

Abstract
Prenatal administration of corticosteroids is common in obstetrics to improve the outcome of premature deliveries. Many pregnant women receive multiple corticosteroid courses. Long-term follow-up studies in humans are limited, but those available suggest detrimental effects on the behavior of those children. Animal data also show adverse effects of prenatal corticosteroids mainly in the hippocampus, a structure sensitive to corticosteroid action. Several molecules involved in neuronal survival, seizure susceptibility, and behavior have been identified as possible targets of prenatal corticosteroid effects. These molecules include hippocampal glucocorticoid receptors, brain-derived neurotrophic factor, corticotropin-releasing hormone, and neuropeptide Y. Prenatal corticosteroid treatment permanently reprograms expression of these molecules. The future goals include development of specific antagonists of corticosteroid activation pathways that would help differentiate between positive main effects and undesired adverse effects of prenatally administered corticosteroids.
Keywords: Betamethasone, Dexamethasone, Hydrocortisone, Antenatal treatment, Seizure susceptibility, Behavioral problems, Neuropeptide Y, Brain-derived neurotrophic factor, glucocorticoid receptors, Mineralocorticoid receptors, Corticotropin
1. Introduction
The hippocampus is very sensitive to regulatory effects of corticosteroids [1–4]. Many excellent reviews on the effects of postnatal corticosteroids in the hippocampus have been published over the past 10 years [3,5–11]. Antenatal/prenatal administration of corticosteroids and its effects on hippocampal development received much less attention [12,13]. However, understanding the effects of prenatal corticosteroid administration is very important because many pregnant women receive corticosteroid therapy, particularly in the last trimester of pregnancy [14]. The purpose of this article is to compare available animal models with the human condition of prenatal corticosteroid administration and to discuss possible mechanistic correlates (derived from the animal models) to the human condition outcome. This review targets mostly behavioral problems and changes in seizure susceptibility associated with alterations of hippocampal function after prenatal corticosteroid exposure.
2. Human conditions
2.1. A single prenatal corticosteroid course
Since 1972, corticosteroid therapy during pregnancy has frequently been used to decrease neonatal mortality by preventing respiratory distress syndrome and intracerebral hemorrhage in the prematurely born neonates [15]. An NIH consensus conference held in 1994 published a statement indicating that the one-course corticosteroid administration during the last trimester of pregnancy is relatively safe and without side effects [16]. This conclusion was based on long-term follow- up trials, which have demonstrated that this therapeutic regimen has no negative long-term neurological and cognitive outcome [16–21].
However, the current trend in clinical practice is:
To treat all women at risk for premature delivery with synthetic corticosteroids (preferentially betamethasone), and
To repeat betamethasone courses if delivery does not occur within 7 days of the initial treatment [22,23].
Thus, the fetuses are frequently exposed to several corticosteroid courses.
2.2. Multiple prenatal corticosteroid courses
The second NIH Consensus Conference “Prenatal Corticosteroids Revisited: Repeated Courses” [15], held in 2000, emphasized that “Animal studies should evaluate the pathophysiologic and metabolic mechanisms of potential benefits and risks, including the effects of repeat corticosteroids on … brain development,” as the prospective data after multiple corticosteroid courses in humans were still insufficient. Although one study in a small sample of children did not find behavioral problems linked to the duration of prenatal corticosteroid therapy [24], others suggest that repeated prenatal courses of corticosteroids may have harmful side effects [25]. Some of these effects are only short-term: Newborns of mothers receiving repeated doses of betamethasone (5–16 doses, 12 mg each, administered twice weekly) demonstrated a transient hypertrophic cardiomyopathy [26]. Other studies have shown long-term effects: Multiple courses of prenatal betamethasone (see Table 1 for a course definition) were associated with increased mortality, decreased fetal growth and birth weight, adrenal suppression at birth, and decreased head circumference [22,27]. Recently, a follow-up study has been published investigating the outcome at 3 and 6 years of age in 541 preterm born infants prenatally exposed to one, two, three or more courses of betamethasone in comparison with the outcome after no corticosteroid exposure [28]. Children who had received three or more courses of therapy were more than three times more likely to have aggressive–destructive behavioral scores above the 90th percentile at both ages than those who had not received prenatal corticosteroids, thus suggesting changes in anxiety [29]. Further, these children displayed greater distractibility at age 3 and hyperactivity at age 6 compared with the no-treatment group. Interestingly, there were no effects of prenatal corticosteroids on intellectual performance.
Table 1.
Prenatal corticosteroid courses in humans
| Drug | One course | Treatment timing | Full term (days) | Timing/terma | Long-term effects of multiple courses | Ref. |
|---|---|---|---|---|---|---|
| DEXb | 4 × 6-mg doses every 6 h | 168–238 days (24–34 weeks) | 270 days (38.5 weeks) | 0.62–0.88 | N/A | [24] |
| BETA | 2 × 12-mg doses in 24 h | 168–238 days (24–34 weeks) | 270 days (38.5 weeks) | 0.62–0.88 | Hyperactivity, distractibility, aggressive–destructive behavior | [28] |
Indicates in the form of a fraction when, during the pregnancy, the corticosteroids were administered if the full term equals 1.00. This fraction makes possible the initial comparison with animal data (Table 2). However, it should be emphasized that experimental animals are born at different stages of maturation. For example, the rat as a precoccious animal is born at a developmental stage corresponding to human premature newborn. Additionally, the dynamics of rat brain development differs from that of humans [115] and is the subject of many comparative studies [114,116,117].
BETA, betamethasone; DEX, dexamethasone; N/A, information not available.
Along with these findings, long-term stress during pregnancy (featuring prolonged elevated maternal corticosteroid levels) is also associated with increased neurological dysfunction, developmental delay (late or poor walking, speech deficits), and behavioral disturbances (such as restlessness, fretfulness, and poor interpersonal skill development) in children [30]. However, in this case the influence of the stressed mother may confound the outcome.
Additional long-term studies in humans after prenatal treatment with multiple corticosteroid courses are not yet available. However, these findings indicate possible long-term (maybe permanent) reprogramming of the brain and especially those structures controlling above-mentioned behaviors [31] such as the hippocampus.
3. Experimental animals
3.1. Rhesus monkeys
Prenatal administration of betamethasone daily on Embryonic Days (E) 120–133 improves the development of fetal lungs and lung volume [32] quite consistently with human indications. However, similar doses of betamethasone have significant adverse effects on the hippocampus: Effects of a single dexamethasone dose administered on E132, or a dexamethasone course of four doses injected at 12-hour intervals beginning on E132, were studied in the hippocampus. Acute effects were determined at E135. Corticosteroids decreased neuronal cell density in all hippocampal areas (subiculum, CA1–CA3, and dentate gyrus granule cells) [33]. Additionally, there was degeneration of neuronal perikarya and dendrites, as well as degeneration of axodendritic synapses of mossy fibers in the CA3 area. Later morphological changes determined on E162 were similar to the findings reported on E135 (Table 2) [33,34].
Table 2.
Effects of repeated prenatal corticosteroids on the nervous systema
| Species | Drug | Dose (mg/kg) | Timing | Term (days) | Timing/termb | Effects | Ref. |
|---|---|---|---|---|---|---|---|
| Rhesus monkey | DEXc | 4 × 0.125–2.5 | E132-133 | 165 | 0.80 | Decreased number of hippocampal neurons, degeneration | [33,34] |
| Sheep | BETA | 4 × 0.5 | E104,111,118,124 | 147 | 0.71–0.84 | Delay in optic nerve myelination | [118] |
| BETA | 4 × 0.5 | E104,111,118,123 | 0.71–0.84 | Delay in brain myeli nation, sciatic nerve growth, and retinal maturation, decreased fetal brain growth | [35,36,118 119] | ||
| Guinea | BETA | 4 × 1.0 | E40,41,50,51 | 68 | 0.59–0.75 | Decreased hippocampal MR | [40] |
| Pig | |||||||
| Rat | BETA | 2 × 0.17–0.34 | E20 | 21–22 | 0.87–0.95 | Decreased [3H]thymidine incorporation in hippocampus | [41] |
| DEX | 7 × 0.1 | E15–21 | 21–22 | 0.68–0.95 | Decreased hippocampal MR and GR | [43] | |
| BETA | 2 × 0.4 | E15 | 21–22 | 0.68–0.71 | Decreased anxiety, Decreased hippocampal GR | Velisek, unpublished | |
| Mouse | BETA | 4–8 × 0.1 | E13–16 | 19 | 0.68–0.84 | No effect on behavioral outcome | |
| E14–15 | 0.74–0.79 | [47] |
3.2. Sheep
Effects of prenatal continuous administration of betamethasone for 2 days into the fetal jugular vein on synapses and synaptogenesis were studied in sheep. A variety of changes were found in several brain regions including the hippocampus [35,36].
3.3. Rodents
There is evidence that corticosteroids are potent regulators of cell development and differentiation already after a single prenatal administration [37–39]. Many studies demonstrate that in rodents, prenatal corticosteroid treatments have multiple effects on hippocampal cell proliferation, neurotransmitter turnover, and receptor expression, as well as on postnatal behaviors. The data in rodents indicate that the effects of prenatal corticosteroids may differ as a function of the corticosteroid used, the treatment paradigm (dose and timing), and the animal model.
3.3.1. Administration of corticosteroids
In guinea pigs, betamethasone administration on E40–41 and E50–51 decreased the expression of hippocampal mineralocorticoid receptors [40]. In rats, betamethasone administration on E20 transiently decreased postnatal weight, as well as decreased early postnatal (PN1 only) [3H]thymidine incorporation in many brain regions including the hippocampus [41], indicating decreases in cell birth rate. Repeated doses of dexamethasone on E17–19 significantly decreased body and brain weight of newborn rats [42]. In the forebrain, norepinephrine and dopamine turnover increased during the prepubertal (Postnatal Days (PN) 20–30) period, followed by a postpubertal (PN 45–55) turnover decrease, compared with controls. Additionally, dexamethasone on E17–19 decreased the postnatal protein/DNA ratio. The authors suggested that this finding is indicative of neuronal replacement by glia in the forebrain [39]. Daily administration of dexamethasone on E16–21 resulted in decreases in both glucocorticoid and mineralocorticoid receptors in the hippocampus [43]. Continuous corticosterone release from pellets on E16–21 induced both short-and long-term changes in spontaneous motor activity, such as increases in motility, rearing, and locomotion in the offspring [44,45]. Prenatal exposure to a daily dose of dexamethasone during the third week (E16–21) of pregnancy altered performance in the open field and in the forced-swim test in the adult offspring [43]. These findings indicate impaired coping of the offspring prenatally exposed to corticosteroids in the stressful environment. In mice, Rayburn et al. [46] determined that a single exposure to prenatal betamethasone impaired performance in a battery of behavioral tests, although multiple prenatal exposures to betamethasone had no effects in these behavioral tests [47].
Our unpublished data indicate that in rats, two doses of betamethasone administered on E15 decreased anxiety as assessed in the elevated plus maze. Betamethasone exposure affects the memory retention index. Findings of anxiolytic effects, as well as no alterations of simple learning due to prenatal corticosteroids, are consistent with previously reported data and also with the human situation [28,43].
3.3.2. Stressful stimuli during pregnancy
Effects of a stressor represented by one 45-minute session of flashing lights during each week of rat pregnancy were studied in rats. The offspring indeed displayed hyperreactivity in the open field test and altered avoidance behavior [48]. Exposure of pregnant rats to 45 minutes restraint in a cylinder three times a day during the third week of gestation was associated with increased corticosteroid levels as well as with enhanced response to the restraint stress in the offspring [49]. The same treatment paradigm worsened working memory in a radial maze and spatial recognition memory in the Y-maze in aged offspring (15–22 months old) [50]. Daily prenatal stress from E15 to delivery leads to decreased neurogenesis of granule cells in the dentate gyrus associated with an impairment of hippocampus-related spatial learning in the water maze in the offspring throughout their lives [51].
These results indicate that increases in naturally occurring corticosteroids due to stress may have some features common with the administration of synthetic corticosteroids (e.g., behavioral problems under stressful conditions). Additionally, prenatal stress resulted in some specific features, such as disturbances of hippocampus-related spatial learning.
4. Prenatal corticosteroids, seizure susceptibility, and epileptogenesis
4.1. Seizure susceptibility
Prolonged prenatal stress or prenatal administration of corticosteroids may significantly alter seizure susceptibility. Such moderate stress as 20 minutes of restraint in a cylinder on E18 enhanced the severity of kainic acid-induced seizures in adult rats [52]. Along with this finding, our preliminary data show that repeated prenatal administration of hydrocortisone, but not betamethasone, on E15 significantly increases susceptibility to kainic acid-induced seizures determined on PN15. The data indicate that increased levels of natural but not synthetic corticosteroids during prenatal brain development result in permanent reprogramming of structures involved in seizure initiation.
4.2. Epileptogenesis
Repeated prenatal administration of betamethasone in rats on E15 significantly altered epileptogenesis determined as progression of dentate gyrus kindling in immature, PN15–16 offspring [53]. To our surprise, however, prenatal exposure to betamethasone delayed kindling development in terms of decreased behavioral seizure scores and shortened hippocampal afterdischarges. Additionally, these effects were sex-specific, more pronounced in female than in male rats. Up to now, there are no data available on epidemiology of prenatal corticosteroid exposure and epilepsy in humans; therefore, correlations between the human and experimental conditions cannot be made.
5. Mechanisms of corticosteroid action in the brain
In tissues, corticosteroids may have genomic and/or nongenomic effects, which can be distinguished by the criteria summarized in Table 3 [54]. In clear-cut cases, fulfillment of just one criterion is sufficient for including or excluding genomic mechanisms (e.g., corticosteroid effects produced in seconds may not be mediated genomically). Thus, prenatal administration of corticosteroids with effects recorded postnatally is consistent with genomic effects.
Table 3.
Criteria for corticosteroid-induced genomic and nongenomic effects [54]
| Criterion | Genomic effects | Nongenomic effects |
|---|---|---|
| 1. Temporal (time lag to effects) | More than 15 min | Considerably earlier than 15 mina |
| 2. MR/GR dependence/independence | Blockade of MR/GR abolishes the effects (receptor dimers only) | Blockade of MR/GR has no effects |
| 3. Genome dependence/independence | Blockade of protein synthesis prevents the effectsb | Blockade of protein synthesis has no effects |
Corticosteroid effects occurring between 10 and 20 min can be attributed to a nongenomic mechanism only if another argument supports this assumption.
However, genomic corticosteroid effects mediated by inhibition of gene expression are not affected.
5.1. Genomic corticosteroid effects in the brain
5.1.1. Receptors and their ligands
Genomic (nuclear) corticosteroid effects are mediated by intracellular glucocorticoid receptors (GRs) and mineralocorticoid receptors (MRs). Both GRs and MRs are localized in the brain and may be co-localized in limbic neurons including hippocampal CA1, dentate granule cells, and amygdala [55–57]. MRs bind aldosterone and corticosterone with a similar affinity. However, there is a 100- to 1000-fold excess of circulating corticosterone compared with aldosterone. Additionally, a corticosterone deactivating enzyme (HSD-2) is missing in the limbic brain [58]. Therefore, limbic brain MRs predominantly see corticosterone. Activation of GRs occurs only with high concentrations of natural corticosteroids (such as at the circadian peak or during a stressful event) [59,60] or after administration of a synthetic analog betamethasone (or dexamethasone), which has high affinity for GRs [61].
5.1.2. Activation of receptors, antagonists, and nuclear effects
Binding of an agonist activates GRs or MRs. The activation is followed by phosphorylation of the receptor and dissociation from the heat shock proteins [62,63]. Phosphorylated receptor–agonist complex translocates to the nucleus and may follow two distinct pathways. In the first pathway, receptor–agonist complexes form homodimers and bind to the hormone response elements in the DNA in synergy with a co-activator [64,65]. The second activation pathway involves activated GR (but not MR)–agonist complex monomers, which interfere with transcription factors in a process that does not require steroid receptor binding to the DNA [63,66]. Antagonists (RU486 = mifepristone for GRs and RU28318 = spironolactone for MRs) follow only the homodimeric pathway, thus binding to the GR/MR instead of an agonist. Receptor–ligand complex homodimers bind to a hormone response element in the DNA. The antagonistic effect is activated by recruiting a co-repressor molecule instead of the co-activator. The co-repressor promotes repression of gene transcription, resulting in antagonism of the GR effects. This mode of action clearly shows that the effects mediated by the second pathway and involving GR–agonist complex monomers cannot be antagonized by current GR antagonists. Thus, development of specific antagonists for GR–agonist complex monomers may help in differentiation of beneficial main and unwanted side effects of prenatal corticosteroids Table 4.
Table 4.
Overview of activated corticosteroid receptors and their actionsa
| GR homodimers | MR homodimers | GR/MR heterodimers | GR monomers | |
|---|---|---|---|---|
| Transcriptional activity | Via DNA GR-responsive element | Weak agonists at DNA GR-responsive element | DNA binding distinct from GR or MR homodimers | Via transcription factors without DNA binding |
| Co-regulator | Required | Required | N/Ab | Not required |
| Anxiety effects | No | N/A | N/A | Yes |
| Ref. | [63,66,68] | [68,120] | [121,122] | [63,66,68] |
See also [11].
N/A, information not available.
6. Target molecules for corticosteroid effects in the hippocampus
Prenatal corticosteroids may reprogram the expression of many molecules significant for neuronal survival, behavior, and seizures. Here, only those molecules are reviewed for which there is accumulating evidence of their connection with corticosteroid effects in the hippocampus as well as with the above-mentioned outcomes
6.1. Corticosteroid receptors
Glucocorticoids themselves can reprogram their receptor system in the hippocampus. Prenatal exposure to dexamethasone induces a decrease in hippocampal GRs [43]; similar decreases were seen after betamethasone (see Figs. 1A and 1B).
Fig. 1.
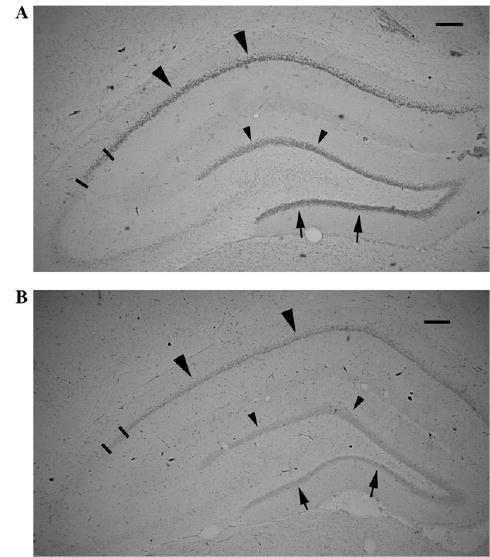
GR expression in the dorsal hippocampus is decreased by prenatal corticosteroid treatment. (A) PN20 control male rat prenatally exposed to saline on E15. GR immunohistochemistry with DAB visualization. Large arrowheads point to the GR expression in the CA1 area; arrows point to the inner blade of the dentate gyrus granule cells. Small arrowheads point to the outer blade of dentate gyrus granule cells. Bars limit the CA2 area. Scale bar = 200 μm. (B) PN20 male rat prenatally exposed to 2 × 0.4 mg/kg betamethasone on E15. Immunohistochemical staining has been run in the same dish as the section shown in (A). Expression of GR in all areas marked by large arrowheads (CA1), arrows (inner granule cell blade), and small arrowheads (outer granule cell blade) as well as in the CA2 (bar limited) is decreased consistently with findings of decreased hippocampal GR after prenatal dexamethasone treatment [43]. Scale bar = 200 μm.
Decrease or absence of nervous system GRs is associated with anxiogenic effects: Mice with conditional knockout of GRs in the nervous system display an impaired behavioral response to stress and have reduced anxiety [67]. On the other hand, a point mutation in the GR preventing dimerization of the activated GR has no effect on anxiety in mice [68]. This indicates that anxiety may be under the control of activated GR monomers. Along with these findings, GR overexpression results in an increased anxiety-related behavior [68].
6.2. Corticotropin-releasing hormone (factor)
Corticotropin-releasing hormone (factor) (CRH (CRF)) is an indivisible part of the (HPA) axis. Parvocellular neurons of the paraventricular nucleus of the hypothalamus produce increased amounts of CRH as a response to acute stress. CRH activates release of ACTH from the anterior pituitary, which promotes release and further synthesis of corticosteroids. Corticosteroids, in turn, provide a negative feedback for release of both CRH and ACTH [65,69], except in the amygdala, where they increase production of CRH. Hippocampal GABAergic interneurons also contain CRH [70,71] (see also Fig. 2).
Fig. 2.

CRH is expressed in the hippocampus. Immunostaining with anti-CRH antibody reveals the presence of CRH-like immunopositivity in the DG hilus of PN20 male rat (arrows point to some positive subgranular cells, and arrowheads to some positive hilar cells) consistent with other studies [70,71]. Bar = 40 μm.
The role of CRH in anxiety has been demonstrated in genetically altered mice: Mice overexpressing CRH demonstrate an increase in anxiogenic behavior measured in the novel environment and in the elevated plus maze [72]. Anxiogenic effects of the overexpressed CRH can be prevented by intracerebroventricular injection of a CRH antagonist. On the other hand, mice with a knockout of the CRH-binding protein (and, therefore, having increased levels of CRH) have increased anxiety [73]. Accordingly, inhibition of CRH release (e.g., by intracerebroventricular atrial natriuretic peptide) is anxiolytic in the open field and in the elevated plus maze test [74].
CRH acts on two specific G-protein-coupled receptors, CRH-R1 and CRH-R2, in the brain [75] (see Table 5). These receptors can be found in many brain nuclei, with specific localization in those responsible for control of fear, anxiety, and emotionality. CRH-R1 is probably responsible for CRH anxiogenic effects, as mice lacking this receptor display reduced anxiety-related behavior and increased exploratory activity [76,77]. Mice with CRH-R2 knockout have, on the other hand, increased anxiety [73]. This finding is followed by use of CRH-R1 (CRF1) receptor antagonist R121919 for testing in human studies for its anxiolytic effects [78].
Table 5.
Localization of CRH receptors
| Receptor | Localization | Ref. |
|---|---|---|
| CRH-R1 (CRF1) | Anterior pituitary, neocortex, basolateral amygdala, hippocampus, cerebellum, forebrain, and brainstem cholinergic nuclei, superior colliculus, substantia nigra | [70,75,76] |
| CRH-R2 (CRF2) | Paraventricular nucleus, lateral septum, cortical, and medial amygdalar nuclei, serotonergic raphe nuclei | [75] |
CRH also has a strong link to seizures [79]. Seizures with features similar to those found in human infantile spasms may be evoked in neonatal rats by intracerebroventricular administration of CRH [80,81]. Accordingly, administration of ACTH (which also suppresses CRH release by negative feedback) is relatively successfully used for the treatment of infantile spasms [82].
6.3. Brain-derived neurotrophic factor
An important corticosteroid regulatory mechanism mediated via brain-derived neurotrophic factor (BDNF) in neuronal death was described in the embryonic hippocampus. It has been shown in neurons from E18 rat hippocampus cultured for 1 day that corticosterone induces neuronal death. This type of neuronal death was associated with a decrease in BDNF, and was prevented by BDNF administration [83]. Similarly, offspring of rats subjected to many different unpredicted stressors during the last week of pregnancy displayed increases in tyrosine kinase B (TrkB), a BDNF receptor, throughout the hippocampus, probably as compensation for corticosteroid-induced suppression of BDNF [84]. Studies in adult rats support this finding by showing that adrenalectomy increases while corticosterone administration decreases BDNF mRNA [84–86]; and repeated stress (high levels of corticosteroids) increases TrkB mRNA [87].
BDNF undoubtedly plays an important role in seizures, epileptogenesis, and neuronal plasticity, although the findings are still controversial. Intrahippocampal administration of BDNF itself has convulsant effects and worsens pilocarpine seizures [88]. Others, however, demonstrate that BDNF infusions may be protective in kindling epileptogenesis [89–91], suggesting treatment paradigm (dose and treatment duration) and probably also model specificity of the BDNF action.
6.4. Neuropeptide Y
Existence of a bilateral relationship between the corticosteroid (or, in general, the HPA axis) system and neuropeptide (NPY) has been demonstrated largely in the hypothalamus, but also in other brain areas. A possible link has been suggested by a study showing co-localization of GR immunoreactivity with NPY immunopositivity in the hypothalamus, locus ceruleus, and subnuclei of tractus solitarii [92]. Glucocorticoids alter NPY expression via GRs: Dexamethasone increases NPY levels in fetal brain cells in cultures, whereas RU486 blocks this effect [93]. Both single and repeated postnatal dexamethasone administration significantly increased NPY content in mediobasal and lateral hypothalamus [94,95]. On the other hand, in adrenalectomized rats hypothalamic levels of NPY are decreased in the arcuate and paraventricular nuclei [96–98], while the infusion of corticosteroids increases NPY expression and synthesis [96,99]. Our unpublished data show that in the hippocampus, prenatal betamethasone exposure on E15 results in long-term NPY increases determined on PN20 (see Fig. 3).
Fig. 3.
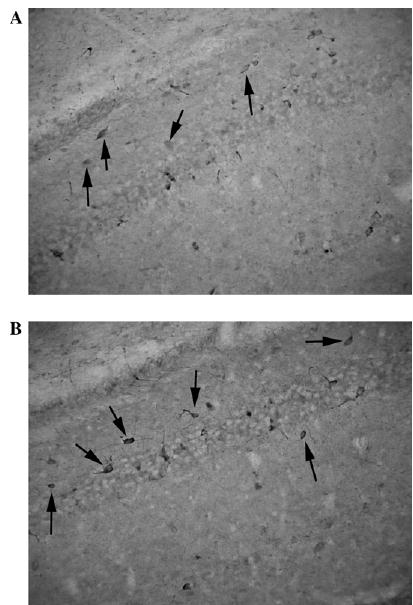
NPY immunopositivity in the CA1 region is increased by prenatal corticosteroid treatment. (A) PN20 control male rat prenatally exposed to saline on E15. Immunointensity is low compared with that of prenatally betamethasone-exposed rat (B). (B) PN20 male rat prenatally exposed to 2 × 0.4 mg/kg betamethasone on E15. Immunointensity is higher compared with the control (A). Immunostaining has been performed in the same dish for both treatments to ensure similar conditions.
The role of NPY in seizures and epileptogenesis is notorious [100]. Increased levels of NPY have anticonvulsant, antiepileptogenic, and neuroprotective effects [101–105]. Thus, it is possible that our findings of decreased kindling epileptogenesis after prenatal betamethasone exposure represent the result of a betamethasone-induced increase in NPY in the hippocampus.
7. Significance
Synthetic corticosteroids are frequently used in repeated courses for prenatal administration to improve lung development if there is a risk of premature delivery. In the United States, approximately 138,000 women annually would present at risk for premature delivery, with 91,915 births between 24 and 34 weeks of gestation [106]. In neonates with birth weight between 501 and 1500 g, use of prenatal corticosteroids reached 79% [14]. Additionally, a European study determined that about 85% of neonates with prenatal corticosteroid therapy receive multiple-course corticosteroid treatments [107]. As shown previously, children with a history of multiple prenatal corticosteroid courses have developmental problems associated with aggressive/destructive behavior, distractability, and hyperactivity [28]. Thus, only in the United States, about 95,000 neonates annually are at potential risk of developing behavioral problems due to repeated prenatal corticosteroid exposure. This is also consistent with findings in experimental animals that early developmental corticosteroids can reprogram the brain [108], including the HPA axis, which may result in predisposition to affective disorders [31,68].
First, it is necessary to determine the exact mechanisms by which prenatal corticosteroids alter postnatal behavior. This will make possible the development of specific, effective, mechanistic therapy regimens for the side effects of prenatal corticosteroid therapy in those situations when prenatal corticosteroid therapy is fully justified and cannot be avoided. These treatments will alleviate potential behavioral problems in children prenatally exposed to betamethasone.
Second, the data indicate that the prenatal programming effects of corticosteroids [12,108], along with early postnatal environmental imprinting [31], are very similar to the organizational effects of sex steroids occurring during a critical period of brain development [109–112]. Thus, it is possible that the effects accomplished by any steroid hormone surge during a critical period of nervous system development are permanently imprinted (programmed, organizational). Data further indicate that prenatal surges of natural corticosteroids may be programming more undesirable side effects than synthetic corticosteroids. These effects are permanent and, therefore, cannot be reverted later [113]. However, by understanding the mechanisms, we may able to design specific treatments, even for limited undesirable side effects of prenatal betamethasone exposure.
Finally it should be emphasized that the data on the outcomes of multiple prenatal corticosteroid courses in humans are inadequate, and most of the available information emanates from animal experiments. Therefore, conclusions from animal studies should be extrapolated to humans only after thorough comparison of the human and animal conditions [114].
8. Conclusions
Studies in experimental animals can reproduce at least some of the behavioral problems occurring in children after multiple prenatal courses of corticosteroids and may provide mechanistic explanations. Currently available data indicate that every corticosteroid action must be judged in a very specific context. First, synthetic corticosteroids have effects different from those of natural corticosteroids and from prolonged stress. There may even be differences between the effects of both principal synthetic corticosteroids, betamethasone and dexamethasone. Second, the doses and timing of the treatment may be critical to the outcome. Prenatal corticosteroid exposure can affect (reprogram) the expression of a variety of molecules such as GRs, CRH, BDNF, and NPY. All these molecules are present and functional in the hippocampus and can be linked to neuronal survival, behavior, and seizures, and additionally may interact with each other. Questions still remain: Which are the primary pathways of the glucocorticoid effects? Which changes seen in these molecules are secondary, compensatory? An increased understanding of the mechanism of action of prenatal corticosteroids in the hippocampus may lead to the discovery of specific treatments preventing undesirable side effects in those children in whom prenatal corticosteroids represent life-saving treatment and cannot be avoided.
Acknowledgments
I thank my wife, Dr. Jana Velíšková, for very critical reading of this article, and Ms. Zunju Hu for outstanding technical assistance. This work was supported by Grant NS-41366 from the NIH.
References
- 1.Yau JL, Olsson T, Morris RG, Meaney MJ, Seckl JR. Glucocorticoids, hippocampal corticosteroid receptor gene expression and antidepressant treatment: relationship with spatial learning in young and aged rats. Neuroscience. 1995;66:571–81. doi: 10.1016/0306-4522(94)00612-9. [DOI] [PubMed] [Google Scholar]
- 2.Cameron HA, McKay RD. Restoring production of hippocampal neurons in old age. Nat Neurosci. 1999;2:894–7. doi: 10.1038/13197. [DOI] [PubMed] [Google Scholar]
- 3.McEwen BS. Corticosteroids and hippocampal plasticity. Ann NY Acad Sci 1994,746:134–42; discussion 142–4, 178–9. [DOI] [PubMed]
- 4.Pavlides C, Watanabe Y, McEwen BS. Effects of glucocorticoids on hippocampal long-term potentiation. Hippocampus. 1993;3:183–92. doi: 10.1002/hipo.450030210. [DOI] [PubMed] [Google Scholar]
- 5.McEwen BS. The brain is an important target of adrenal steroid actions: a comparison of synthetic and natural steroids. Ann NY Acad Sci. 1997;823:201–13. doi: 10.1111/j.1749-6632.1997.tb48392.x. [DOI] [PubMed] [Google Scholar]
- 6.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–22. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 7.McEwen BS, Magarinos AM. Stress effects on morphology and function of the hippocampus. Ann NY Acad Sci. 1997;821:271–84. doi: 10.1111/j.1749-6632.1997.tb48286.x. [DOI] [PubMed] [Google Scholar]
- 8.Brown ES, Rush AJ, McEwen BS. Hippocampal remodeling and damage by corticosteroids: implications for mood disorders. Neuropsychopharmacology. 1999;21:474–84. doi: 10.1016/S0893-133X(99)00054-8. [DOI] [PubMed] [Google Scholar]
- 9.Sapolsky RM. Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress. 1996;1:1–19. doi: 10.3109/10253899609001092. [DOI] [PubMed] [Google Scholar]
- 10.Lee AL, Ogle WO, Sapolsky RM. Stress and depression: possible links to neuron death in the hippocampus. Bipolar Disord. 2002;4:117–28. doi: 10.1034/j.1399-5618.2002.01144.x. [DOI] [PubMed] [Google Scholar]
- 11.Joels M. Corticosteroid actions in the hippocampus. J Neuroendocrinol. 2001;13:657–69. doi: 10.1046/j.1365-2826.2001.00688.x. [DOI] [PubMed] [Google Scholar]
- 12.Matthews SG. Antenatal glucocorticoids and programming of the developing CNS. Pediatr Res. 2000;47:291–300. doi: 10.1203/00006450-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Aghajafari F, Murphy K, Matthews S, Ohlsson A, Amankwah K, Hannah M. Repeated doses of antenatal corticosteroids in animals: a systematic review. Am J Obstet Gynecol. 2002;186:843–9. doi: 10.1067/mob.2002.121624. [DOI] [PubMed] [Google Scholar]
- 14.Fanaroff AA, Hack M, Walsh MC. The NICHD neonatal research network: changes in practice and outcomes during the first 15 years. Semin Perinatol. 2003;27:281–7. doi: 10.1016/s0146-0005(03)00055-7. [DOI] [PubMed] [Google Scholar]
- 15.Antenatal corticosteroids revisited: repeat courses. NIH Consens Statement 2000,17:1–10 [cited 2001 October 29]. Available from: http://odp.od.nih.gov/consensus/cons/112/112_statement.htm [PubMed]
- 16.Effect of corticosteroids for fetal maturation on perinatal outcomes. NIH Consens Statement 1994;12:1–24. [PubMed]
- 17.NIH Consensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. Effect of corticosteroids for fetal maturation on perinatal outcomes. [see comments] JAMA. 1995;273:413–8. doi: 10.1001/jama.1995.03520290065031. [DOI] [PubMed] [Google Scholar]
- 18.MacArthur BA, Howie RN, Dezoete JA, Elkins J. Cognitive and psychosocial development of 4-year-old children whose mothers were treated antenatally with betamethasone. Pediatrics. 1981;68:638–43. [PubMed] [Google Scholar]
- 19.MacArthur BA, Howie RN, Dezoete JA, Elkins J. School progress and cognitive development of 6-year-old children whose mothers were treated antenatally with betamethasone. Pediatrics. 1982;70:99–105. [PubMed] [Google Scholar]
- 20.Schmand B, Neuvel J, Smolders-de Haas H, Hoeks J, Treffers PE, Koppe JG. Psychological development of children who were treated antenatally with corticosteroids to prevent respiratory distress syndrome. Pediatrics. 1990;86:58–64. [PubMed] [Google Scholar]
- 21.Smolders-de Haas H, Neuvel J, Schmand B, Treffers PE, Koppe JG, Hoeks J. Physical development and medical history of children who were treated antenatally with corticosteroids to prevent respiratory distress syndrome: a 10- to 12-year follow-up. Pediatrics. 1990;86:65–70. [PubMed] [Google Scholar]
- 22.French NP, Hagan R, Evans SF, Godfrey M, Newnham JP. Repeated antenatal corticosteroids: size at birth and subsequent development. Am J Obstet Gynecol. 1999;180:114–21. doi: 10.1016/s0002-9378(99)70160-2. [DOI] [PubMed] [Google Scholar]
- 23.Ng PC, Wong GW, Lam CW, Lee CH, Fok TF, Wong MY, et al. Effect of multiple courses of antenatal corticosteroids on pituitary–adrenal function in preterm infants. Arch Dis Child Fetal Neonatal Ed. 1999;80:F213–6. doi: 10.1136/fn.80.3.f213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorp JA, O’Connor M, Belden B, Etzenhouser J, Hoffman EL, Jones PG. Effects of phenobarbital and multiple-dose corticosteroids on developmental outcome at age 7 years. Obstet Gynecol. 2003;101:363–73. doi: 10.1016/s0029-7844(02)02509-7. [DOI] [PubMed] [Google Scholar]
- 25.Seckl JR. Antenatal glucocorticoid therapy: a caveat to the applause. Clin Sci (Colch) 2000;98:127–8. [PubMed] [Google Scholar]
- 26.Yunis KA, Bitar FF, Hayek P, Mroueh SM, Mikati M. Transient hypertrophic cardiomyopathy in the newborn following multiple doses of antenatal corticosteroids. Am J Perinatol. 1999;16:17–21. doi: 10.1055/s-2007-993830. [DOI] [PubMed] [Google Scholar]
- 27.Banks BA, Cnaan A, Morgan MA for the North American Thyrotropin-Releasing Hormone Study Group. Multiple courses of antenatal corticosteroids and outcome of premature neonates. Am J Obstet Gynecol. 1999;181:709–17. doi: 10.1016/s0002-9378(99)70517-x. [DOI] [PubMed] [Google Scholar]
- 28.French NP, Hagan R, Evans SF, Mullan A, Newnham JP. Repeated antenatal corticosteroids: effects on cerebral palsy and childhood behavior. Am J Obstet Gynecol. 2004;190:588–95. doi: 10.1016/j.ajog.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Pedder J. Psychoanalytic views of aggression: some theoretical problems. Br J Med Psychol. 1992;65(Pt 2):95–106. doi: 10.1111/j.2044-8341.1992.tb01690.x. [DOI] [PubMed] [Google Scholar]
- 30.Stott DH. Follow-up study from birth of the effects of prenatal stresses. Dev Med Child Neurol. 1973;15:770–87. doi: 10.1111/j.1469-8749.1973.tb04912.x. [DOI] [PubMed] [Google Scholar]
- 31.Brunson KL, Avishai-Eliner S, Hatalski CG, Baram TZ. Neurobiology of the stress response early in life: evolution of a concept and the role of corticotropin releasing hormone. Mol Psychiatry. 2001;6:647–56. doi: 10.1038/sj.mp.4000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson JW, Mitzner W, Beck JC, et al. Long-term effects of betamethasone on fetal development. Am J Obstet Gynecol. 1981;141:1053–64. doi: 10.1016/s0002-9378(16)32697-7. [DOI] [PubMed] [Google Scholar]
- 33.Uno H, Lohmiller L, Thieme C, et al. Brain damage induced by prenatal exposure to dexamethasone in fetal rhesus macaques: I. Hippocampus Dev Brain Res. 1990;53:157–67. doi: 10.1016/0165-3806(90)90002-g. [DOI] [PubMed] [Google Scholar]
- 34.Uno H, Eisele S, Sakai A, et al. Neurotoxicity of glucocorticoids in the primate brain. Horm Behav. 1994;28:336–48. doi: 10.1006/hbeh.1994.1030. [DOI] [PubMed] [Google Scholar]
- 35.Schwab M, Antonow-Schlorke I, Kuhn B, et al. Effect of antenatal betamethasone treatment on microtubule-associated proteins MAP1B and MAP2 in fetal sheep. J Physiol. 2001;530:497–506. doi: 10.1111/j.1469-7793.2001.0497k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antonow-Schlorke I, Kuhn B, Muller T, et al. Antenatal betamethasone treatment reduces synaptophysin immunoreactivity in presynaptic terminals in the fetal sheep brain. Neurosci Lett. 2001;297:147–50. doi: 10.1016/s0304-3940(00)01605-0. [DOI] [PubMed] [Google Scholar]
- 37.De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 38.Seckl JR. Physiologic programming of the fetus. Clin Perinatol 1998,25:939–62. vii. [PubMed]
- 39.Slotkin TA, Lappi SE, McCook EC, Tayyeb MI, Eylers JP, Seidler FJ. Glucocorticoids and the development of neuronal function: effects of prenatal dexamethasone exposure on central noradrenergic activity. Biol Neonate. 1992;61:326–36. doi: 10.1159/000243761. [DOI] [PubMed] [Google Scholar]
- 40.Owen D, Matthews SG. Glucocorticoids and sex-dependent development of brain glucocorticoid and mineralocorticoid receptors. Endocrinology. 2003;144:2775–84. doi: 10.1210/en.2002-0145. [DOI] [PubMed] [Google Scholar]
- 41.Scheepens A, van de Waarenburg M, van den Hove D, Blanco CE. A single course of prenatal betamethasone in the rat alters postnatal brain cell proliferation but not apoptosis. J Physiol. 2003;552:163–75. doi: 10.1113/jphysiol.2003.043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlos RQ, Seidler FJ, Slotkin TA. Fetal dexamethasone exposure alters macromolecular characteristics of rat brain development: a critical period for regionally selective alterations? Teratology. 1992;46:45–59. doi: 10.1002/tera.1420460108. [DOI] [PubMed] [Google Scholar]
- 43.Welberg LA, Seckl JR, Holmes MC. Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: possible implications for behaviour. Neuroscience. 2001;104:71–9. doi: 10.1016/s0306-4522(01)00065-3. [DOI] [PubMed] [Google Scholar]
- 44.Diaz R, Fuxe K, Ogren SO. Prenatal corticosterone treatment induces long-term changes in spontaneous and apomorphine-mediated motor activity in male and female rats. Neuroscience. 1997;81:129–40. doi: 10.1016/s0306-4522(97)00141-3. [DOI] [PubMed] [Google Scholar]
- 45.Mickley GA, Ferguson JL, Mulvihill MA, Nemeth TJ. Progressive behavioral changes during the maturation of rats with early radiation-induced hypoplasia of fascia dentata granule cells. Neurotoxicol Teratol. 1989;11:385–93. doi: 10.1016/0892-0362(89)90012-3. [DOI] [PubMed] [Google Scholar]
- 46.Rayburn WF, Christensen HD, Gonzalez CL. A placebo-controlled comparison between betamethasone and dexamethasone for fetal maturation: differences in neurobehavioral development of mice offspring. Am J Obstet Gynecol 1997,176:842–50; discussion 850–1. [DOI] [PubMed]
- 47.Rayburn WF, Christensen HD, Gonzalez CL, Rayburn LA, Stewart JD. Effect of in utero exposure to betamethasone on motivation/anxiety testing in mice offspring. Neurotoxicol Teratol. 1998;20:475–81. doi: 10.1016/s0892-0362(97)00131-1. [DOI] [PubMed] [Google Scholar]
- 48.Fride E, Dan Y, Feldon J, Halevy G, Weinstock M. Effects of prenatal stress on vulnerability to stress in prepubertal and adult rats. Physiol Behav. 1986;37:681–7. doi: 10.1016/0031-9384(86)90172-1. [DOI] [PubMed] [Google Scholar]
- 49.Barbazanges A, Piazza PV, Le Moal M, Maccari S. Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J Neurosci. 1996;16:3943–9. doi: 10.1523/JNEUROSCI.16-12-03943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vallee M, MacCari S, Dellu F, Simon H, Le Moal M, Mayo W. Long-term effects of prenatal stress and postnatal handling on age-related glucocorticoid secretion and cognitive performance: a longitudinal study in the rat. Eur J Neurosci. 1999;11:2906–16. doi: 10.1046/j.1460-9568.1999.00705.x. [DOI] [PubMed] [Google Scholar]
- 51.Lemaire V, Koehl M, Le Moal M, Abrous DN. Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc Natl Acad Sci USA. 2000;97:11032–7. doi: 10.1073/pnas.97.20.11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frye CA, Bayon LE. Prenatal stress reduces the effectiveness of the neurosteroid 3-alpha, 5-alpha-THP to block kainic-acid-induced seizures. Dev Psychobiol. 1999;34:227–34. [PubMed] [Google Scholar]
- 53.Velíšek L. Prenatal betamethasone exposure suppresses kindling epileptogenesis in immature rats. In: Corcoran ME, Moshé SL, editors. Kindling 6, in press.
- 54.Makara GB, Haller J. Non-genomic effects of glucocorticoids in the neural system: evidence, mechanisms and implications. Prog Neurobiol. 2001;65:367–90. doi: 10.1016/s0301-0082(01)00012-0. [DOI] [PubMed] [Google Scholar]
- 55.van Steensel B, Brink M, van der Meulen K, et al. Localization of the glucocorticoid receptor in discrete clusters in the cell nucleus. J Cell Sci. 1995;108(Pt 9):3003–11. doi: 10.1242/jcs.108.9.3003. [DOI] [PubMed] [Google Scholar]
- 56.van Steensel B, Jenster G, Damm K, Brinkmann AO, van Driel R. Domains of the human androgen receptor and glucocorticoid receptor involved in binding to the nuclear matrix. J Cell Biochem. 1995;57:465–78. doi: 10.1002/jcb.240570312. [DOI] [PubMed] [Google Scholar]
- 57.Helm KA, Han JS, Gallagher M. Effects of cholinergic lesions produced by infusions of 192 IgG-saporin on glucocorticoid receptor mRNA expression in hippocampus and medial prefrontal cortex of the rat. Neuroscience. 2002;115:765–74. doi: 10.1016/s0306-4522(02)00487-6. [DOI] [PubMed] [Google Scholar]
- 58.de Kloet ER. Hormones, brain and stress. Endocr Regul. 2003;37:51–68. [PubMed] [Google Scholar]
- 59.Reul JM, van den Bosch FR, de Kloet ER. Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: functional implications. J Endocrinol. 1987;115:459–67. doi: 10.1677/joe.0.1150459. [DOI] [PubMed] [Google Scholar]
- 60.Reul JM, van den Bosch FR, de Kloet ER. Differential response of type I and type II corticosteroid receptors to changes in plasma steroid level and circadian rhythmicity. Neuroendocrinology. 1987;45:407–12. doi: 10.1159/000124766. [DOI] [PubMed] [Google Scholar]
- 61.Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–11. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 62.Munck A, Mendel DB, Smith LI, Orti E. Glucocorticoid receptors and actions. Am Rev Respir Dis. 1990;141:S2–10. [PubMed] [Google Scholar]
- 63.Beato M, Sanchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17:587–609. doi: 10.1210/edrv-17-6-587. [DOI] [PubMed] [Google Scholar]
- 64.Meijer OC. Coregulator proteins and corticosteroid action in the brain. J Neuroendocrinol. 2002;14:499–505. doi: 10.1046/j.1365-2826.2002.00795.x. [DOI] [PubMed] [Google Scholar]
- 65.De Kloet ER. Hormones and the stressed brain. Ann NY Acad Sci. 2004;1018:1–15. doi: 10.1196/annals.1296.001. [DOI] [PubMed] [Google Scholar]
- 66.McEwan IJ, Wright AP, Gustafsson JA. Mechanism of gene expression by the glucocorticoid receptor: role of protein–protein interactions. Bioessays. 1997;19:153–60. doi: 10.1002/bies.950190210. [DOI] [PubMed] [Google Scholar]
- 67.Tronche F, Kellendonk C, Kretz O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 68.Muller M, Holsboer F, Keck ME. Genetic modification of corticosteroid receptor signalling: novel insights into pathophysiology and treatment strategies of human affective disorders. Neuropeptides. 2002;36:117–31. doi: 10.1054/npep.2002.0896. [DOI] [PubMed] [Google Scholar]
- 69.Dallman MF, Akana SF, Strack AM, et al. Chronic stress-induced effects of corticosterone on brain: direct and indirect. Ann NY Acad Sci. 2004;1018:141–50. doi: 10.1196/annals.1296.017. [DOI] [PubMed] [Google Scholar]
- 70.Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: pre-and postsynaptic location and release by stress. Neuroscience. 2004;126:533–40. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yan XX, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8:231–43. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW. Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci. 1994;14:2579–84. doi: 10.1523/JNEUROSCI.14-05-02579.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Contarino A, Gold LH. Targeted mutations of the corticotropin-releasing factor system: effects on physiology and behavior. Neuropeptides. 2002;36:103–16. doi: 10.1054/npep.2002.0899. [DOI] [PubMed] [Google Scholar]
- 74.Strohle A, Jahn H, Montkowski A, et al. Central and peripheral administration of atriopeptin is anxiolytic in rats. Neuroendocrinology. 1997;65:210–5. doi: 10.1159/000127274. [DOI] [PubMed] [Google Scholar]
- 75.Strohle A, Holsboer F. Stress responsive neurohormones in depression and anxiety. Pharmacopsychiatry. 2003;36(Suppl 3):S207–14. doi: 10.1055/s-2003-45132. [DOI] [PubMed] [Google Scholar]
- 76.Timpl P, Spanagel R, Sillaber I, et al. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–6. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- 77.Smith GW, Aubry JM, Dellu F, et al. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- 78.Zobel AW, Nickel T, Kunzel HE, et al. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34:171–81. doi: 10.1016/s0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 79.Baram TZ, Mitchell WG, Snead OC, Horton EJ, Saito M. Brain–adrenal axis hormones are altered in the CSF of infants with massive infantile spasms. Neurology. 1992;42:1171–5. doi: 10.1212/wnl.42.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baram TZ, Schultz L. Corticotropin-releasing hormone is a rapid and potent convulsant in the infant rat. Dev Brain Res. 1991;61:97–101. doi: 10.1016/0165-3806(91)90118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baram TZ, Schultz L. ACTH does not control neonatal seizures induced by administration of exogenous corticotropin-releasing hormone. Epilepsia. 1995;36:174–8. doi: 10.1111/j.1528-1157.1995.tb00977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kellaway P, Frost JD, Hrachovy RA. Infantile spasms. In: Morselli PL, Pippenger CE, Penry JK, editors. Antiepileptic drug therapy in pediatrics. New York: Raven Press; 1983. p. 115–36.
- 83.Nitta A, Ohmiya M, Sometani A, et al. Brain-derived neurotrophic factor prevents neuronal cell death induced by corticosterone. J Neurosci Res. 1999;57:227–35. doi: 10.1002/(SICI)1097-4547(19990715)57:2<227::AID-JNR8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 84.Schaaf MJ, de Jong J, de Kloet ER, Vreugdenhil E. Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813:112–20. doi: 10.1016/s0006-8993(98)01010-5. [DOI] [PubMed] [Google Scholar]
- 85.Chao HM, Sakai RR, Ma LY, McEwen BS. Adrenal steroid regulation of neurotrophic factor expression in the rat hippocampus. Endocrinology. 1998;139:3112–8. doi: 10.1210/endo.139.7.6114. [DOI] [PubMed] [Google Scholar]
- 86.Schaaf MJ, Hoetelmans RW, de Kloet ER, Vreugdenhil E. Corticosterone regulates expression of BDNF and trkB but not NT-3 and trkC mRNA in the rat hippocampus. J Neurosci Res. 1997;48:334–41. [PubMed] [Google Scholar]
- 87.Nibuya M, Takahashi M, Russell DS, Duman RS. Repeated stress increases catalytic TrkB mRNA in rat hippocampus. Neurosci Lett. 1999;267:81–4. doi: 10.1016/s0304-3940(99)00335-3. [DOI] [PubMed] [Google Scholar]
- 88.Scharfman HE, Goodman JH, Sollas AL, Croll SD. Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol. 2002;174:201–14. doi: 10.1006/exnr.2002.7869. [DOI] [PubMed] [Google Scholar]
- 89.Larmet Y, Reibel S, Carnahan J, Nawa H, Marescaux C, Depaulis A. Protective effects of brain-derived neurotrophic factor on the development of hippocampal kindling in the rat. Neuro Report. 1995;6:1937–41. doi: 10.1097/00001756-199510020-00027. [DOI] [PubMed] [Google Scholar]
- 90.Reibel S, Depaulis A, Larmet Y. BDNF and epilepsy—the bad could turn out to be good. Trends Neurosci. 2001;24:318–9. doi: 10.1016/s0166-2236(00)01869-5. [DOI] [PubMed] [Google Scholar]
- 91.Reibel S, Larmet Y, Le BT, Carnahan J, Marescaux C, Depaulis A. Brain-derived neurotrophic factor delays hippocampal kindling in the rat. Neuroscience. 2000;100:777–88. doi: 10.1016/s0306-4522(00)00351-1. [DOI] [PubMed] [Google Scholar]
- 92.Harfstrand A. Brain neuropeptide Y mechanisms: basic aspects and involvement in cardiovascular and neuroendocrine regulation. Acta Physiol Scand Suppl. 1987;565:1–83. [PubMed] [Google Scholar]
- 93.Barnea A, Cho G, Hajibeigi A, Aguila MC, Magni P. Dexamethasone-induced accumulation of neuropeptide-Y by aggregating fetal brain cells in culture: a process dependent on the developmental age of the aggregates. Endocrinology. 1991;129:931–8. doi: 10.1210/endo-129-2-931. [DOI] [PubMed] [Google Scholar]
- 94.Corder R, Pralong F, Turnill D, Saudan P, Muller AF, Gaillard RC. Dexamethasone treatment increases neuropeptide Y levels in rat hypothalamic neurones. Life Sci. 1988;43:1879–86. doi: 10.1016/s0024-3205(88)80005-5. [DOI] [PubMed] [Google Scholar]
- 95.McKibbin PE, Cotton SJ, McCarthy HD, Williams G. The effect of dexamethasone on neuropeptide Y concentrations in specific hypothalamic regions. Life Sci. 1992;51:1301–7. doi: 10.1016/0024-3205(92)90020-p. [DOI] [PubMed] [Google Scholar]
- 96.Pralong FP, Corder R, Gaillard RC. The effects of chronic glucocorticoid excess, adrenalectomy and stress on neuropeptide Y in individual rat hypothalamic nuclei. Neuropeptides. 1993;25:223–31. doi: 10.1016/0143-4179(93)90107-l. [DOI] [PubMed] [Google Scholar]
- 97.White BD, Dean RG, Martin RJ. Adrenalectomy decreases neuropeptide Y mRNA levels in the arcuate nucleus. Brain Res Bull. 1990;25:711–5. doi: 10.1016/0361-9230(90)90047-4. [DOI] [PubMed] [Google Scholar]
- 98.Akabayashi A, Watanabe Y, Wahlestedt C, McEwen BS, Paez X, Leibowitz SF. Hypothalamic neuropeptide Y, its gene expression and receptor activity: relation to circulating corticosterone in adrenalectomized rats. Brain Res. 1994;665:201–12. doi: 10.1016/0006-8993(94)91339-0. [DOI] [PubMed] [Google Scholar]
- 99.Dean RG, White BD. Neuropeptide Y expression in rat brain: effects of adrenalectomy. Neurosci Lett. 1990;114:339–44. doi: 10.1016/0304-3940(90)90587-y. [DOI] [PubMed] [Google Scholar]
- 100.Vezzani A, Sperk G, Colmers WF. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci. 1999;22:25–30. doi: 10.1016/s0166-2236(98)01284-3. [DOI] [PubMed] [Google Scholar]
- 101.Woldbye DP, Larsen PJ, Mikkelsen JD, Klemp K, Madsen TM, Bolwig TG. Powerful inhibition of kainic acid seizures by neuropeptide Y via Y5- like receptors. Nat Med. 1997;3:761–4. doi: 10.1038/nm0797-761. [DOI] [PubMed] [Google Scholar]
- 102.Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, Palmiter RD. Knock-out mice reveal a critical antiepileptic role for neuropeptide Y. J Neurosci. 1997;17:8927–36. doi: 10.1523/JNEUROSCI.17-23-08927.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–21. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 104.Richichi C, Lin EJ, Stefanin D, et al. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. 2004;24:3051–9. doi: 10.1523/JNEUROSCI.4056-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smialowska M, Wieronska JM, Szewczyk B. Neuroprotective effect of NPY on kainate neurotoxicity in the hippocampus. Pol J Pharmacol. 2003;55:979–86. [PubMed] [Google Scholar]
- 106.Caughey AB, Parer JT. Recommendations for repeat courses of antenatal corticosteroids: a decision analysis. Am J Obstet Gynecol 2002,186:1221–6; discussion 1226–9. [DOI] [PubMed]
- 107.Empana JP, Anceschi MM, Szabo I, Cosmi EV, Breart G, Truffert P. Antenatal corticosteroids policies in 14 European countries: factors associated with multiple courses. The EUR-AIL survey Acta. Paediatr. 2004;93:1318–22. [PubMed] [Google Scholar]
- 108.Welberg LAM, Seckl JR. Prenatal stress, glucocorticoids and the programming of the brain. J Neuroendocrinol. 2001;13:113–28. doi: 10.1046/j.1365-2826.2001.00601.x. [DOI] [PubMed] [Google Scholar]
- 109.Kelly DD. Sexual differentiation of the nervous system. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of neural science. 3rd ed. New York: Elsevier; 1991. p. 959–73.
- 110.Breedlove SM. Sexual differentiation of the human nervous system. Annu Rev Psychol. 1994;45:389–418. doi: 10.1146/annurev.ps.45.020194.002133. [DOI] [PubMed] [Google Scholar]
- 111.MacLusky NJ, Naftolin F. Sexual differentiation of the central nervous system. Science. 1981;211:1294–303. doi: 10.1126/science.6163211. [DOI] [PubMed] [Google Scholar]
- 112.Gorski RA. Gonadal hormones and the perinatal development of neuroendocrine function. In: Martini L, Ganong WF, editors. Frontiers in neuroendocrinology. New York: Oxford Univ. Press; 1971. p. 237–90.
- 113.Jost A, Magre S. Testicular development phases and dual hormonal control of sexual organogenesis. In: Serio Mea, editor. Sexual differentiation: basic and clinical aspects. New York: -Raven Press; 1984. p. 1–15.
- 114.Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–24. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Velíšek L, Moshé SL. Effects of brief seizures during development. In: Sutula T, Pitkanen A, editors. Do seizures damage the brain. Amsterdam: Elsevier; 2002. p. 355–64.
- 116.Gottlieb A, Keydor I, Epstein HT. Rodent brain growth stages: an analytical review. Biol Neonate. 1977;32:166–76. doi: 10.1159/000241012. [DOI] [PubMed] [Google Scholar]
- 117.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 118.Dunlop SA, Archer MA, Quinlivan JA, Beazley LD, Newnham JP. Repeated prenatal corticosteroids delay myelination in the ovine central nervous system. J Matern Fetal Med. 1997;6:309–13. doi: 10.1002/(SICI)1520-6661(199711/12)6:6<309::AID-MFM1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 119.Huang WL, Beazley LD, Quinlivan JA, Evans SF, Newnham JP, Dunlop SA. Effect of corticosteroids on brain growth in fetal sheep. Obstet Gynecol. 1999;94:213–8. doi: 10.1016/s0029-7844(99)00265-3. [DOI] [PubMed] [Google Scholar]
- 120.Hellal-Levy C, Fagart J, Souque A, Rafestin-Oblin ME. Mechanistic aspects of mineralocorticoid receptor activation. Kidney Int. 2000;57:1250–5. doi: 10.1046/j.1523-1755.2000.00958.x. [DOI] [PubMed] [Google Scholar]
- 121.Liu W, Wang J, Sauter NK, Pearce D. Steroid receptor heterodimerization demonstrated in vitro and in vivo. Proc Natl Acad Sci USA. 1995;92:12480–4. doi: 10.1073/pnas.92.26.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Trapp T, Rupprecht R, Castren M, Reul JM, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptor: a new principle of glucocorticoid action in the CNS. Neuron. 1994;13:1457–62. doi: 10.1016/0896-6273(94)90431-6. [DOI] [PubMed] [Google Scholar]