

Abstract
Background: CHOP is a transcriptional regulator involved in apoptosis caused by endoplasmic reticulum (ER) stress. We previously reported that CHOP as well as other ER stress response genes is induced in the liver of a murine model of intragastric ethanol feeding. This study was undertaken to determine the role of CHOP in hepatocellular apoptosis and liver injury in this model.
Methods: CHOP wild-type (+/+) mice and CHOP null (-/-) mice were fed alcohol for four weeks with glucose as control. Hematoxylin-eosin staining, TUNEL, and caspase 3 staining of liver tissues were performed for assessment of fatty liver, necroinflammation, and apoptosis. Total RNA was extracted for microarray and reverse transcription-PCR analyses, and proteins were used for western blotting.
Results: Significant increased liver/body ratio, steatosis, liver triglyceride levels, and plasma homocysteine concentrations were observed in alcohol-fed mice as compared with controls in both genotypes. There was no significant difference between wild-type and CHOP null (-/-) mice in the parameters related to fatty liver. Alcohol-induced increased serum alanine aminotransferase levels and necroinflammatory foci were not significantly reduced in CHOP null (-/-) mice. However, apoptosis was present in alcohol-fed wild-type mice but virtually absent in alcohol-fed CHOP null (-/-) mice. The ER stress response indicated by increased Grp78 mRNA was observed in both types of mice fed alcohol. Of 12,423 transcripts analyzed for ≥ two-fold changes, several related to apoptosis were influenced by CHOP: Gadd45 and cathepsin B were up-regulated in ethanol-fed wild-type mice but not in CHOP null (-/-) mice, whereas Jun D and Bcl-xL were down-regulated in ethanol-fed wild-type mice but not in ethanol-fed CHOP null (-/-) mice.
Conclusions: CHOP null (-/-) mice have remarkable absence of hepatocellular apoptosis in response to alcohol feeding but no protection against hyperhomocysteinemia, ER stress, and fatty liver. Thus, CHOP up-regulation occurs downstream of and contributes to one manifestation of ER stress, namely, apoptosis. Microarray studies confirmed by PCR analysis and western blotting indicate that genes affected by CHOP are both proapoptotic and antiapoptotic and CHOP induction by ethanol may tip the balance of cell survival and death toward apoptosis.
WE PREVIOUSLY IDENTIFIED the endoplasmic reticulum (ER) stress response in the murine model of intragastric alcohol feeding and linked this response to alcohol-induced hyperhomocysteinemia associated with down-regulation of methionine synthase (Ji et al., 2004; Ji and Kaplowitz, 2003). The reversal of hyperhomocysteinemia by feeding betaine, which provides a methyl donor for conversion of homocysteine to methionine, abrogated ER stress along with the pathologic features of alcoholic liver disease, namely, fatty liver, necroinflammation, and apoptosis. It is believed that excess homocysteine leads to homocysteinylation of proteins causing their accumulation or malfolding in the ER (Werstuck et al., 2001; Zhang et al., 2001). This triggers the unfolded protein response, which is an adaptive, protective mechanism. However, prolonged or severe malfolding leads to a more extensive and complex response, referred to as ER stress, which includes an attempt to destroy the malfunctioning cells by apoptosis. Several potential mechanisms for ER stress-induced apoptosis have been proposed, but the one considered essential involves the participation of CHOP (Ji and Kaplowitz, 2004; Oyadomari and Mori, 2004).
CHOP has been described as a growth arrest and DNA damage-inducible gene that encodes a C/EBP-homologous protein of 29 kDa. CHOP is a transcription factor that when bound to its partner C/EBPβ causes transcription of certain genes and acts as a dominant regulative of C/EBP dimers (Ron and Habener, 1992). CHOP is up-regulated in ER stress mainly through the activation of PERK and consequent up-regulation of ATF4 (Fawcett et al., 1999; Harding et al., 2000; Luo et al., 2003). CHOP -/- cells are resistant to ER stress-induced apoptosis (Zinszner, 1998). However, the mechanism of how CHOP promotes apoptosis is not well understood. CHOP has been reported to regulate DOC1, a carbonic anhydrase that catalyzes the reversible hydration of CO2 to H2CO3 (Sok et al., 1999) causing intracellular acidification. Because the membrane pore-forming activity of the proapoptotic regulator Bax is pH dependent, CHOP- and DOC1-dependent intracellular acidification may contribute to apoptosis by increasing the pore-forming activity of Bax. Overexpression of CHOP led to down-regulation of BclII expression, depletion of cellular GSH, and exaggerated production of ROS in several cell lines. Restoration of BclII expression in CHOP-overexpressing cells led to replenishment of GSH and a reduction in levels of ROS and protected cells from ER stress-induced cell death (McCullough et al., 2001). Overexpression of CHOP was also linked to enhanced c-Jun N-terminal protein kinase activation and to increased sensitivity to thapsigargin-induced cell death (Li and Holbrook, 2004). Most recently, CHOP was shown to be responsible for ER stress-mediated induction of the death receptor-DR5 in cancer cells (Yamaguchi and Wang, 2004), further suggesting a critical role of CHOP in regulating apoptosis during ER stress.
In the present study, we examined the role of CHOP in the pathogenesis of liver injury in the murine model of intragastric ethanol feeding by comparing the pathologic and gene expression changes in CHOP null (-/-) and CHOP wild-type (+/+) mice.
MATERIALS AND METHODS
Ethanol-Fed Animals
Mice (C57BL/6) were bred from two breeding pairs of CHOP heterozygous mutant (provided by David Ron, School of Medicine, New York University). The intragastric ethanol infusion model was described previously (Ji and Kaplowitz, 2003). Briefly, mice underwent aseptic surgery during general anesthesia with ketamine and xylazine for implantation of a long-term gastrostomy catheter. After a one-week acclimatization period with infusion of a control high fat diet, ethanol infusion was initiated at a dosage of 18 g/kg/day, which was increased by 1.5 g every two days until it reached 29 g/kg/day. At the initial ethanol dose, total caloric intake derived from a diet and ethanol was set at 533 cal/kg, and the caloric percentages of ethanol, dietary carbohydrate (dextrose), protein (lactalbumin hydrolysate), and fat (corn oil) were 24.3%, 15.7%, 25%, and 35%, respectively. The highest ethanol dose at the end of four weeks accounted for 34% of calories. Adequate vitamin and salt mix were included at the recommended amounts by the Committee on Animal Nutrition of the National Research Council (AIN-76A, 4.42 g/liter and 15.4 g/liter, respectively; Dyets, Inc., PA). The animals were treated in accordance with the Guide for Care and Use of Laboratory Animals (NIH, Bethesda, MD, Publication 86-23, 1985).
Hematoxylin-Eosin Staining and TUNEL
Detailed procedures were described previously (Ji et al., 2003, 2004). Briefly, at the time of death, small pieces of liver tissue were harvested and fixed immediately in 10% buffered formalin phosphate and 3% paraformaldehyde (Sigma, St. Louis, MO). After paraffin embedding, 5-μm transverse sections were prepared and stained with hematoxylin-eosin. Liver sections were coded and fat graded on a scale of zero to four, with zero representing normal liver architecture (Ji and Kaplowitz, 2003). Apoptotic hepatocytes were detected by the TUNEL procedures with a TACS TdT Kit (R&D Systems, Inc.) and by immunohistochemical analysis with antiactivated caspase 3 antibodies. Clusters of inflammatory cells replacing areas of parenchyma in hematoxylin-eosin-stained sections were described as necroinflammatory foci. Quantitation of hepatic apoptosis and necroinflammatory foci was described previously (Ji et al., 2004; Matsumaru et al., 2003).
DNA Isolation and Genotyping With PCR Analysis
Genomic DNA was extracted from the tails of F1 mice using the QIAGEN DNeasy Tissue Kit (Valencia, CA). CHOP wild-type (+/+) and CHOP null (-/-) mice were identified by PCR analysis using two pairs of primers: CHOP forward, 5′-CACTACTCTTGACCCTGCGT-3′; CHOP reverse, 5′-GGAGAGACAGACAGGAGGTGAT-3′; PGK-Neo forward, 5′-CGGGTAGGGGAGGCGCTT-3′; PKG-Neo reverse, 5′-CAAGGAACGCCCGTCGTG-3′. The Tag PCR Master Mix Kit from QIAGEN was used for PCR analysis.
RNA Isolation and One-Step Reverse Transcription-PCR (RT-PCR) Analysis. Total hepatic RNA was isolated from fresh liver tissues using the RNeasy Mini Kit from QIAGEN following the manufacturer’s instructions and with the addition of 500 units of a RNase inhibitor (RNAguard, Amersham Phamacia Biotech) to each starting material (usually 300 mg of liver tissue). RNA was stored at -80°C until use. The QIAGEN OneStep RT-PCR Kit was used for RT-PCR analysis. The primer sequences used are listed in Table 1.
Table 1.
Pathological Features of CHOP (+/+ vs -/-) Mice Fed Ethanol
| Genotype |
CHOP (+/+) |
CHOP (-/-) |
||
|---|---|---|---|---|
| Treatment | Control (n = 3) | Ethanol (n = 4) | Control (n = 4) | Ethanol (n = 4) |
| Liver/body | 0.053 ± 0.001 | 0.071 ± 0.002* | 0.056 ± 0.004 | 0.067 ± 0.003* |
| Steatosis score | 0.17 ± 0.03 | 2.5 ± 0.58* | 0.13 ± 0.25 | 2.1 ± 0.25* |
| Triglycerides (mg/mg protein) | 0.065 ± 0.016 | 0.305 ± 0.019* | 0.052 ± 0.010 | 0.29 ± 0.018* |
| ALT (U) | 14 ± 0.6 | 112 ± 14* | 13 ± 1.7 | 87 ± 37* |
| Hcy (μM) | 2.6 ± 0.75 | 18.3 ± 1.93 | 2.9 ± 0.22 | 17.3 ± 3.65* |
| Necroinflammatory foci1 | 0 | 0.5 ± 0.6* | 0 | 0.3 ± 0.5* |
| Apoptotic nuclei2 | 0.3 ± 0.6 | 5.5 ± 1.3* | 0 | 0 |
number of necrotic foci counted in five microscope fields of each section of liver tissue with x200 original magnification.
number of apoptotic nuclei counted in five microscope fields of each section of liver tissue with x200 original magnification.
p < 0.05 compared to control.
The PCR optimal cycle number for each gene was determined empirically to obtain detectable but nonsaturating PCR product. To quantitate mRNA abundance of genes, the intensity of the unknown sample was determined with the PhosphorImage (Molecular Dynamics, Sunnyvale, CA) following the manufacturer’s instructions, and the relative expression was compared and normalized to the expression of β-actin in that same sample. The increase/decrease and SEM were calculated from analysis for all mice from each group. The absolute value for basal gene expression between genes was not compared.
Microarray Gene Analysis
Mouse gene chip-MG-U74Av2 from Affymetrix (Santa Clara, CA) was used for large-scale gene profiling. The gene expression analysis was provided by the Microarray Core Facility Service of the USC Research Center for Liver Diseases. Two samples from each group were analyzed. Changes of genes with a two-fold cutoff value were reported. About 60% of the changes could be confirmed by RT-PCR analysis and western blotting using the criterion.
Western Blotting
Proteins were extracted according to a previously reported method (Ji and Kaplowitz, 2003; Matsumaru et al., 2003). Briefly, 40 μg of protein/lane was run in 12% denatured polyacrylamide gel and electrophoresed on a nitrocellulose membrane for detection by primary and secondary antibodies. Antibodies to GRP78, CHOP, Jun D, Gadd45, Bcl-xL, and DR5 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). After incubation with primary antibody, the membrane was washed and incubated with corresponding horseradish peroxidase-labeled secondary antibody (Santa Cruz Biotechnology, Inc.) or alkaline phosphatase-labeled antibody (Cell Signaling Technology) for 45 min. Proteins were visualized using LumiGLO Reagent (Cell Signaling Technology) on CL-Xposure films (PIERCE, Rockford, IL) at an optimized time point. Relative signal intensity was quantified with the PhosphorImage.
Serum alanine aminotransferase and homocysteine levels were analyzed using previously described methods (Ji and Kaplowitz, 2003). The levels of triglycerides and cholesterol in mouse sera and livers were determined using the Sigma Diagnostics Triglyceride and Infinity Cholesterol Reagent.
Statistical Analysis
Experiments were performed routinely with four to six mice per group with values presented as mean ± SD. All the studies were replicated with representative data shown. Statistical analysis was performed using the Student’s t test for unpaired data or ANOVA and the Tukey-Kamer multiple comparisons test post hoc. p < 0.05 was considered significant.
RESULTS
Pathologic Features of Alcohol-Fed Mice Deficient in CHOP
Twenty-nine F1 mice were obtained from the breeding, of which seven were wild-type (+/+) indicated by presence of a CHOP fragment and absence of a PGK-Neo fragment (Ron et al., 1992), 14 were CHOP heterozygous (±), and eight were CHOP null (-/-) indicated by absence of the CHOP fragment and presence of the PGK-Neo fragment. Wild-type and null mice were then used for the intragastric ethanol infusion model. Significant increases in liver/body ratio, steatosis score, liver triglyceride levels, and alanine aminotransferase concentration were observed in both genotypes in response to ethanol exposure (Table 1; Fig. 1A). There was no difference between wild-type and CHOP null mice in these parameters. Compared with controls, plasma homocysteine levels were increased by seven-fold in wild-type mice and six-fold in CHOP null mice. Necroinflammatory foci were observed in both genotypes fed ethanol (Table 1). Using TUNEL and activated caspase 3 staining, increased hepatic apoptosis was detected in wild-type mice but not in CHOP null mice in response to ethanol treatment (Table 1; Fig. 1B, C).
Fig. 1.
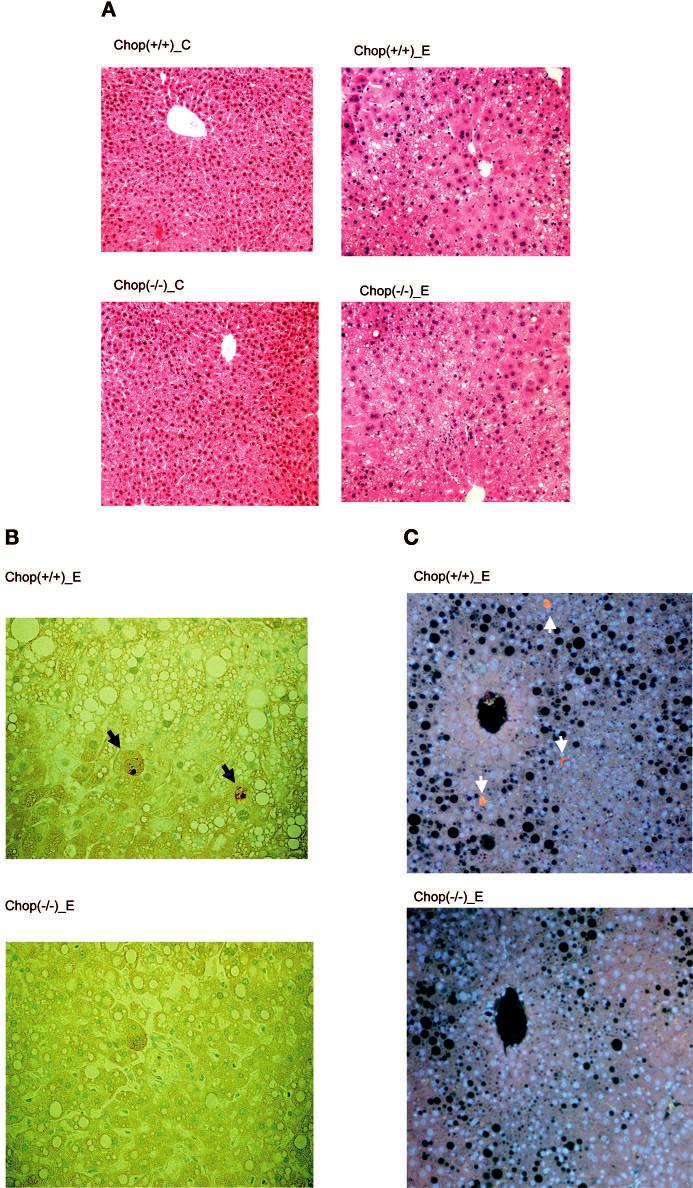
Light microscopic appearance of liver tissues from control and ethanol-fed CHOP mice at four weeks. CHOP (+/+), CHOP wild-type mice; CHOP (-/-), CHOP null mice; C, pair-fed mice; E, ethanol-fed mice. A) Hematoxylin-eosin staining (magnification, ×100) shows alcohol-induced fat accumulation and necroinflammatory foci. B) Alcohol-induced apoptosis (TUNEL; magnification, ×200). The arrowheads identify apoptotic hepatocytes in tissue from wild-type mice; normal nuclei stain green. C) Alcohol-induced apoptosis in tissue from wild-type mice identified by antiactive caspase 3 antibodies (immunohistochemical analysis; magnification, ×200). Normal nuclei stain blue, and the black holes are fat. The pair-fed controls had no identifiable caspase 3-positive cells (data not shown). Arrows indicate apoptotic cells in B and C.
Gene Expression Profile in Alcohol-Fed Mice Deficient in CHOP
To identify genes that are regulated after ethanol treatment, we analyzed a total of 12,423 genes for up/down expression with a two-fold cutoff value. Seventeen genes were up-regulated and 12 genes were down-regulated in both wild-type and CHOP null mice fed ethanol (Table 2). The overall pattern showed a few major differences in extent of changes. For example, CYP2b10 and 2b13 and GSTμ3 were more greatly induced in CHOP -/- mice, and several genes such as immunoglobulin κ chain variable 28, regulator of G protein signaling 3, and phospholipid transfer protein were more greatly repressed in wild-type animals. However, the significance of these differences between null and wild type is uncertain, and most of the changes were comparable. Regarding other insights into the pathogenesis of alcohol injury independent of genotype, the expected induction of Cyp2e1 was observed, and confirmation of induction of the ER stress markers Grp78 and ER stress-responsive SREBP-1 was observed. Of interest NIP3 was induced, confirming our previous identification of the induction of this BclII family member, which promotes necrosis (Ji and Kaplowitz, 2003), and betaine-homocysteine methyltransferase and methionine adenosyltransferase 1α were down-regulated, which may contribute to hyperhomocysteinemia (Avila et al., 2000; Barak et al., 1996, 2003; Ji et al., 2004; Lu et al., 2002). Furthermore, many of the changes in Table 1 agree with findings of previous studies using this approach. For example, up-regulation of CYP2c29 and CYP2b9 and carbonyl reductase and down-regulation of alcohol dehydrogenase and fatty acid synthase were reported (Deaciuc et al, 2004a, 2004b; Ji and Kaplowitz, 2003; Seth et al., 2003).
Table 2.
Genes that Were Up- or Down-Regulated in Both Wild Type and CHOP (-/-) Mice Fed Ethanol
| Fold of change |
|||
|---|---|---|---|
| Gene ID | Gene name | Wild type | CHOP (-/-) |
| X81579 | Insulin-like growth factor binding protein 1 IGFBP-1 | 10.9 | 4.9 |
| X61800 | CCAAT/enhancer binding protein (C/EBP), δ | 6.2 | 3.8 |
| J04596 | GRO1 oncogene | 4.7 | 28.4 |
| M21856 | Cytochrome P450, 2b10 | 4.5 | 15.7 |
| M60358 | Cytochrome P450, 2b13 | 3.4 | 2.7 |
| D17674 | Cytochrome P450, 2c29 | 3.4 | 4.4 |
| AF009605 | Phosphoenolpyruvate carboxykinase 1 | 3.1 | 2.6 |
| M21855 | Cytochrome P450, 2b9 | 2.9 | 2.8 |
| X73230 | Arylsulfatase A (ASA) | 2.8 | 7.3 |
| J03953 | Glutathione S-transferase, μ3 | 2.6 | 19.8 |
| X01026 | Cytochrome P450, 2e1 | 2.6 | 2.1 |
| U31966 | Carbonyl reductase 1 | 2.5 | 3.4 |
| X57349 | Transferrin receptor | 2.5 | 2.2 |
| AF041054 | BCL2/adenovirus E1B 19 kDa-interacting protein 1, NIP3 | 2.1 | 3.6 |
| AJ002387 | Glucose-regulated protein, 78kD | 2.1 | 2.6 |
| U49861 | Deiodinase, iodothyronine, type I | 2.1 | 4.9 |
| AI843895 | Sterol regulatory element binding factor 1 | 2.0 | 2.4 |
| M18237 | Immunoglobulin kappa chain variable 28 | −27.4 | −2.2 |
| AI844739 | Regulator of G–protein signaling 3 | −12.7 | −3.3 |
| U87147 | Flavin containing monooxygenase 3 | −12.4 | −15.7 |
| U28960 | Phospholipid transfer protein | −10.1 | −5.1 |
| U88327 | Cytokine inducible SH2-containing protein 2 | −9.4 | −10.9 |
| Y14004 | Cytosolic acyl-CoA thioesterase 1 | −6.3 | −2.6 |
| D42048 | Squalene epoxidase | −5.1 | −2.0 |
| X13135 | Fatty acid synthase | −4.6 | −2.4 |
| AF033381 | Betaine-homocysteine methyltransferase | −4.4 | −2.1 |
| AI046345 | Alcohol dehydrogenase 4 | −3.8 | −2.2 |
| L13622 | Methionine adenosyltransferase I, α | −3.8 | −2.2 |
| U96116 | Hydroxysteroid (17-β) dehydrogenase 10 | −3.1 | −2.3 |
To identify candidate genes that are specifically regulated by CHOP, we list in Table 3 hepatic genes that responded differently to ethanol feeding in CHOP null mice versus wild-type mice. Four genes were up-regulated in wild-type mice but down-regulated in CHOP null mice fed ethanol. Growth arrest and DNA damage-inducible 45 and cathepsin B that promote apoptosis were among the down-regulated genes. Ten genes were down-regulated in wild type but up regulated in CHOP null mice fed ethanol. Apoptosis-related genes such as topoisomerase (DNA) III, Bcl-xL, Jun D, and Jun B were among those that were up-regulated (Deaciuc et al., 2004a, 2004b; Seth et al., 2003).
Table 3.
Genes that Were Differentially Regulated in Wild Type and in CHOP (-/-) Mice Fed Ethanol
| Fold of change |
|||
|---|---|---|---|
| Gene ID | Gene name | Wild type | CHOP (-/-) |
| U94828 | Regulator of G-protein signaling 16 | 5.9 | −2.0 |
| AF055638 | Growth arrest and DNA-damage-inducible 45γ | 4.7 | −3.4 |
| L06047 | Glutathione S-transferase α | 2.4 | −2.1 |
| M65270 | Cathepsin B | 2.2 | −2.9 |
| L11333 | Esterase 31 | −11.2 | 2.3 |
| AV141027 | Cytochrome P450, 7b1 | −4.0 | 4.2 |
| AB013603 | Topoisomerase (DNA) III | −3.6 | 2.2 |
| AI842328 | Calmodulin 3 | −3.4 | 2.8 |
| L35049 | Bcl-xL | −3.0 | 2.4 |
| J04696 | Glutathione S-transferase, μ2 | −2.8 | 6.7 |
| J04509 | Jun-D | −2.2 | 2.1 |
| U20735 | Jun-B | −2.1 | 3.9 |
| AF037371 | Cytochrome c oxidase | −2.1 | 2.7 |
| M21285 | Stearoyl-Coenzyme A desaturase 1 | −2.7 | 2.0 |
RT-PCR Analysis and Western Blotting of Selective ER Stress and CHOP-Targeted Genes
The ER stress response was detected in both wild-type and CHOP null mice exposed to alcohol (Fig. 2). The mRNA of Grp78 was increased in wild-type and CHOP null mice in response to alcohol treatment. The mRNA of CHOP was increased in wild-type mice but, as expected, was absent in CHOP null mice in response to ethanol treatment.
Fig. 2.
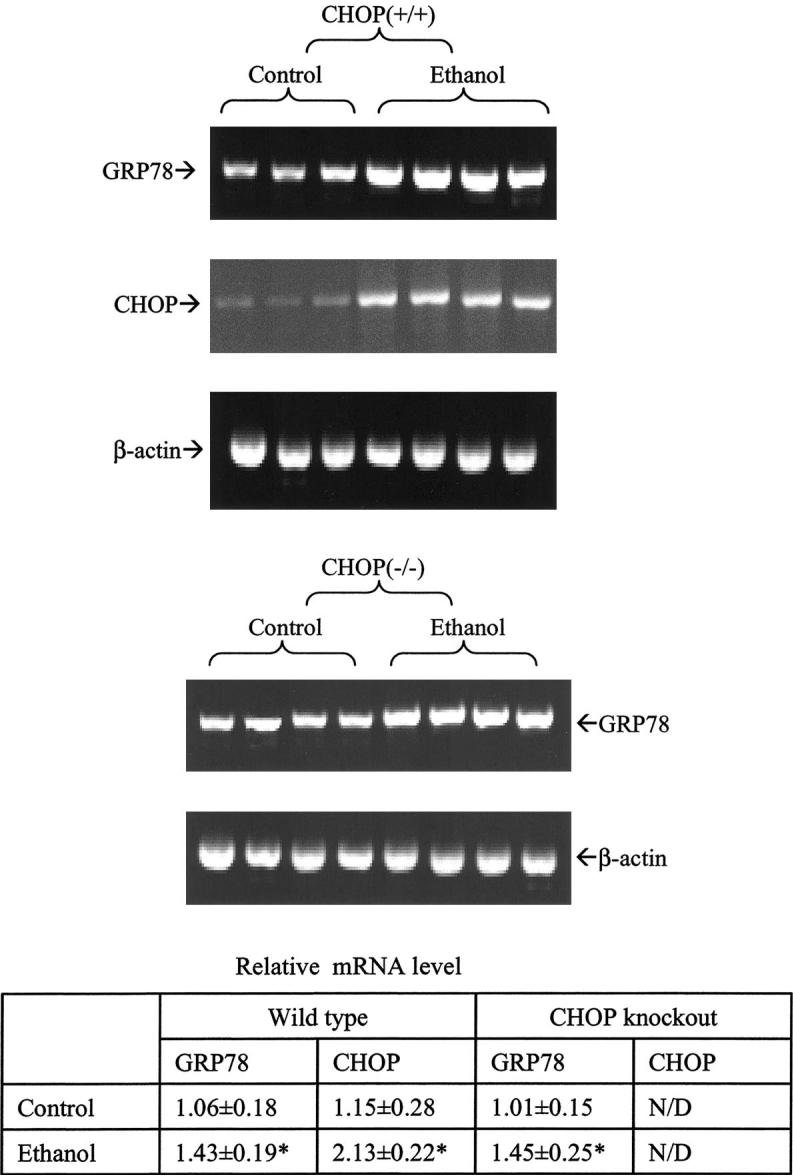
Endoplasmic reticulum (ER) stress response in mice fed alcohol for four weeks. GRP78, glucose-regulated protein; CHOP (+/+), CHOP wild-type mice; CHOP (-/-), CHOP null mice. Upper panel, reverse transcription-PCR analysis; lower panel, quantitation of mRNA levels. * p < 0.05 as compared with control (n = 3 or 4).
RT-PCR analysis and western blotting were used to confirm the possible CHOP-targeted genes revealed by the microarray analysis. RT-PCR analysis confirmed induction of mRNA of Gadd45 and cathepsin B in wild-type but not CHOP null mice in response to alcohol treatment (Fig. 3). Both Jun D and Bcl-xL were down-regulated in ethanol-treated wild-type but not in CHOP null mice. On the western blots (Fig. 4), CHOP and Gadd45 proteins were increased in wild-type but not in CHOP null mice in response to ethanol exposure. Both Jun D and Bcl-xL proteins were decreased in wild type but not in CHOP null mice exposed to ethanol.
Fig. 3.

Reverse transcription-PCR analysis of genes regulated by CHOP in ethanol-fed mice. The table shows densitometric values for three mice per group normalized to the expression of β-actin (means ± SD). C, control; E, alcohol treated; Gadd45, growth arrest and DNA damage-inducible protein 45; DR5, death receptor 5 or trail receptor 2. *, p < 0.05.
Fig. 4.
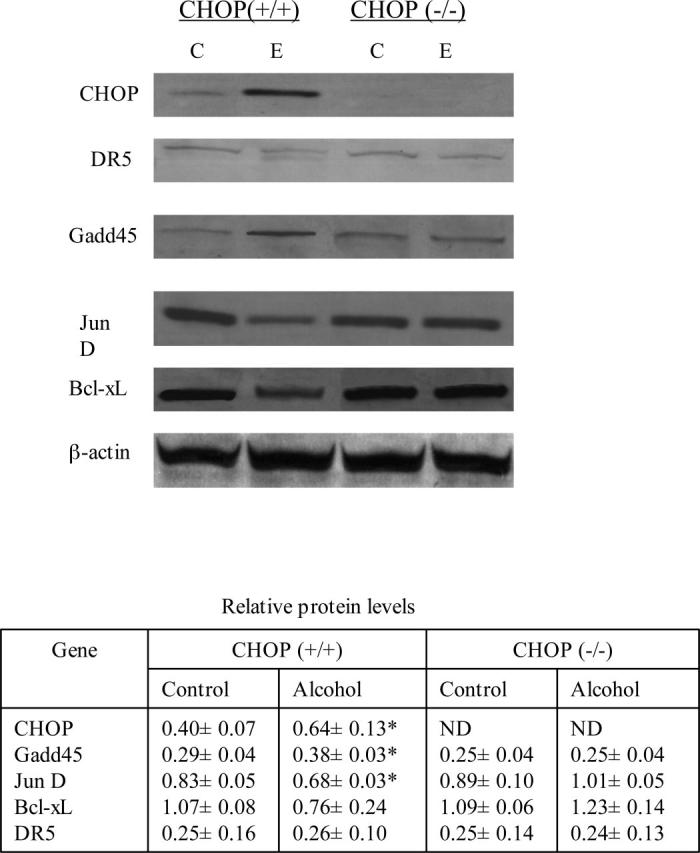
Western blots of proteins regulated by CHOP in ethanol-fed mice. The table shows densitometric values for three mice per group normalized to the expression of β-actin (means ± SD). C, control; E, alcohol treated; Gadd45, growth arrest and DNA damage-inducible protein 45; DR5, death receptor 5 or trail receptor 2. * p < 0.05.
DR5/trail receptor 2 is involved in CHOP-regulated apoptosis in some carcinoma cells (Yamaguchi and Wang, 2004). Because it is not included in the microarray analysis gene list, we examined the DR5 expression with both RT-PCR analysis and western blotting. The abundance of mRNA was low in liver samples, and no change was detected in either wild-type or CHOP null mice. A trace amount of DR5 protein was detected on western blots. In addition, we were not able to detect liver expression of the carbonic anhydrase DOC1, which responds to CHOP in certain cell types (Sok et al., 1999), in any of the groups.
DISCUSSION
Apoptosis is a feature of both experimental and human alcoholic liver injury (Baroni et al., 1994; Benedetti et al., 1988; Deaciuc, 2001; Kaplowitz and Tsukamoto, 1996; Minana et al., 2002; Neuman, 2001). Modest increases in apoptosis have been observed in both, but apoptosis at a single “freeze frame” may not represent the magnitude of its contribution because of rapid removal. Therefore, the stress of moderate apoptosis on hepatocellular turnover may contribute to loss of functional mass of liver, particularly when regeneration is impaired as in the case of alcohol feeding. Furthermore, the increased turnover of hepatocytes may contribute to the susceptibility to carcinogenesis. Although the specific role of apoptosis in alcoholic liver disease remains an open question, elucidation of its mechanism is worthy of attention. In this regard, we suggested that ER stress may be an important mode of apoptosis in alcoholic liver injury (Ji and Kaplowitz, 2003, 2004). CHOP is a transcription factor that is up-regulated in response to ER stress. In various models of ER stress, CHOP has been identified as an important contributor to apoptosis, although the precise details of this action are not well understood (Oyadomari and Mori, 2004).
In view of the proposed role of CHOP in ER stress-induced apoptosis, we examined the role of CHOP by comparing the effects of alcohol feeding in CHOP null mice with those in wild-type littermates; we observed a striking absence of apoptosis in CHOP null mice in response to alcohol, whereas we confirmed moderate apoptosis in wild-type mice. However, the absence of CHOP did not decrease the number of necroinflammatory foci, suggesting that the latter is due to a different mediator induced by ER stress. We previously showed that inhibition of ER stress by preventing alcohol-induced hyperhomocysteinemia with betaine feeding diminished both apoptosis and necroinflammation in this model (Ji and Kaplowitz, 2003). Furthermore, the hyperhomocysteinemia and ER stress-dependent fatty liver and induction of Grp78 (ER stress marker) were not altered in alcohol-fed CHOP null mice. This indicates that CHOP is only one of several important consequences of ER stress and plays a particularly important role in the apoptosis observed in this condition.
As noted before, the mechanism by which CHOP promotes apoptosis is not well understood. To gain insight into this mechanism considering that CHOP is a transcription regulator, we performed microarray analysis of liver gene expression. Our strategy was to screen by microarray analysis using pairs of individual control and alcohol-fed wild-type and CHOP null mice using two-fold changes to identify candidates. The changes in candidate genes known to influence apoptosis were confirmed in three individual mice from each group using semiquantitative RT-PCR analysis and western blotting. At both the mRNA level and the protein level, several apoptosis-related genes were identified that were influenced by CHOP. Gadd45γ (Artuso et al., 1995; Kearsey et al., 1995) is a transcription factor that has been shown to be induced or repressed by NF-κB and to protect or promote apoptosis in various cancer cells (Dong et al., 2004; Kokkinakis et al., 2004; Zerbini et al., 2004, 2005). It was up-regulated in ethanol-fed wild-type mice but not in ethanol-fed CHOP null mice. Jun D is a prosurvival transcription factor that coordinately regulates NF-κB-dependent expression of IAP-1, a caspase inhibitor (Lamb et al., 2003). It was down-regulated by ethanol feeding in wild-type mice but not CHOP knock-out mice. Bcl-xL is an antiapoptosis member of the Bcl-2 family (Boise et al., 1993; Tsujimoto et al., 1997). Interestingly, its expression was decreased in ethanol-fed wild-type mice but not in CHOP null mice. Somewhat analogous to this finding, in other experimental systems, CHOP has been shown to inhibit BclII expression (Li and Holbrook, 2004; Mikula et al., 2003). Cathepsin B has been implicated in the apoptotic pathway in response to TNF (Guicciardi et al., 2000). We observed an increase in mRNA only in ethanol-fed wild-type mice but could not confirm this at the protein level due to lack of antibodies. Thus, the microarray analysis and confirmation of RT-PCR analysis and western blotting findings provided new insights such as ethanol-induced up-regulation of proapoptotic Gadd45γ and cathepsin B and down-regulation of anti-apoptotic Bcl-xL and Jun D, all of which were abrogated in the absence of CHOP, indicating that these changes are either directly or indirectly due to CHOP. At present, it is not clear if any of these changes in wild-type mice represent the key factors responsible for apoptosis in the alcohol-fed mice, and more research is required. Because the changes in expression of these genes in response to ethanol were of moderate magnitude, it may be that the interplay of all or some may create an imbalance in pro- versus antiapoptosis.
We have been seeking evidence to support our hypothesis that alcohol-induced ER stress is one of the major causes of the early alcohol-induced liver injury, including SREBP-regulated fat accumulation, CHOP-regulated apoptosis, and calcium-induced oxidative stress and hepatic inflammation. Although contribution of alcohol-induced ER stress to liver damage requires further elucidation, alcohol-induced apoptosis is a well known component of alcohol-induced liver damage both experimentally and in humans. Although the importance of apoptosis versus necrosis is unclear, generally necrosis is more inflammatory. However, there is evidence that persistent apoptosis of parenchymal cells is sufficient to induce fibrotic responses, suggesting a mechanistic link between apoptosis and fibrosis (Takehara et al., 2004). Therefore, prevention of apoptosis in alcoholics could slow down or prevent alcohol-induced liver fibrosis. Alternatively, it is even conceivable that ER stress-induced apoptosis is a protective phenomenon, providing a means to remove damaged cells with much less collateral damage. Because our experiments were subacute (≤ one month), we do not know whether inhibition of CHOP-mediated apoptosis will have positive, negative, or no impact on the progression of alcoholic liver disease. The present study with microarray screening identified additional changes other than the ER stress response, such as up-regulation of insulin-like growth factor binding protein 1, CYP2E1, carbonyl reductase 1, and transferrin receptor as well as down-regulation of regulator of G protein signaling, flavin containing monooxygenase, and methionine adenosyltransferase 1, which suggest that alcohol-induced response is complex. Further investigation is needed to define the contribution of each factor or pathway to alcohol-induced disease.
In summary, the absence of CHOP prevented ethanol-induced apoptosis but did not affect the other manifestations of alcoholic liver injury, such as fatty liver, up-regulation of SREBP and Grp78, necroinflammatory foci, and hyperhomocysteinemia. Microarray analysis, RT-PCR analysis, and western blotting revealed candidate genes that may be regulated by CHOP and contribute to apoptosis in alcoholic livers.
ACKNOWLEDGMENTS
The authors thank the Animal Core of USC/UCLA Research Center for Alcoholic Liver and Pancreatic Diseases for providing mice for the intragastric infusion model, the Morphology Core for TUNEL and immunohistochemical analysis, and the Microarray Subcore of the USC Research Center for Liver Diseases.
Footnotes
Dr. Cheng Ji, Ph.D. GI/Liver Division, USC Keck School of Medicine, HMR-101, 2011 Zonal Avenue, Los Angeles, CA 90033; Fax: 323 442 5425; E-mail: chengji@usc.edu
Supported by grants from the US National Institute on Alcohol Abuse and Alcoholism (R01AA014428-02 to NK and CJ; and P50AA11999), the National Institute of Diabetes and Digestive and Kidney Diseases (P30DK048522-10), and the Robert E. and May R. Wright Foundation (263 to CJ).
REFERENCES
- Artuso M, Esteve A, Bresil H, Vuillaume M, Hall J. The role of the ataxia telangiectasia gene in the p53, WAF1/CIP1(p21)- and GADD45-mediated response to DNA damage produced by ionising radiation. Oncogene. 1995;11:1427–1435. [PubMed] [Google Scholar]
- Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, Prieto J, Lu SC, Caballeria J, Rodes J, Mato JM. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- Barak AJ, Beckenhauer HC, Mailliard ME, Kharbanda KK, Tuma DJ. Betaine lowers elevated s-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. J Nutr. 2003;133:2845–2848. doi: 10.1093/jn/133.9.2845. [DOI] [PubMed] [Google Scholar]
- Barak AJ, Beckenhauer HC, Tuma DJ. Betaine, ethanol, and the liver: A review. Alcohol. 1996;13:395–398. doi: 10.1016/0741-8329(96)00030-4. [DOI] [PubMed] [Google Scholar]
- Baroni GS, Marucci L, Benedetti A, Mancini R, Jezequel AM, Orlandi F. Chronic ethanol feeding increases apoptosis and cell proliferation in rat liver. J Hepatol. 1994;20:508–513. doi: 10.1016/s0168-8278(05)80498-2. [DOI] [PubMed] [Google Scholar]
- Benedetti A, Brunelli E, Risicate R, Cilluffo T, Jezequel AM, Orlandi F. Subcellular changes and apoptosis induced by ethanol in rat liver. J Hepatol. 1988;6:137–143. doi: 10.1016/s0168-8278(88)80024-2. [DOI] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Deaciuc IV, Arteel GE, Peng X, Hill DB, McClain CJ. Gene expression in the liver of rats fed alcohol by means of intragastric infusion. Alcohol. 2004a;33:17–30. doi: 10.1016/j.alcohol.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Deaciuc IV, D’Souza NB, de Villiers WJS, Burikhanov R, Sarphie TG, Hill DB, McClain CJ. Inhibition of caspases in vivo protects the rat liver against alcohol-induced sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res. 2001;25:935–943. [PubMed] [Google Scholar]
- Deaciuc IV, Doherty DE, Burikhanov R, Lee EY, Stromberg AJ, Peng X, de Villiers WJ. Large-scale gene profiling of the liver in a mouse model of chronic, intragastric ethanol infusion. J Hepatol. 2004b;40:219–227. doi: 10.1016/j.jhep.2003.10.021. [DOI] [PubMed] [Google Scholar]
- Dong YG, Chen DD, He JG, Guan YY. Effects of 15-deoxy-delta12,14-prostaglandin J2 on cell proliferation and apoptosis in ECV304 endothelial cells. Acta Pharmacol Sin. 2004;25:47–53. [PubMed] [Google Scholar]
- Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- Ji C, Deng Q, Kaplowitz N. Role of TNF-alpha in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology. 2004;40:442–451. doi: 10.1002/hep.20309. [DOI] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol. 2004;10:1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplowitz N, Tsukamoto H. Oxidative stress and liver disease. Prog Liver Dis. 1996;14:131–159. [PubMed] [Google Scholar]
- Kearsey JM, Coates PJ, Prescott AR, Warbrick E, Hall PA. Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene. 1995;11:1675–1683. [PubMed] [Google Scholar]
- Kokkinakis DM, Liu X, Chada S, Ahmed MM, Shareef MM, Singha UK, Yang S, Luo J. Modulation of gene expression in human central nervous system tumors under methionine deprivation-induced stress. Cancer Res. 2004;64:7513–7525. doi: 10.1158/0008-5472.CAN-04-0592. [DOI] [PubMed] [Google Scholar]
- Lamb JA, Ventura JJ, Hess P, Flavell RA, Davis RJ. JunD mediates survival signaling by the JNK signal transduction pathway. Mol Cell. 2003;11:1479–1489. doi: 10.1016/s1097-2765(03)00203-x. [DOI] [PubMed] [Google Scholar]
- Li J, Holbrook NJ. Elevated gadd153/chop expression and enhanced c-Jun N-terminal protein kinase activation sensitize aged cells to ER stress. Exp Gerontol. 2004;39:735–744. doi: 10.1016/j.exger.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Lu SC, Tsukamoto H, Mato JM. Role of abnormal methionine metabolism in alcoholic liver injury. Alcohol. 2002;27:155–162. doi: 10.1016/s0741-8329(02)00226-4. [DOI] [PubMed] [Google Scholar]
- Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translational block: Activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 2003;278:37375–37385. doi: 10.1074/jbc.M303619200. [DOI] [PubMed] [Google Scholar]
- Matsumaru K, Ji C, Kaplowitz N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology. 2003;37:1425–1434. doi: 10.1053/jhep.2003.50230. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikula M, Gotzmann J, Fischer AN, Wolschek MF, Thallinger C, Schulte-Hermann R, Beug H, Mikulits W. The proto-oncoprotein c-Fos negatively regulates hepatocellular tumorigenesis. Oncogene. 2003;22:6725–6738. doi: 10.1038/sj.onc.1206781. [DOI] [PubMed] [Google Scholar]
- Minana JB, Gomez-Cambronero L, Lloret A, Pallardo FV, Del Olmo J, Escudero A, Rodrigo JM, Pelliin A, Vina JR, Vina J, Sastre J. Mitochondrial oxidative stress and CD95 ligand: A dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology. 2002;35:1205–1214. doi: 10.1053/jhep.2002.32969. [DOI] [PubMed] [Google Scholar]
- Neuman MG. Apoptosis in diseases of the liver. Crit Rev Clin Lab Sci. 2001;38:109–166. doi: 10.1080/20014091084182. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Seth D, Leo MA, McGuinness PH, Lieber CS, Brennan Y, Williams R, Wang XM, McCaughan GW, Gorrell MD, Haber PS. Gene expression profiling of alcoholic liver disease in the baboon (Papio hamadryas) and human liver. Am J Pathol. 2003;163:2303–2317. doi: 10.1016/S0002-9440(10)63587-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sok J, Wang XZ, Batchvarova N, Kuroda M, Harding H, Ron D. CHOP-dependent stress-inducible expression of a novel form of carbonic anhydrase VI. Mol Cell Biol. 1999;19:495–504. doi: 10.1128/mcb.19.1.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehara T, Tatsumi T, Suzuki T, Rucker EB, 3rd, Hennighausen L, Jinushi M, Miyagi T, Kanazawa Y, Hayashi N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S, Eguchi Y, Kamiike W, Matsuda H. Bcl-2 and Bcl-xL block apoptosis as well as necrosis: Possible involvement of common mediators in apoptotic and necrotic signal transduction pathways. Leukemia. 1997;3:S380–S382. [PubMed] [Google Scholar]
- Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR, Austin RC. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107:1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- Zhang C, Cai Y, Adachi MT, Oshiro S, Aso T, Kaufman RJ, Kitajima S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J Biol Chem. 2001;276:35867–35874. doi: 10.1074/jbc.M100747200. [DOI] [PubMed] [Google Scholar]
- Zerbini LF, Libermann TA. Life and death in cancer: GADD45 alpha and gamma are critical regulators of NF-kappaB mediated escape from programmed cell death. Cell Cycle. 2005;4:18–20. doi: 10.4161/cc.4.1.1363. [DOI] [PubMed] [Google Scholar]
- Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, Wei W, Joseph M, Gu X, Grall F, Goldring MB, Zhou JR, Libermann TA. NF-kappa B-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proc Natl Acad Sci USA. 2004;101:13618–13623. doi: 10.1073/pnas.0402069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]