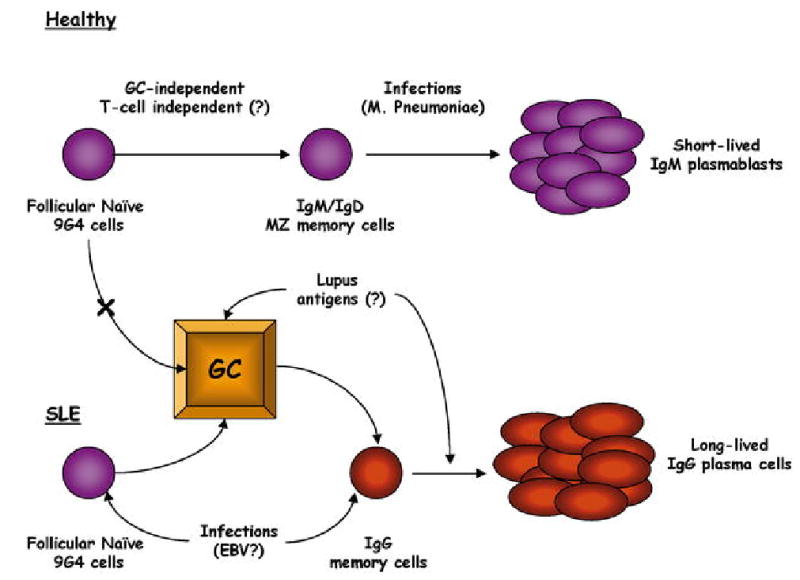
Hypothetical model for the regulation of autoreactive B cells. In healthy subjects, autoreactive 9G4 B cells are functionally sequestered within the innate immune system and participate in beneficial immune responses in a GC-independent, T cell-independent manner that prevents their accumulation in the long-lived IgG compartments. In contrast, in SLE intrinsically abnormal B cells respond to a number of possible stimuli (infections, excessive BAFF, excessive IFN-α, abnormal T cell regulation) by participating in productive GC reactions and expanding into the IgG post-GC memory and plasma cell compartments
Acknowledgments
This work was supported in part by grants to J.A. (NIAMS K08AR048303 and the Lupus Foundation of America) and I.S. (RO1 AI049660-01A1 and U19 AI56390).
References
- 1.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 2.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 3.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J Immunol. 1998;160:51. [PubMed] [Google Scholar]
- 4.Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000;1:475. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 5.Yu P, Wang Y, Chin RK, et al. B cells control the migration of a subset of dendritic cells into B cell follicles via CXC chemokine ligand 13 in a lymphotoxin-dependent fashion. J Immunol. 2002;168:5117. doi: 10.4049/jimmunol.168.10.5117. [DOI] [PubMed] [Google Scholar]
- 6.Schaerli P, Willimann K, Lang AB, et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boes M, Prodeus AP, Schmidt T, et al. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. 1998;188:2381. doi: 10.1084/jem.188.12.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ochsenbein AF, Fehr T, Lutz C, et al. Control of early viral and bacterial distribution and disease by natural antibodies. Science. 1999;286:2156. doi: 10.1126/science.286.5447.2156. [DOI] [PubMed] [Google Scholar]
- 9.van Essen D, Dullforce P, Gray D. Role of B cells in maintaining helper T-cell memory. Phil Trans Royal Soc London Series B. 2000;355:351. doi: 10.1098/rstb.2000.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fillatreau S, Sweenie CH, McGeachy MJ, et al. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 11.Boes M, Schmidt T, Linkemann K, et al. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. 2000;97:1184. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian J, Zekzer D, Hanssen L, et al. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 13.Leadbetter EA, Rifkin IR, Hohlbaum AM, et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 14.Fields ML, Erikson J. The regulation of lupus-associated autoantibodies: immunoglobulin transgenic models. Curr Opin Immunol. 2003;15:709. doi: 10.1016/j.coi.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Pugh-Bernard AE, Silverman GJ, Cappione AJ, et al. Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J Clin Invest. 2001;108:1061. doi: 10.1172/JCI12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a “natural immune memory”. Immunol Rev. 2000;175:70. [PubMed] [Google Scholar]
- 17.Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev. 2004;197:179. doi: 10.1111/j.0105-2896.2004.0109.x. [DOI] [PubMed] [Google Scholar]
- 18.Lopes-Carvalho T, Kearney JF. Development and selection of marginal zone B cells. Immunol Rev. 2004;197:192. doi: 10.1111/j.0105-2896.2004.0112.x. [DOI] [PubMed] [Google Scholar]
- 19.Wardemann H, Boehm T, Dear N, et al. B-1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J Exp Med. 2002;195:771. doi: 10.1084/jem.20011140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruetzmann S, Rosado MM, Weber H, et al. Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med. 2003;197:939. doi: 10.1084/jem.20022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasaian MT, Casali P. Autoimmunity-prone B-1 (CD5 B) cells, natural antibodies and self recognition. Autoimmunity. 1993;15:315. doi: 10.3109/08916939309115755. [DOI] [PubMed] [Google Scholar]
- 22.Zandvoort A, Lodewijk ME, de Boer NK, et al. CD27 expression in the human splenic marginal zone: the infant marginal zone is populated by naive B cells. Tissue Antigen. 2001;58:234. doi: 10.1034/j.1399-0039.2001.580403.x. [DOI] [PubMed] [Google Scholar]
- 23.Weller S, Braun MC, Tan BK, et al (2004) Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a pre-diversified immunoglobulin repertoire. Blood:2004 [DOI] [PMC free article] [PubMed]
- 24.Pascual V, Liu YJ, Magalski A, et al. Analysis of somatic mutation in five B cell subsets of human tonsil. J Exp Med. 1994;180:329. doi: 10.1084/jem.180.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bohnhorst J, Bjorgan MB, Thoen JE, et al. Bm1-bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the b cell subpopulations in patients with primary sjogren’s syndrome. J Immunol. 2001;167:3610. doi: 10.4049/jimmunol.167.7.3610. [DOI] [PubMed] [Google Scholar]
- 26.Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes; CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188:1679. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weller S, Faili A, Garcia C, et al. CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci USA. 2001;98:1166. doi: 10.1073/pnas.98.3.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cocca BA, Seal SN, D’Agnillo P, et al. Structural basis for autoantibody recognition of phosphatidylserine-beta 2 glycoprotein I and apoptotic cells. Proc Natl Acad Sci USA. 2001;98:13826. doi: 10.1073/pnas.241510698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potter KN, Li YC, Capra JD. The cross-reactive idiotopes recognized by the monoclonal antibodies 9G4 and LC1 are located in framework region 1 of two non-overlapping subsets of human VH4 family encoded antibodies. Scand J Immunol. 1994;40:43. doi: 10.1111/j.1365-3083.1994.tb03431.x. [DOI] [PubMed] [Google Scholar]
- 30.Potter KN, Hobby P, Klijn S, et al. Evidence for involvement of a hydrophobic patch in framework region 1 of human V4-34-encoded Igs in recognition of the red blood cell I antigen. J Immunol. 2002;169:3777. doi: 10.4049/jimmunol.169.7.3777. [DOI] [PubMed] [Google Scholar]
- 31.Zheng NY, Wilson K, Wang X, et al. Human immunoglobulin selection associated with class switch and possible tolerogenic origins for C delta class-switched B cells. J Clin Invest. 2004;113:1188. doi: 10.1172/JCI20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mockridge CI, Rahman A, Buchan S, et al. Common patterns of B cell perturbation and expanded V4-34 immunoglobulin gene usage in autoimmunity and infection. Autoimmunity. 2004;37:9. doi: 10.1080/08916930310001624656. [DOI] [PubMed] [Google Scholar]
- 33.Chapman CJ, Spellerberg MB, Smith GA, et al. Autoanti-red cell antibodies synthesized by patients with infectious mononucleosis utilize the VH4-21 gene segment. J Immunol. 1993;151:1051. [PubMed] [Google Scholar]
- 34.Cappione AJ, Pugh-Bernard AE, Anolik JH, et al. Lupus IgG VH4.34 antibodies bind to a 220-kDa glycoform of CD45/B220 on the surface of human B lymphocytes. J Immunol. 2004;172:4298. doi: 10.4049/jimmunol.172.7.4298. [DOI] [PubMed] [Google Scholar]
- 35.Stevenson FK, Smith GJ, North J, et al. Identification of normal B-cell counterparts of neoplastic cells which secrete cold agglutinins of anti-I and anti-i specificity. Br J Haematol. 1989;72:9. doi: 10.1111/j.1365-2141.1989.tb07643.x. [DOI] [PubMed] [Google Scholar]
- 36.Chapman C, Spellerberg M, Hamblin T, et al. Pattern of usage of the VH4-21 gene by B lymphocytes in a patient with EBV infection indicates ongoing mutation and class switching. Ann N Y Acad Sci. 1995;764:195. doi: 10.1111/j.1749-6632.1995.tb55827.x. [DOI] [PubMed] [Google Scholar]
- 37.Silberstein LE, Jefferies LC, Goldman J, et al. Variable region gene analysis of pathologic human autoantibodies to the related i and I red blood cell antigens. Blood. 1991;78:2372. [PubMed] [Google Scholar]
- 38.Pascual V, Victor K, Spellerberg M, et al. VH restriction among human cold agglutinins. The VH4-21 gene segment is required to encode anti-I and anti-i specificities. J Immunol. 1992;149:2337. [PubMed] [Google Scholar]
- 39.Parr TB, Johnson TA, Silberstein LE, et al. Anti-B cell autoantibodies encoded by VH 4-21 genes in human fetal spleen do not require in vivo somatic selection. Eur J Immunol. 1994;24:2941. doi: 10.1002/eji.1830241204. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Spellerberg MB, Stevenson FK, et al. The I binding specificity of human VH 4-34 (VH 4-21) encoded antibodies is determined by both VH framework region 1 and complementarity determining region 3. J Mol Biol. 1996;256:577. doi: 10.1006/jmbi.1996.0110. [DOI] [PubMed] [Google Scholar]
- 41.Schutte ME, van Es JH, Silberstein LE, et al. VH4.21-encoded natural autoantibodies with anti-i specificity mirror those associated with cold hemagglutinin disease. J Immunol. 1993;151:6569. [PubMed] [Google Scholar]
- 42.Andris JS, Miller AB, Abraham SR, et al. Variable region gene segment utilization in rhesus monkey hybridomas producing human red blood cell-specific antibodies: predominance of the VH4 family but not VH4-21 (V4-34) Mol Immunol. 1997;34:237. doi: 10.1016/s0161-5890(97)00021-7. [DOI] [PubMed] [Google Scholar]
- 43.Sanz I, Hwang LY, Hasemann C, et al. Polymorphisms of immunologically relevant loci in human disease. Autoimmunity and human heavy chain variable regions. Ann N Y Acad Sci. 1988;546:133. doi: 10.1111/j.1749-6632.1988.tb21628.x. [DOI] [PubMed] [Google Scholar]
- 44.Williams C, Weigel L, Sanz I, et al (1991) Small human VH gene families show remarkably little polymorphism. In: Cazenave P (ed) Anti-idiotypic vaccines. Progress in vaccinology. Springer, New York, p 22
- 45.van Dijk KW, Sasso EH, Milner EC. Polymorphism of the human Ig VH4 gene family. J Immunol. 1991;146:3646. [PubMed] [Google Scholar]
- 46.Weng NP, Snyder JG, Yu-Lee LY, et al. Polymorphism of human immunoglobulin VH4 germ-line genes. Eur J Immunol. 1992;22:1075. doi: 10.1002/eji.1830220430. [DOI] [PubMed] [Google Scholar]
- 47.Wang H, Clarke SH. Positive selection focuses the VH12 B-cell repertoire towards a single B1 specificity with survival function. Immunol Rev. 2004;197:51. doi: 10.1111/j.0105-2896.2004.0098.x. [DOI] [PubMed] [Google Scholar]
- 48.Eda S, Yamanaka M, Beppu M. Carbohydrate-mediated phagocytic recognition of early apoptotic cells undergoing transient capping of CD43 glycoprotein. J Biol Chem. 2004;279:5967. doi: 10.1074/jbc.M310805200. [DOI] [PubMed] [Google Scholar]
- 49.Feizi T, Monger E. Search for I antigen in human tissues. Nature. 1967;216:1025. doi: 10.1038/2161025a0. [DOI] [PubMed] [Google Scholar]
- 50.Feizi T. Immunochemical studies of mammalian glycoproteins with blood group I activity. Clin Sci Mol Med. 1973;45:17P. doi: 10.1042/cs045017p. [DOI] [PubMed] [Google Scholar]
- 51.Feizi T, Turberville C, Westwood JH. Blood-group precursors and cancer-related antigens. Lancet. 1975;II:391. doi: 10.1016/s0140-6736(75)92899-8. [DOI] [PubMed] [Google Scholar]
- 52.Feizi T. The I and i antigens on certain normal and pathologic tissues. Rev Fran Trans Immuno-Hematol. 1978;21:165. doi: 10.1016/s0338-4535(78)80039-7. [DOI] [PubMed] [Google Scholar]
- 53.Feizi T. The blood group Ii system: a carbohydrate antigen system defined by naturally monoclonal or oligoclonal autoantibodies of man. Immunol Commun. 1981;10:127. doi: 10.3109/08820138109050693. [DOI] [PubMed] [Google Scholar]
- 54.Thompson KM, Sutherland J, Barden G, et al. Human monoclonal antibodies against blood group antigens preferentially express a VH4-21 variable region gene-associated epitope. Scand J Immunol. 1991;34:509. doi: 10.1111/j.1365-3083.1991.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 55.Pascual V, Victor K, Lelsz D, et al. Nucleotide sequence analysis of the V regions of two IgM cold agglutinins. Evidence that the VH4-21 gene segment is responsible for the major cross-reactive idiotype. J Immunol. 1991;146:4385. [PubMed] [Google Scholar]
- 56.Grillot-Courvalin C, Brouet JC, Piller F, et al. An anti-B cell autoantibody from Wiskott-Aldrich syndrome which recognizes i blood group specificity on normal human B cells. Eur J Immunol. 1992;22:1781. doi: 10.1002/eji.1830220717. [DOI] [PubMed] [Google Scholar]
- 57.Smith G, Spellerberg M, Boulton F, et al. The immunoglobulin VH gene, VH4-21, specifically encodes autoanti-red cell antibodies against the I or i antigens. Vox Sanguinis. 1995;68:231. doi: 10.1111/j.1423-0410.1995.tb02578.x. [DOI] [PubMed] [Google Scholar]
- 58.Silberstein LE, George A, Durdik JM, et al. The V4-34 encoded anti-i autoantibodies recognize a large subset of human and mouse B-cells. Blood Cells Mol Dis. 1996;22:126. doi: 10.1006/bcmd.1996.0020. [DOI] [PubMed] [Google Scholar]
- 59.Thomas MD, Clough K, Melamed MD, et al. A human monoclonal antibody encoded by the V4-34 gene segment recognises melanoma-associated ganglioside via CDR3 and FWR1. Hum Antibodies. 1999;9:95. [PubMed] [Google Scholar]
- 60.Bhat NM, Bieber MM, Spellerberg MB, et al. Recognition of auto- and exoantigens by V4-34 gene encoded antibodies. Scand J Immunol. 2000;51:134. doi: 10.1046/j.1365-3083.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 61.Stevenson FK, Longhurst C, Chapman CJ, et al. Utilization of the VH4-21 gene segment by anti-DNA antibodies from patients with systemic lupus erythematosus. J Autoimmun. 1993;6:809. doi: 10.1006/jaut.1993.1066. [DOI] [PubMed] [Google Scholar]
- 62.Thorpe SJ, Turner CE, Stevenson FK, et al. Human monoclonal antibodies encoded by the V4-34 gene segment show cold agglutinin activity and variable multireactivity which correlates with the predicted charge of the heavy-chain variable region. Immunology. 1998;93:129. doi: 10.1046/j.1365-2567.1998.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhat NM, Bieber MM, Chapman CJ, et al. Human antilipid A monoclonal antibodies bind to human B cells and the i antigen on cord red blood cells. J Immunol. 1993;151:5011. [PubMed] [Google Scholar]
- 64.Spellerberg MB, Chapman CJ, Mockridge CI, et al. Dual recognition of lipid A and DNA by human antibodies encoded by the VH4-21 gene: a possible link between infection and lupus. Hum Antibodies Hybridomas. 1995;6:52. [PubMed] [Google Scholar]
- 65.Bieber MM, Bhat NM, Teng NN. Anti-endotoxin human monoclonal antibody A6H4C5 (HA-1A) utilizes the VH4.21 gene. Clin Infect Dis. 1995;21:S186. doi: 10.1093/clinids/21.supplement_2.s186. [DOI] [PubMed] [Google Scholar]
- 66.Steinberg AD, Gourley MF, Klinman DM, et al. NIH conference. Systemic lupus erythematosus. Ann Intern Med. 1991;115:548. doi: 10.7326/0003-4819-115-7-548. [DOI] [PubMed] [Google Scholar]
- 67.Krieg AM. CpG DNA: a pathogenic factor in systemic lupus erythematosus? J Clin Immunol. 1995;15:284. doi: 10.1007/BF01541318. [DOI] [PubMed] [Google Scholar]
- 68.Ronnblom L, Alm GV. An etiopathogenic role for the type I IFN system in SLE. Trends Immunol. 2001;22:427. doi: 10.1016/s1471-4906(01)01955-x. [DOI] [PubMed] [Google Scholar]
- 69.Isenberg D, Spellerberg M, Williams W, et al. Identification of the 9G4 idiotope in systemic lupus erythematosus. Br J Rheumatol. 1993;32:876. doi: 10.1093/rheumatology/32.10.876. [DOI] [PubMed] [Google Scholar]
- 70.Isenberg DA, McClure C, Farewell V, et al. Correlation of 9G4 idiotope with disease activity in patients with systemic lupus erythematosus. Ann Rheum Dis. 1998;57:566. doi: 10.1136/ard.57.9.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Vollenhoven RF, Bieber MM, Powell MJ, et al. VH4-34 encoded antibodies in systemic lupus erythematosus: a specific diagnostic marker that correlates with clinical disease characteristics. J Rheumatol. 1999;26:1727. [PubMed] [Google Scholar]
- 72.Feizi T, Taylor-Robinson D. Cold agglutinin anti-I and Mycoplasma pneumoniae. Immunology. 1967;13:405. [PMC free article] [PubMed] [Google Scholar]
- 73.Loomes LM, Uemura K, Childs RA, et al. Erythrocyte receptors for Mycoplasma pneumoniae are sialylated oligosaccharides of Ii antigen type. Nature. 1984;307:560. doi: 10.1038/307560a0. [DOI] [PubMed] [Google Scholar]
- 74.Pugh-Bernard AE, Cappione A, Anolik J, et al. From cold-agglutinin disease to systemic lupus erythematosus: lessons in human B-cell tolerance and Its breakdown. Trans Med Hemother. 2004;31:84. [Google Scholar]
- 75.Ciaffoni S, Luzzati R, Roata C, et al. Presence and significance of cold agglutinins in patients with HIV infection. Haematologica. 1992;77:233. [PubMed] [Google Scholar]
- 76.Riboldi P, Gaidano G, Schettino EW, et al. Two acquired immunodeficiency syndrome-associated Burkitt’s lymphomas produce specific anti-i IgM cold agglutinins using somatically mutated VH4-21 segments. Blood. 1994;83:2952. [PMC free article] [PubMed] [Google Scholar]
- 77.Bhat NM, Lee LM, Vollenhoven RV, et al. VH4-34 encoded antibody in systemic lupus erythematosus: effect of isotype. J Rheumatol. 2002;29:2114. [PubMed] [Google Scholar]
- 78.del Rincon I, Zeidel M, Rey E, et al. Delineation of the human systemic lupus erythematosus anti-smith antibody response using phage-display combinatorial libraries. J Immunol. 2000;165:7011. doi: 10.4049/jimmunol.165.12.7011. [DOI] [PubMed] [Google Scholar]
- 79.Bhat NM, Bieber MM, Stevenson FK, et al. Rapid cytotoxicity of human B lymphocytes induced by VH4-34 (VH4.21) gene-encoded monoclonal antibodies. Clin Exp Immunol. 1996;105:183. doi: 10.1046/j.1365-2249.1996.d01-733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bleesing JJ, Brown MR, Novicio C, et al. A composite picture of TCRalpha/beta(+) CD4(−)CD8(−) T cells (alpha/beta-DNTCs) in humans with autoimmune lymphoproliferative syndrome. Clin Immunol. 2002;104:21. doi: 10.1006/clim.2002.5225. [DOI] [PubMed] [Google Scholar]
- 81.Bave U, Alm GV, Ronnblom L. The Combination of apoptotic U937 cells and lupus IgG is a potent IFN-alpha inducer. J Immunol. 2000;165:3519. doi: 10.4049/jimmunol.165.6.3519. [DOI] [PubMed] [Google Scholar]
- 82.Blanco P, Palucka AK, Gill M, et al. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 83.Majeti R, Xu Z, Parslow TG, et al. An inactivating point mutation in the inhibitory wedge of CD45 causes lymphoproliferation and autoimmunity. Cell. 2000;103:1059. doi: 10.1016/s0092-8674(00)00209-9. [DOI] [PubMed] [Google Scholar]
- 84.Cyster JG, Healy JI, Kishihara K, et al. Regulation of B-lymphocyte negative and positive selection by tyrosine phosphatase CD45. Nature. 1996;381:325. doi: 10.1038/381325a0. [DOI] [PubMed] [Google Scholar]
- 85.Williams GW, Bluestein HG, Steinberg AD. Brain-reactive lymphocytotoxic antibody in the cerebrospinal fluid of patients with systemic lupus erythematosus: correlation with central nervous system involvement. Clin Immunol Immunopathol. 1981;18:126. doi: 10.1016/0090-1229(81)90016-7. [DOI] [PubMed] [Google Scholar]
- 86.Kraj P, Rao SP, Glas AM, et al. The human heavy chain Ig V region gene repertoire is biased at all stages of B cell ontogeny, including early pre-B cells. J Immunol. 1997;158:5824. [PubMed] [Google Scholar]
- 87.Pascual V, Widhopf G, Capra JD. The human VH repertoire: a restricted set of VH genes may be the target of immune regulation. Int Rev Immunol. 1992;8:147. doi: 10.3109/08830189209055570. [DOI] [PubMed] [Google Scholar]
- 88.Cappione A, Anolik J, Zheng B, et al. Attenuation of B cell receptor signaling in human autoreactive B cells. Implications for SLE. Arthritis Rheum. 2003;48:S270. [Google Scholar]
- 89.Goodnow CC, Cyster JG, Hartley SB, et al. Self-tolerance checkpoints in B lymphocyte development. Adv Immunol. 1995;59:279. doi: 10.1016/s0065-2776(08)60633-1. [DOI] [PubMed] [Google Scholar]
- 90.Brunswick M, June CH, Finkelman FD, et al. Surface immunoglobulin-mediated B-cell activation in the absence of detectable elevations in intracellular ionized calcium: a model for T-cell-independent B-cell activation. Proc Natl Acad Sci USA. 1989;86:6724. doi: 10.1073/pnas.86.17.6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anolik J, Cappione A, Sanz I. Biochemical and DNA microarray analysis of BCR-mediated signaling in a population of autoreactive human B-lymphocytes. Arthritis Rheum. 2002;46:S588. [Google Scholar]
- 92.Healy JI, Dolmetsch RE, Lewis RS, et al. Quantitative and qualitative control of antigen receptor signalling in tolerant B lymphocytes. Novartis Found Symp. 1998;215:137. doi: 10.1002/9780470515525.ch10. [DOI] [PubMed] [Google Scholar]
- 93.Vora KA, Wang LC, Rao SP, et al. Cutting edge: Germinal centers formed in the absence of B cell-activating factor belonging to the TNF family exhibit impaired maturation and function. J Immunol. 2003;171:547. doi: 10.4049/jimmunol.171.2.547. [DOI] [PubMed] [Google Scholar]
- 94.Rahman ZS, Rao SP, Kalled SL, et al. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med. 2003;198:1157. doi: 10.1084/jem.20030495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seo SJ, Fields ML, Buckler JL, et al. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 2002;16:535. doi: 10.1016/s1074-7613(02)00298-4. [DOI] [PubMed] [Google Scholar]
- 96.Rathmell JC, Cooke MP, Ho WY, et al. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376:181. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- 97.Sobel ES, Katagiri T, Katagiri K, et al. An intrinsic B cell defect is required for the production of autoantibodies in the lpr model of murine systemic autoimmunity. J Exp Med. 1991;173:1441. doi: 10.1084/jem.173.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.James JA, Neas BR, Moser KL, et al. Systemic lupus erythematosus in adults is associated with previous Epstein-Barr virus exposure. Arthritis Rheum. 2001;44:1122. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 99.Leadbetter EA, Rifkin IR, Marshak-Rothstein A. Toll-like receptors and activation of autoreactive B cells. Curr Dir Autoimmun. 2003;6:105. doi: 10.1159/000066858. [DOI] [PubMed] [Google Scholar]
- 100.Pascual V, Banchereau J, Palucka AK. The central role of dendritic cells and interferon-alpha in SLE. Curr Opin Rheumatol. 2003;15:548. doi: 10.1097/00002281-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 101.Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 102.Zhou T, Zhang J, Carter R, et al. BLyS and B cell autoimmunity. Curr Dir Autoimmun. 2003;6:21. doi: 10.1159/000066854. [DOI] [PubMed] [Google Scholar]
- 103.Pugh-Bernard A, Hocknell K, Cappione A, et al. VH4-34 anti-I/i autoantibodies recognize apoptotic cells. Arthritis Rheum. 2002;46:S126. [Google Scholar]
- 104.Devitt A, Moffatt OD, Raykundalia C, et al. Human CD14 mediates recognition and phagocytosis of apoptotic cells. Nature. 1998;392:505. doi: 10.1038/33169. [DOI] [PubMed] [Google Scholar]
- 105.Chan VW, Mecklenbrauker I, Su I, et al. The molecular mechanism of B cell activation by toll-like receptor protein RP-105. J Exp Med. 1998;188:93. doi: 10.1084/jem.188.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miura Y, Shimazu R, Miyake K, et al. RP105 is associated with MD-1 and transmits an activation signal in human B cells. Blood. 1998;92:2815. [PubMed] [Google Scholar]
- 107.Feizi T. Structural and biological aspects of blood group I and i antigens on glycolipids and glycoproteins. Rev Franc Trans Immuno-Hematol. 1980;23:563. doi: 10.1016/s0338-4535(80)80160-7. [DOI] [PubMed] [Google Scholar]
- 108.Shinkai K, Locksley RM. CD1, tuberculosis, and the evolution of major histocompatibility complex molecules. J Exp Med. 2000;191:907. doi: 10.1084/jem.191.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sieling PA, Chatterjee D, Porcelli SA, et al. CD1-restricted T cell recognition of microbial lipoglycan antigens. Science. 1995;269:227. doi: 10.1126/science.7542404. [DOI] [PubMed] [Google Scholar]
- 110.Shamshiev A, Donda A, Carena I, et al. Self glycolipids as T-cell autoantigens. Eur J Immunol. 1999;29:1667. doi: 10.1002/(SICI)1521-4141(199905)29:05<1667::AID-IMMU1667>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 111.Sugita M, Brenner MB. T lymphocyte recognition of human group 1 CD1 molecules: implications for innate and acquired immunity. Semin Immunol. 2000;12:511. doi: 10.1006/smim.2000.0277. [DOI] [PubMed] [Google Scholar]
- 112.Prendergast MM, Lastovica AJ, Moran AP. Lipopolysaccharides from Campylobacter jejuni O:41 strains associated with Guillain-Barre syndrome exhibit mimicry of GM1 ganglioside. Infect Immun. 1998;66:3649. doi: 10.1128/iai.66.8.3649-3655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brossay L, Chioda M, Burdin N, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med. 1998;188:1521. doi: 10.1084/jem.188.8.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.D’Andrea A, Goux D, De Lalla C, et al. Neonatal invariant Valpha24+ NKT lymphocytes are activated memory cells. Eur J Immunol. 2000;30:1544. doi: 10.1002/1521-4141(200006)30:6<1544::AID-IMMU1544>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 115.Benlagha K, Bendelac A. CD1d-restricted mouse V alpha 14 and human V alpha 24 T cells: lymphocytes of innate immunity. Semin Immunol. 2000;12:537. doi: 10.1006/smim.2000.0276. [DOI] [PubMed] [Google Scholar]
- 116.Park SH, Benlagha K, Lee D, et al. Unaltered phenotype, tissue distribution and function of Valpha14(+) NKT cells in germ-free mice. Eur J Immunol. 2000;30:620. doi: 10.1002/1521-4141(200002)30:2<620::AID-IMMU620>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 117.Porcelli S, Morita CT, Brenner MB. CD1b restricts the response of human CD4−8− T lymphocytes to a microbial antigen. Nature. 1992;360:593. doi: 10.1038/360593a0. [DOI] [PubMed] [Google Scholar]
- 118.Gumperz JE, Brenner MB. CD1-specific T cells in microbial immunity. Curr Opin Immunol. 2001;13:471. doi: 10.1016/s0952-7915(00)00243-0. [DOI] [PubMed] [Google Scholar]
- 119.Zeng D, Dick M, Cheng L, et al. Subsets of transgenic T cells that recognize CD1 induce or prevent murine lupus: role of cytokines. J Exp Med. 1998;187:525. doi: 10.1084/jem.187.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kawano T, Nakayama T, Kamada N, et al. Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res. 1999;59:5102. [PubMed] [Google Scholar]
- 121.Sharif S, Arreaza GA, Zucker P, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med. 2001;7:1057. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- 122.Singh AK, Wilson MT, Hong S, et al. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801. doi: 10.1084/jem.194.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chan OT, Hannum LG, Haberman AM, et al. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mohan C, Morel L, Yang P, et al. Accumulation of splenic B1a cells with potent antigen-presenting capability in NZM2410 lupus-prone mice. Arthritis Rheum. 1998;41:1652. doi: 10.1002/1529-0131(199809)41:9<1652::AID-ART17>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 125.Oliver AM, Martin F, Kearney JF. IgM(high)CD21(high) lymphocytes enriched in the splenic marginal zone generate effector cells more rapidly than the bulk of follicular B cells. J Immunol. 1999;162:7198. [PubMed] [Google Scholar]
- 126.Datta SK, Patel H, Berry D. Induction of a cationic shift in IgG anti-DNA autoantibodies. Role of T helper cells with classical and novel phenotypes in three murine models of lupus nephritis. J Exp Med. 1987;165:1252. doi: 10.1084/jem.165.5.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double-negative (CD4-/CD8-) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol. 1989;143:103. [PubMed] [Google Scholar]
- 128.Porcelli S, Yockey CE, Brenner MB, et al. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med. 1993;178:1. doi: 10.1084/jem.178.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Devi BS, Van Noordin S, Krausz T, et al. Peripheral blood lymphocytes in SLE—hyperexpression of CD154 on T and B lymphocytes and increased number of double negative T cells. J Autoimmun. 1998;11:471. doi: 10.1006/jaut.1998.0213. [DOI] [PubMed] [Google Scholar]
- 130.Zeng D, Lee MK, Tung J, et al. Cutting edge: A role for CD1 in the pathogenesis of lupus in NZB/NZW mice. J Immunol. 2000;164:5000. doi: 10.4049/jimmunol.164.10.5000. [DOI] [PubMed] [Google Scholar]
- 131.Straus SE, Sneller M, Lenardo MJ, et al. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med. 1999;130:591. doi: 10.7326/0003-4819-130-7-199904060-00020. [DOI] [PubMed] [Google Scholar]
- 132.Anolik J, Barnard J, Cappione A, Pugh-Bernard A, Felger R, Looney J, Sanz I (2004) Rituximab normalizes peripheral B-cell abnormalities in SLE. Arthritis Rheum (in press) [DOI] [PubMed]