

SUMMARY
Heterotrimeric guanine nucleotide-binding proteins (G proteins) transmit signals from membrane bound G protein-coupled receptors (GPCRs) to intracellular effector proteins. The Gq subfamily of Gα subunits couples GPCR activation to the enzymatic activity of phospholipase C-β (PLC-β). Regulators of G protein signaling (RGS) proteins bind to activated Gα subunits, including Gαq, and regulate Gα signaling by acting as GTPase activating proteins (GAPs), increasing the rate of the intrinsic GTPase activity, or by acting as effector antagonists for Gα subunits. GPCR kinases (GRKs) phosphorylate agonist-bound receptors in the first step of receptor desensitization. The amino-termini of all GRKs contain an RGS homology (RH) domain (Siderovski et al., Curr. Bio., 1996) and binding of the GRK2 RH domain to Gαq attenuates PLC-β activity (Carman et al., J. Biol. Chem. 1999). The RH domain of GRK2 interacts with Gαq/11 through a novel Gα binding surface termed the “C” site (Sterne-Marr et al., J. Biol. Chem. 2003). Here, molecular modeling of the Gαq-GRK2 complex and site-directed mutagenesis of Gαq were used to identify residues in Gαq that interact with GRK2. The model identifies Pro185 in Switch I of Gαq as being at the crux of the interface, and mutation of this residue to lysine disrupts Gαq binding to the GRK2-RH domain. Switch III also appears to play a role in GRK2 binding because the mutations Gαq-V240A, Gαq-D243A, both residues within switch III, and Gαq-Q152A, a residue that structurally supports switch III, are defective in binding GRK2. Furthermore, GRK2-mediated inhibition of Gαq-Q152A-R183C-stimulated inositol phosphate release is reduced in comparison to Gαq-R183C. Interestingly, the model also predicts that residues in the helical domain of Gαq interact with GRK2. In fact, the mutants Gαq-K77A, Gαq-L78D, Gαq-Q81A and Gαq-R92A have reduced binding to the GRK2-RH domain. Finally, while the mutant Gαq-T187K has greatly reduced binding to RGS2 and RGS4 it has little to no effect on binding to GRK2. Thus the RH domain A and C sites for Gαq interaction rely on contacts with distinct regions and different switch I residues in Gαq.
INTRODUCTION
G protein coupled receptors (GPCRs)1 are heptahelical integral membrane proteins responsible for the transmission of extracellular signals, such as light, neurotransmitters and hormones, to intracellular signaling pathways. Agonist-bound GPCRs directly interact with heterotrimeric (αβγ) G proteins and catalyze nucleotide exchange on Gα subunits (1). Several mechanisms are in place to ensure the appropriate level of response to an agonist. Receptors become desensitized to agonist stimulation upon phosphorylation by GPCR kinases (GRKs) and subsequent binding of arrestins (2–5). The Gα subunit has intrinsic GTPase activity that returns the G protein to the inactive GDP bound state, promoting reassociation with Gβγ (6).
A third mechanism, which accounts for the rapid desensitization observed in cellular signaling systems such as phototransduction in the eye (7), is attributed to GTPase activating proteins (GAPs), that bind Gα subunits and accelerate the GTPase reaction. GAPs for Gα subunits include effector molecules, such as phospholipase C-β, and regulators of G protein signaling (RGS) proteins (8,9). There are over 30 RGS proteins identified, all of which contain an approximately 130 residue domain called the RGS homology (RH) domain (10,11). RGS proteins act as GAPs by binding to Gα and stabilizing the transition state of GTP hydrolysis (11). The crystal structures of the RH domains from RGS4, RGS9, axin, GRK2, p115RhoGEF and PDZRhoGEF have been determined (11–16). A typical RH domain consists of nine helices organized into a bundle subdomain of helices α4-α7 and a terminal subdomain of helices α1-α3 and α8- α9 (11). The RH domains of RGS proteins contact Gα subunits with a discontinuous surface composed of loops between helices 3/4, 5/6 and 7/8, that has been defined as the A site (17). In contrast, the RH domain of axin, which is not known to bind Gα subunits, binds the adenomatous polyposis coli protein (APC) at a cleft formed between the terminal and bundle subdomains (13). This has been defined as the B site (17).
The RH domain of GRK2 is responsible for specifically binding to active forms of Gαq, Gα11 and Gα14, but not Gαs, Gαi, Gα12/13 or Gα16 (18–21). The structure of full length GRK2 indicates that its RH domain assumes a fold similar to other RGS proteins with the addition of two α helices contributed by residues 513–547, which follow its protein kinase domain in the primary sequence (14). Previously, we have shown that the binding site for Gαq on the RH domain of GRK2 is distinct from both the RH domain A and B sites (22), and consists primarily of the solvent-exposed surface of the α5 helix. This site is now referred to as the C site (22).
The residues on Gαq required for association with the C site of the GRK2 RH domain are not known. Interestingly, a G188S mutation in Gαq has no effect on the GRK2-Gαq interaction (22), even though this mutation prevents the interaction of Gαq with other RGS proteins (23,24), suggesting that the Gαq residues critical for interaction with GRK2 are different from those used to bind RGS proteins. However the activationdependent association of Gαq with GRK2 requires that at least part of the interface involves the switch regions of Gαq (18). In support of this, GRK2 binds chimeric proteins that contain the GTPase domain of Gαq and the helical domain of Gα16, the only Gαq family member that does not interact with the RH domain of GRK2, but not reciprocal chimeras, in an activation dependent manner (19).
In this study we use a molecular modeling approach to identify residues on Gαq that may interact with GRK2. Site-directed mutagenesis followed by GST-pulldown and cellular inositol phosphate assays indicate that contact sites for GRK2 on Gαq include the Switch I and III regions as well as residues in the helical domain of Gαq. Some of these residues are distinct from those that are important for interactions with RGS2 and RGS4. In addition, our previous mutational studies of the GRK2 C site were based upon a homology model of the GRK2 RH domain (22). Because the structure of full-length GRK2 indicates that the α5 helix, the major point of Gαq contact, is significantly longer than modeled (14), we have further refined the C site based on the Gαq:RH model and the Gαq mutagenesis studies described herein.
EXPERIMENTAL PROCEDURES
Materials
HEK-293 cells were from American Type Culture Collection (CRL- 1573). FuGENE 6 transfection reagent was from Roche Molecular Biochemicals. Super Signal West Pico ECL reagents were from Pierce. Myo-[3H] Inositol was obtained from Perkin Elmer Life Sciences. Cell culture media were from Mediatech Cellgro. GRK2 mouse monoclonal antibody was from Upstate Biotechnology. HRP-conjugated antimouse secondary antibody was from Promega. Ultima Flo AF and Ultima Gold scintillation cocktails were from Packard Chemical. All other chemicals and reagents were from Sigma Chemical Co. and Fisher Scientific.
Expression Plasmids and Mutagenesis
pcDNA3-Gαq-R183C (Gαq-RC)with an internal EE epitope tag was provided by Dr. C. Berlot. EE tagged Gαq and Gαq-Q209L (Gαq-QL) have been described previously (19). All Gαq, Gαq QL and Gαq RC point mutants were created in the background of EE tagged protein using the sequential PCR method (25). The GST-RGS4 expression plasmid was provided by Dr. R. Neubig and pcDNA3-RGS2 was provided by Dr. D. Siderovski. GST-RGS2 was created by using PCR to engineer a 5’ BamH1 site and a 3’ XhoI site onto RGS2 and then subcloning the RGS2 fragment into the BamH1 and XhoI sites of pGEX-5X1. GRK2 constructs have been described previously (18,22). GRK2 mutants were prepared by sequential PCR or by QuikChange Mutagenesis (Stratagene).
Cell Culture and Transfection
HEK-293 cells were maintained in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum and 100 units/ml penicillinstreptomycin at 37 ºC in 5% CO2. Cells in 6 well plates were transfected with 1 μg of DNA and 3 μl of FuGENE, while 3 μg of DNA and 9 μl of FuGENE were used for the transfection of cells in 6 cm dishes.
Molecular Modeling
Gαq was homology modeled with the SWISS-MODEL server (26) using as a template the structure of Gαi (49% sequence identity) in complex with RGS4 (11), which represents the most complete atomic model of an activated Gα subunit. The model was subsequently verified by ERRAT (27). As expected, the regions of the model evaluated as most unreliable (over the 95% confidence level) are the effector binding loops of Gαq, which vary greatly among the four Gα subfamilies. However, the three switch regions of the G protein represent the most likely binding site for GRK2 given its requirement for activated Gαq (18). The three switch regions of the model are distant from the effector loops, include some of the most highly conserved residues among Gα subunits, and are found in essentially the same conformation in all crystal structures that involve an activated Gα subunit. Evaluation by ERRAT also suggested that these regions were modeled reliably.
To model the GRK2-Gαq complex, automated docking programs were tested, but not used because they generally fail to accurately model changes, such as those of side chains, upon complex formation (28,29). We instead imposed several strict constraints based on experimental data to manually dock Gαq with GRK2. The resulting model of their complex at the very least should predict which regions of Gαq could be responsible for both complex formation and specificity. The first constraint was to limit the GRK2- interaction surface of Gαq to its three switch regions and to the αA helix and the αB-αC loop in the helical domain. This constraint derives from the facts that the formation of the GRK2-Gαq complex is dependent on the active conformation of the G protein, and therefore presumably the conformation of its switch regions (18), and that the adjacent αA helix and αB-αC loop have also been shown to contribute to the binding of RH domains in other Gα subunits (12,30). The second constraint was to limit the Gαqinteraction surface of GRK2 to solvent exposed residues on the α5 and α6 helices of its RH domain, which were previously identified to be important for complex formation with Gαq (22). The third constraint was to fix the relative orientation of Gαq and GRK2 to the plane of a common cell membrane, as each of their orientations with respect to a cellular membrane is relatively well known from prior crystal structures and electrostatic calculations. The orientation of Gαq with respect to the plane of the plasma membrane, defined by the GRK2-Gβγ structure (14), was fixed by docking it against the Gβγ subunits present in the GRK2-Gβγ complex in a manner similar to Gαi in the Gαi1Gβ1γ2 structure (31). Gαq was then translated and rotated along the plane of the membrane until its GRK2-interaction surface was adjacent to the Gαq-interaction surface of GRK2. The model was manually adjusted to optimize the packing of residues at the protein interface, and then minimized using simulated annealing in CNS(32) to relieve any bad contacts between side chains. Harmonic restraints were imposed on the Cα positions during refinement to keep the backbone relatively fixed (0.28 Å root mean squared deviation between initial and final coordinates of Gαq). The modeled interface buries 2100 Å2 of surface area, which is on par with or larger than those observed in crystal structures of other RH domain-Gα complexes (e.g. 1,800 Å2 in the RGS9-Gαt/i1 complex(12)). The final model of the complex was verified by the program PROCHECK(33), ERRAT, and VERIFY3D, which indicated that the residues involved in the interface were consistent with a reasonably packed and complementary structure.
Inositol Phosphate Production Assay
HEK-293 cells were transfected with 0.1 μg of EE GαqRC or EE Gαq RC mutant constructs, 0.2 μg of myc-His tagged Gβ1, 0.1 μg of Gγ2, the indicated amounts of GRK2-K220R or RGS2 and pcDNA3 up to a total of 1 μg DNA. Twenty-four hours after transfection, cells were replated on 4 wells of a 24- well plate and 3 wells were labeled for 16 h with 2 μCi/ml [3H]-inositol. Inositol phosphate production was determined as previously described (22). Results are the average of at least three experiments done in triplicate and represented as Percent Control. Control is the level of inositol phosphate produced in the absence of cotransfected GRK2-K220R or RGS2. Graphing and statistical analysis, as described in Figure legends, was performed using GraphPad Prism.
The fourth well of replated transfected cells was used to monitor whether the coexpression of GRK2 or RGS2 had any effect on the expression of Gα subunits. Cells were lysed with 50 μl of SDS sample buffer, vigorously homogenized and boiled for 5 minutes. 20 μl of the sample was then subjected to 12 % SDS-PAGE and transferred to PVDF. The PVDF was then probed with 2 μg/ml of EE monoclonal antibody followed by horseradish peroxidase-conjugated secondary antibody (1:10,000 dilution). Pierce SuperSignal West Pico reagents were used to visualize immunoblots. Blots from assays with GRK2-K220R were stripped with a 50 mM glycine buffer, pH 2.0, and reprobed with a GRK2 specific monoclonal antibody followed by horseradish peroxidaseconjugated secondary antibody (1:10,000 dilution) to determine the effect of transfecting increasing amounts of GRK2-K220R cDNA on GRK2 expression.
Purification of GST fusion proteins
GST-GRK2-(45–178), GST-RGS2 or GSTRGS4 were expressed in BL-21 cells and purified using glutathione Sepharose 4B beads (from Amersham Life Sciences) essentially as described in Sterne-Marr et al (22) for GST-GRK2-(45–178). The glutathione Sepharose bound GST-GRK2-(45–178), GSTRGS2 or GST-RGS4 is washed three times in lysis buffer to remove any glycerol before being added to the lysates.
GST-GRK2-(45–178), GST-RGS2 and GST-RGS4 Interaction Assays
HEK-293 cells were transfected in 6 cm dishes with 2.0 μg of Gαq or mutant Gαq cDNA, 0.2 μg of myc-His tagged Gβ1, 0.1 μg of Gγ2, and pcDNA3 up to a total of 3.0 μg of DNA. 24 h after transfection cells were washed with cold PBS and lysed with 0.3 ml of lysis buffer (20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM dithiothreitol, 100 mM NaCl, 5 mM MgCl2, 0.7 % Triton X-100, 1 mM phenylmethylsulfonyl fluoride, and 5 μg/ml leupeptin and aprotinin). After 1 h of lysis at 4ºC, cells were centrifuged for 3 min at full speed in a microcentrifuge. For Gαq-RC and Gαq-QL assays, 200 μl of the supernatant was removed to a new tube and incubated with 8 μg of GST-GRK2-(45–178), GSTRGS2 or GST-RGS4, all pre-bound to glutathione Sepharose beads, for 1-2 h at 4°C. The remaining supernatant, denoted “L” for lysates, was saved for subsequent immunoblot analysis alongside pull down samples. After incubation of the lysates with the GSTGRK2-(45–178), GST-RGS2 or GST-RGS4 bound beads, the samples are pelleted at low speed in a microcentrifuge for 3 min and the beads are washed 3 times with lysis buffer. Proteins were then eluted from beads in 50 μl of SDS sample buffer and boiled for 5 minutes.
For GST pull-down assays carried out in the absence or presence of AlF4−, 250 μl of the Gαq-containing supernatant (described above) was removed and split equally into two tubes. To one of the tubes AlCl3 (25 μM), NaF (5 mM) and MgCl2 (1 mM) were 12 added. GST-GRK2-(45–178), GST-RGS2 or GST-RGS4 (8 μg) bound beads were then added to each tube and incubated for 4–5 h at 4 ºC. The remaining supernatant, denoted “L” for lysate, was saved for subsequent immunoblot analysis alongside samples. After incubation of the lysates with the GST-GRK2-(45–178), GST-RGS2 or GST-RGS4 bound beads, the samples are pelleted at low speed in a microcentrifuge for 3 min and the beads are washed 3 times with lysis buffer. Proteins were then eluted from beads in 50 μl of SDS sample buffer and boiled for 5 minutes.
In all cases, 20 μl of each pull-down sample was subjected to 12 % SDS-PAGE and transferred to PVDF, which was probed with 2 μg/ml EE monoclonal antibody followed by horseradish peroxidase-conjugated secondary antibody (1:10,000 dilution). A portion of the initial lysate that represents 4% of the protein present in the lysates was also analyzed on immunoblots alongside GST pull-down samples and indicated by “L” in figures. Pierce SuperSignal West Pico reagents were used to visualize immunoblots. For graphical representation of pull-down assays images of western blots were acquired using a Kodak DC-40 digital camera and the net intensity of each band was calculated using Kodak Digital Science 1D Image Analysis Software. The percent of the mutant Gαq that was pulled-down by the GST-fusion protein was calculated and compared to the control, which is the percent of Gαq that interacted with the GST-fusion protein and displayed as percent control + S.D. Graphing and statistical analysis, as described in Figure legends, was performed using GraphPad Prism.
Pull-down Assays with GRK2 RH Domain Mutants
Mutants of the GRK2 RH domain were assayed as GST-GRK2-(45–178) fusions using bovine brain extract as a source of WT Gαq as described (22). To allow comparison of severely defective Gαq- binding mutants, a more sensitive assay was developed by using 20 μg/ml fusion protein and 500 μg/ml bovine brain extract protein in the pull-down assays.
RESULTS
Molecular modeling of the G αq:GRK2 interface
The binding surface for Gαq has been localized primarily to the α5 helix of the GRK2 RH domain and is distinct from the protein-binding surfaces used by other RH domains (11,13,22). Accordingly, this interaction surface has been termed the C site, following the nomenclature proposed by Zhong and Neubig (17). GRK2 only interacts with activated Gαq (18) suggesting the involvement of at least one Gαq switch region in the interaction. However, the C site of the RH domain may bind residues on Gαq that are distinct from those that interact with RGS proteins such as RGS2 and RGS4. We have already shown that the RGS-resistant Switch I mutant, Gαq-G188S, retains association with GRK2 (22).
In order to predict which Gαq residues could interact with the RH domain of GRK2, a homology model of Gαq was manually docked with the RH domain from the GRK2-Gβγ crystal structure(14) by imposing several specific constraints required by prior biochemical and structural analyses (see methods). The docking model predicts that the long α5 helix of the GRK2 RH domain docks into the cleft formed between the helical domain and the Ras-like domain of Gαq and engages primarily switch I and III of the Ras-like domain (Figure 1 A). This binding mode would be substantially different from those observed for the complexes of Gαi and Gαt with RGS4 (11) and RGS9 (12), respectively (Figure 1 B), although in each case the switch regions of Gα provide the primary interaction site. In particular, the model predicts that Pro185 of Gαq is at the crux of the interface with its side chain packing against Asp110, Met114 and Leu118 of GRK2 (Figure 1 C–D). Indeed, both Asp110 and Met114 have previously been shown to be important for the interaction of GRK2 with Gαq (22). Residues within the αA helix of Gαq were also predicted to be in close proximity to the GRK2 RH domain, and could explain why residues such as Val137 of GRK2, which is quite distant from Asp110, Met114 and Leu118 of GRK2, has an effect on Gαq binding when mutated to alanine. Therefore, residues in both the switch I and III regions and adjacent regions of the helical domain of Gαq were targeted for mutagenesis.
Figure 1.
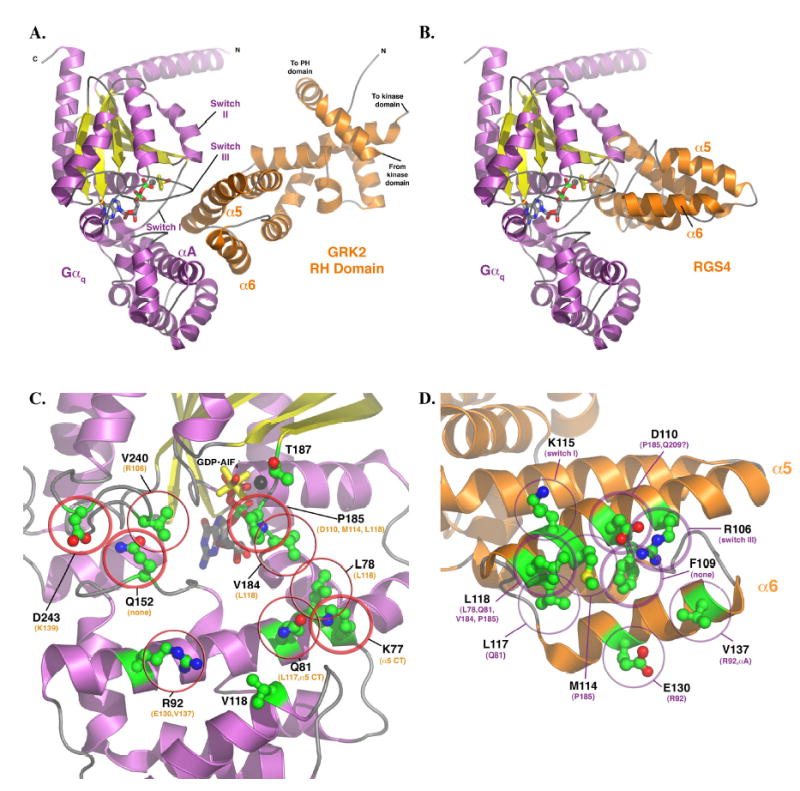
(A) Model of the Gαq-GRK2 RH domain complex and their interacting surfaces. The model of Gαq was homology modeled based on the AlF4− bound structure of Gαi in complex with RGS4 (11) and then docked with the RH domain as described in Experimental Procedures. The switch regions and the αA helix of Gαq (purple and yellow) are labeled, as are the α5 and α6 helices of the GRK2 RH domain. These structural elements constitute the principal interaction surfaces of each protein. The proposed plane of the plasma membrane runs along the top of the complex, as shown in the figure. The switch regions of Gαq are delineated by V182 to Y192 (switch I), V204- T224 (switch II), and D236-R247 (switch III). (B) Model of Gαq in complex with RGS4, based on the atomic structure of Gαi-RGS4 (11). The RH domains of GRK2 and RGS4 both interact with the switch regions of the G protein, but the surface of the RH domain used in the contact is unique. In the RGS4 complex, the α5 helix faces out of the page, while in the Gαq-GRK2 complex it forms the principal contact surface. Panels (C) and (D) represent views of Gαq and GRK2, respectively, as if the complex shown in panel (A) were opened like a book. (C) The GRK2-interacting surface of Gαq. The residues shown as ball-and-stick models with green carbons are those mutated and analyzed in this study. Thick circles indicate residues that had a dramatic effect upon mutation (as per Table 1), thin circles indicate an “intermediate” effect, and no circles indicate no effect, at least upon GRK2 binding and inhibition of IP3 release. The residues listed in orange are those that each Gαq residue is predicted to contact. The black sphere represents Mg2+. (D) The Gαq-interacting surface of the GRK2 RH domain. The residues shown as balland- stick models with green carbons are those mutated and analyzed in this (L118 and E130) and our previous study (22). Thick circles indicate residues that had a dramatic effect upon mutation, thin circles indicate an “intermediate” effect, and no circles indicate no effect, at least upon Gαq binding. The residues listed in purple are those that each GRK2 residue is predicted to contact. All panels were created using PyMOL (38). The coordinates of the model of the Gαq-GRK2 RH domain complex are available in a pdb file as Supplementary Data.
Identification of G αq switch residues that interact with the RH domain of GRK2
To test the effects of point mutations in the switch I and III regions of Gαq, they were transiently over-expressed in HEK-293 cells, and GST-GRK2-(45–178) was then used to pull-down mutant Gαq in lysates from the transfected cells. Each point mutant was made in the background of an otherwise wild-type (non-activated) Gαq and a constitutively active Gαq-Q209L (Gαq-QL). Selected mutants were also generated in the constitutively active Gαq-R183C (Gαq-RC). Point mutants in the wild-type background were activated by the addition of AlF4−, which binds Gα·GDP and occupies the space normally filled by the γ phosphate of GTP. This causes the Gα subunit to assume a conformation that is thought to mimic the transition state of GTP hydrolysis (34). In contrast, Gαq-QL and Gαq-RC are constitutively active because Glu209 and Arg183 are involved in the hydrolysis of the γ phosphate of GTP and their mutation to leucine and cysteine, respectively, greatly decreases the rate of this reaction (34). Initially, binding of the point mutants to GST-GRK2-(45–178) was examined in the AlF4− and Gαq-QL backgrounds. However, we also wanted to examine the effects of the mutations on the ability of GRK2 to inhibit the Gαq-mediated formation of inositol phosphate in cells. Unfortunately, the cotransfection of GRK2-K220R, a kinase deficient mutant of GRK2 (35), caused a marked decrease in the expression of several mutants, particularly K77P, Q152A, P185K, and T187K (data not shown). Previously, we had observed that relatively small amounts of co-transfected GRK2 were able to inhibit inositol phosphate production stimulated by constitutively active Gαq-RC (data not shown). We therefore generated several of the mutants, K77A, Q81A, R92A, Q152A, P185K and T187K, in the Gαq-RC background and assessed their ability to bind GRK2. The effect of each mutation on binding to the RH domain of GRK2 was tested, and the results are summarized in Table 1 and described below.
Table 1.
Effect of Gαq Point Mutants on RH domain binding. The table summarizes the effects of the Gαq point mutants on binding to GRK2, RGS4 and RGS2. Pull-down assays were performed with GST-GRK2-(45–178), GST-RGS4 or GST-RGS2 on cell lysates that had been transfected with Gαq containing the different point mutations, as described in Experimental Procedures. There is no detectable binding of Gαq or Gαq mutants to GST alone. Binding to AlF4− activated forms of the mutants and to mutants in the Gαq-RC background was assessed for GRK2, RGS4 and RGS2; however, RGS4 does not bind to Gαq-QL so the effects of the point mutants in the QL form on binding could only be tested with GRK2 and RGS2. (+++) indicates that similar amounts (81–100% of control as described in Experimental Procedures) of the Gαq point mutant and wt Gαq bound to GRK2, RGS4 or RGS2, (++) indicates 51–80% of control, (+) indicates 21–50% of control and (−) indicates 0–20% of control. ND- not determined. Data are from 2 to 6 experiments.
| GST-GRK2-(45–178)
|
GST-RGS2
|
GST-RGS4
|
||||||
|---|---|---|---|---|---|---|---|---|
| Gαq construct1 | AlF4− | QL | RC | AlF4− | QL | RC | AlF4− | RC |
| wt-Gαq | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| K77A2 | +++ | − | +++ | +++ | − | +++ | +++ | +++ |
| K77P | −3 | − | ND | ND | ND | ND | − | ND |
| L78D | ++4 | +++ | ND | ND | ND | ND | +++ | ND |
| Q81A2 | ++ | +++ | +++ | + | +++ | + | ++ | +4 |
| R92A2 | +++ | + | +++ | + | + | ++ | − | +++ |
| V118A | +++ | +++ | ND | ND | ND | ND | +++ | ND |
| Q152A2 | + | + | ++ | + | ++ | ++ | − | +++ |
| V184D | ++ | +++ | ND | ND | ND | ND | − | ND |
| P185K2 | − | − | − | +++ | − | − | +++ | ND |
| T187K2 | +++ | +++ | +++ | − | − | + | − | +4 |
| V240A | ++ | +++ | ND | ND | ND | ND | +++ | ND |
| D243A | − | +++ | ND | ND | ND | ND | ++ | ND |
Mutants are expressed at levels similar to Gαq-wt, Gαq-Q209L or Gαq- R183C, respectively, with the following exceptions: K77A-QL (39% of Gαq-QL), P185K (59% of Gαq), P185K-QL (72% of Gαq-QL), P185K-RC (30% of Gαq-RC) and Q152A-RC (75% of Gαq-RC).
The statistical significance of the difference between the indicated mutants and control is indicated in Figures 2 and 4 for GRK2 and RGS2, respectively.
For mutants indicated by a (−), statistical analysis could not be performed because there generally was no observable pull-down.
Binding of Gαq-L78D to GSTGRK2- RH is significantly different than binding of Gαq to GST-GRK2-RH (p < 0.001) and binding of Gαq-Q81A-RC and Gαq-T187K-RC to GST-RGS4 is statistically different then binding of Gαq-RC to GST-RGS4 (p < 0.001).
Our model of the GRK2-Gαq interaction predicts that Pro185 is buried within the interface, and therefore will represent a critical specificity determinant. As expected, mutation of Pro185 to lysine, the corresponding residue in Gαi, has a profound negative effect on GRK2 binding (Figure 2 A, B and C). Binding of P185K-QL and P185K-RC (Figure 2 B and C) to the GRK2 RH domain is completely attenuated while binding of P185K in the presence of AlF4− is less then 20 % of wild type (Figure 2 A). The mutation of Pro185, located in Switch I between the helical domain and the GTPase domain of Gαq, does appear to decrease the expression of Gαq by approximately 40% (data not shown). However, as will be discussed later, this mutant retains its ability to bind to RGS proteins when activated by AlF4−. Therefore, by mutating Gαq Pro185 we disrupt GRK2 binding, as predicted by the model of their complex.
Figure 2.
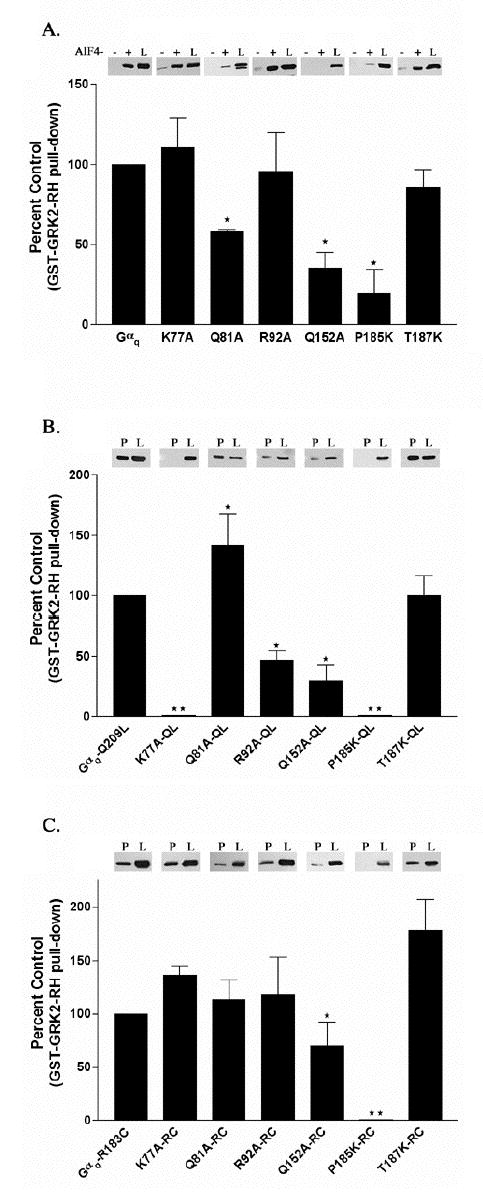
Interaction of GST-GRK2-(45–178) with Gαq point mutants activated by AlF4−, the Q209L or the R183C mutation. (A) HEK-293 cells were transfected with EE tagged versions of Gαq point mutants and Gβ and Gγ constructs. Cells were lysed and binding to GST-GRK2-(45–178) in the presence (+) or the absence (−) of AlF4− was determined as described in Experimental Procedures. The (+) and (−) lanes represent 40% of the Gαq or Gαq mutant pulled down from the 125 μl of lysate. In these experiments we detect little to no binding of GST-GRK2-(45–178) to Gαq or Gαq point mutants in the absence of AlF4−. Underneath the representative western blot the percent of each Gαq mutant pulled down by GST-GRK2-(45–178) in the presence of AlF4− is compared to the control, which is the percent of Gαq pulled down by GST-GRK2 (45–178) in the presence of AlF4−, and is represented graphically as the percent of control + S.D. (B) HEK-293 cells were transfected with EE tagged versions of Gαq-Q209L point mutants and Gβ and Gγ constructs. Cells were lysed and binding of the QL mutants to GST-GRK2-(45–178) was determined as described in Experimental Procedures. Results are plotted as described in A. (C) HEK-293 cells were transfected with EE tagged versions of Gαq-R183C point mutants and Gβ and Gγ constructs. Cells were lysed and binding of the RC mutants to GST-GRK2-(45–178) was determined as described in Experimental Procedures. The lanes labeled “P” represent 40% of the Gαq or Gαq mutant that was present in the pulldown from 200 μl of lysate. The lanes in A, B and C labeled “L” represent 4% of total Gαq or Gαq mutant available in the lysate for pull-down. Results are plotted as described in A. The (⋆) indicates that the amount of the marked Gαq mutant pulled-down is significantly different (p < 0.05) by one-way ANOVA followed by a Dunnett post-test, than the amount of Gαq pulled-down by GST-GRK2. The (⋆⋆) indicates that statistical analysis could not be performed on the binding of GST-GRK2-RH to the K77A-QL, P185K-QL or P185K-RC mutants because there was no detectable pull-down. The data are averages from three to six independent experiments.
Two additional residues in the Switch I region of Gαq, Val184 and Thr187, were also targeted by site-directed mutagenesis. Mutation of Val184 to aspartic acid is predicted to lessen favorable contacts with Leu118 of GRK2. The V184D mutant has a modest effect (< 80 % of control) on AlF4− activated Gαq binding to GRK2, but this effect is lost in the Gαq-QL background. Similarly, mutation of Thr187 to lysine, the corresponding residue in Gα12/13, was also predicted to destabilize the GRK2-Gαq interface, perhaps by creating unfavorable contacts with the side chains of Lys115 and Thr111 of GRK2. However, GRK2 binding to Gαq is unaffected by the T187K mutation, regardless of whether it is in the context of Gαq·GDP·AlF4−, Gαq-RC or Gαq-QL (Figure 2 A, B and C). Substititution of residues at these positions may be permitted because they exist at the periphery of the interface and thus are partially solvent-exposed in the model. They could thereby accommodate longer side chains.
The model also predicts that the GRK2 RH domain interacts with the backbone of the Switch III residue Val240 and that there is a potential salt-bridge between Asp243 of Gαq and Lys139 of GRK2. The Gαq-V240A and D243A mutants reduce or eliminate, respectively, AlF4−-dependent binding to GRK2 (Table 1). However, both V240A-QL and D243A-QL interact with GRK2 to the same extent as Gαq-QL. This suggests that Switch III is more critical for binding in AlF4−-activated Gαq than the QL and RC conformations of the enzyme. The structural basis for these differences is not clear, but may be due to subtle conformational changes in the three switches when bound to either GTP or a transition state complex.
Gln152 is a highly conserved alpha-helical domain residue whose side-chain makes specific hydrogen bonds within the Ras-like domain of Gα subunits, principally with the backbone of Switch III and with a conserved arginine residue that likewise supports Switch III. Because Gln152 is changed to histidine in Gα16, which does not bind GRK2, and because of its proximity to the modeled RH domain, it was also targeted by sitedirected mutagenesis. Q152A·GDP·AlF4−, Gαq-Q152A-QL and the Q152A-RC have reduced binding to the GRK2 RH domain (Table 1 and Figure 2 A and C). These results with the Q152A, V240A and D243A mutants confirm a role for Switch III in binding GRK2.
Identification of G αq helical domain residues that interact with the RH domain of GRK2
Several residues within the helical domain of Gαq are likewise predicted to be involved in the interaction with GRK2 (Figure 1). Leu78 is predicted to interact with GRK2 Leu118 and the L78D mutant had a slight effect (< 80 % of control) on the AlF4−- dependent binding to the GRK2 RH domain (Table 1). Two residues in the αA helix of Gαq, Lys77 and Gln81, are expected to interact with the carboxyl terminus of the GRK2 α5 helix. The Q81A mutant of Gαq has reduced binding to the GRK2 RH in the presence of AlF4− (Table 1 and Figure 2 A). However, Q81A-QL and Q81A-RC (Table 1 and Figure 2 B and 2 C) retain the ability to bind to the GRK2 RH domain. Replacement of Lys77 with a proline, the analogous Gα16 residue, disrupts AlF4−-dependent binding to GRK2 and binding to GRK2 in the Gαq-QL background (Table 1). In contrast, the K77A mutant only has an effect on binding to GRK2 in the Gαq-QL background (Table 1 and Figure 2 A, B and C). These data support a role for the α-helical domain of Gαq in dictating the specificity and the affinity of the GRK2-Gαq interaction.
There are additional residues in the helical domain that are in close proximity to the GRK2 RH domain in the model but that are not conserved in Gα16. The V118A mutation has no effect on GRK2 binding (Table 1). Mutation of Arg92, whose aliphatic side chain is predicted to interact with Val137 of GRK2, to alanine does not have an effect on AlF4− dependent binding or binding to Gαq-RC (Table 1 and Figure 2 A and C); however, reduced binding to GRK2 is seen with R92A-QL (Table 1 and Figure 2 B). In addition to the interactions between the aliphatic portion of the Gαq Arg92 side-chain and GRK2 Val137, our model predicts that its guanidino group forms a salt-bridge with GRK2 Glu130. To test this idea, E130A was introduced into GST-GRK2-(45–178). Similar to another GRK2-α6 mutant V137A, E130A shows a modest deficiency in its ability to bind Gαq/11 in a GST-pull-down assay (data not shown).
Gαq-Q152A-RC is less sensitive than G αq-RC to GRK2-mediated inhibition of inositol phosphate production
We then tested the interaction of GRK2 with Gαq-RC mutants in intact cells. We have previously used co-transfection of Gβγ to stabilize the expression of Gα subunits in the presence of RGS proteins (19). In addition, very low amounts of GRK2-K220R, 5 ng of cDNA, are able to inhibit signaling from Gαq RC (Figure 3 A and B). The ability of GRK2-K220R to inhibit Gαq-RC signaling is not affected by the co-expression of Gβγ (data not shown). These conditions allowed us to detect differences in the sensitivity of the point mutants to GRK2 inhibition. Unfortunately, even under these conditions, the expression of the P185K-RC mutant was inversely proportional to the amount of GRK2-K220R transfected (data not shown). Therefore it was not included in these experiments. Even so, the expressed P185K-RC is functional because Gαq-P185K-RC still activates PLCβ and binds Gβγ (data not shown).
Figure 3.
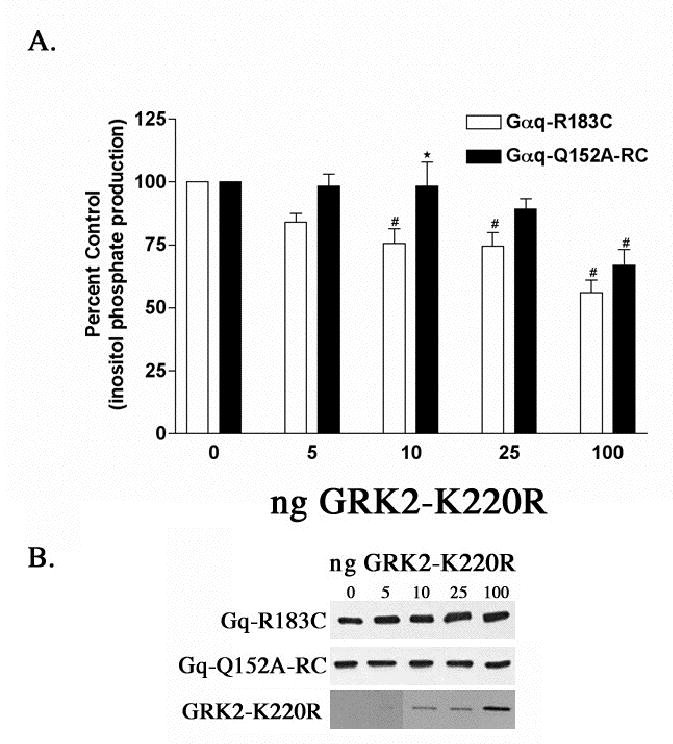
Effect of the Q152A point mutation in Gαq-RC on the ability of GRK2 to inhibit inositol phosphate production. (A) HEK-293 cells were transfected with 0.1 μg of the constitutively active Gαq-R183C or Gαq-Q152A-RC and 0.2 μg of myc, His-tagged Gβ and 0.1 μg of Gγ and increasing amounts of GRK2-K220R and empty vector up to a total of 1.0 μg of DNA. 24 hrs after transfection the cells were labeled with 2 μCi/ml myo-[3H]inositol and 16 hours later inositol phosphate production was determined, as described in Experimental Procedures. The results shown are averages from five independent experiments each done in triplicate and displayed as percent control ± S.D. The control is the inositol phosphate production stimulated by Gαq-R183C or Gαq- Q152A-RC in the absence of any co-expressed GRK2-K220R. A (⋆) denotes a statistically significant difference (p < 0.05) by two-way ANOVA followed by a Bonferroni post-test, between the indicated Gαq-Q152A-RC bar and the Gαq-RC bar transfected with the same amount of GRK2-K220R. A (#) indicates a statistically significant difference (p < 0.05) by one-way ANOVA followed by a Dunnett post-test, between the indicated bar and the control, either Gαq-RC or Gαq-Q152A-RC in the absence of cotransfected GRK2-K220R. (B) Western blots of total cellular lysates from a representative inositol phosphate experiment from (A) probed with the EE monoclonal antibody showing that increasing GRK2-K220R expression does not effect expression of Gαq-RC or Gαq-Q152A-RC. The bottom panel of Figure 4 B shows the level of GRK2- K220R overexpression. The bands corresponding to 10, 25 and 100 ng of GRK2-K220R transfected can be seen after very short exposures; however, the GRK2 band corresponding to 5 ng of cDNA transfected is barely visible, even after long exposures, suggesting that comparatively low levels of GRK2 expression can significantly inhibit Gαq signaling.
The Gαq mutant, other than Gαq-P185K, that consistently inhibited the interaction with the RH domain of GRK2 in pull-down experiments is Gαq-Q152A (Table 1 and Figure 2). Q152A-RC also showed resistance to GRK2-K220R inhibition of inositol phosphate production (Figure 3 A). Although the differences in the inhibition of Gαq-RC versus Q152A-RC signaling by GRK2-K220R are small, the effects are reproducible. For example, the transfection of 10 ng of GRK2-K220R led to 25% inhibition of the inositol phosphate stimulated by Gαq-RC, while the same amount of GRK2-K220R did not inhibit Q152A-RC (Figure 3 A). The difference between Gαq-RC and Q152A-RC is less pronounced at higher levels of GRK2-K220R expression, suggesting that this mutant lowers the affinity of but does not totally disrupt the interaction between GRK2 and Gαq (Figure 3 A). This would agree with GST-GRK2 pull-down data that shows a marked decrease in binding to Q152A in the presence of AlF4− and the Q209L mutation and a smaller decrease in binding to Q152A-RC (Table 1 and Figure 2 A, B and C).
Two of the remaining Gαq mutants tested for inhibition of inositol phosphate production by GRK2 showed only minor defects in GRK2 interaction. Although the Q81A mutant binds to the RH domain of GRK2 in both the QL and RC form (Figure 2 B and C), in the presence of AlF4− the binding of Q81A is reduced (Table 1 and Figure 2 A). In the inositol phosphate assays with Gαq-Q81A-RC and Gαq-T187K-RC there are small differences in comparison to R183C in the ability of low levels of GRK2 to inhibit signaling (data not shown). However at higher concentrations of GRK2-K220R, Gαq- Q81A-RC and Gαq-T187K-RC are inhibited to a level that is similar to Gαq-RC (data not shown). The R92A-RC mutant is inhibited in a manner that is essentially identical to R183C (data not shown). Importantly, Figure 3 B shows that expression of GRK2- K220R, even at a high level, does not decrease the expression of Gαq-RC or the Gαq- Q152A-RC mutant, indicating that the observed differences in inositol phosphate production are due to differences in binding of GRK2 to Gαq-RC relative to Gαq-Q152ARC, and not to differences in expression levels. In general, the data from the inositol phosphate assays are consistent with the pull-down assays with the RH domain of GRK2.
The A sites of RGS2 and RGS4 bind different surfaces of G αq than the C site of GRK2
We have previously shown that the binding surface for Gαq on the GRK2 RH domain is distinct from that of RGS4, and in this set of experiments we wanted to determine the effect of the mutations made in Gαq on binding to RGS2 and RGS4 (22). There is little or no difference in the ability of RGS2 versus RGS4 to bind each of the Gαq mutants (Table 1), and Figure 4 presents GST pull-down data with RGS2.
Figure 4.
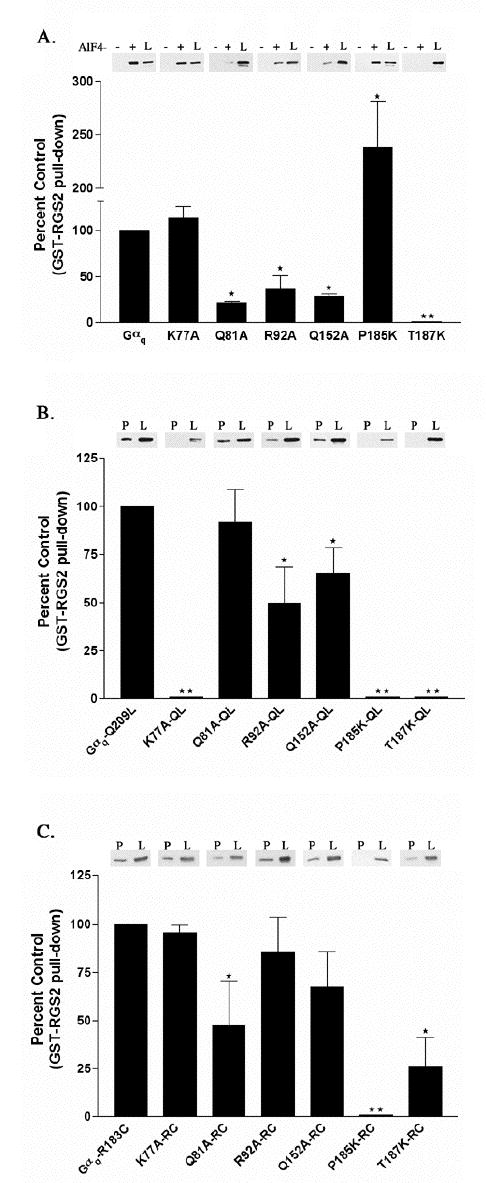
Interaction of GST-RGS2 with Gαq point mutants activated by AlF4−, the Q209L or the R183C mutation. (A) HEK-293 cells were transfected with EE tagged versions of Gαq point mutants and Gβ and Gγ constructs. Cells were lysed and binding to GST-RGS2 in the presence (+) or the absence (−) of AlF4− was determined as described in Experimental Procedures. The (+) and (−) lanes represent 40% of the Gαq or Gαq mutant pulled down from the 125 μl of lysate. In these experiments we detect little to no binding of GST- RGS2 to Gαq or Gαq point mutants in the absence of AlF4−. Underneath the representative western blot the percent of each Gαq mutant pulled down by GST- RGS2 in the presence of AlF4− is compared to the control, which is the percent of Gαq pulled down by GST-RGS2 in the presence of AlF4−, and is represented graphically as the percent of control ± S.D. (B) HEK-293 cells were transfected with EE tagged versions of Gαq-Q209L point mutants and Gβ and Gγ constructs. Cells were lysed and binding of the QL mutants to GST-RGS2 was determined as described in Experimental Procedures. Results are plotted as described in A. (C) HEK-293 cells were transfected with EE tagged versions of Gαq-R183C point mutants and Gβ and Gγ constructs. Cells were lysed and binding of the RC mutants to GST-RGS2 was determined as described in Experimental Procedures. The lanes labeled “P” in B and C represent 40% of the Gαq or Gαq mutant that was present in the pull-down from 200 μl of lysate. The lanes in A, B and C labeled “L” represent 4% of total Gαq or Gαq mutant available in the lysate for pull-down. Results are plotted as described in A. The (⋆) indicates that the amount of the marked Gαq mutant pulled-down is significantly different (p < 0.05) by one-way ANOVA followed by a Dunnett post-test, than the amount of Gαq pulled-down by GST-RGS2. The (⋆⋆) indicates that statistical analysis could not be performed on the binding of GST-RGS2 to the K77A-QL, P185K-QL, P185K-RC, T187K or T187K-QL mutants because there was no detectable pull-down. The data are averages from three to six independent experiments.
Mutation of residues in the Switch I region of Gαq interfere with binding to RGS2 and RGS4. The mutation that has the most profound effect on binding to GRK2, P185K, also does not bind to RGS2 and RGS4 in the context of the RC mutation (Table 1 and Figure 4 C). However P185K activated by AlF4− does bind to RGS4 and RGS2 (Table 1 and Figure 4 A). Finally, the conserved Thr residue (position 187 in Gαq and 182 in Gαi), which is completely buried in the Gαi-RGS4 interaction, is essential for binding to RGS2 and RGS4 (11). Substitution of T187 with a lysine drastically reduces binding to RGS2 and RGS4 in all active forms (Table 1 and Figure 4 A, B and C) but has no affect on binding to GRK2 (Table 1 and Figure 2). In addition, we have previously shown that the RGS resistant mutant Gαq-G188S binds to GRK2 (22). Therefore, there are substantial differences between the surface of Gαq bound by the A site of typical RGS proteins and the C site of the GRK2 RH domain, as predicted by their modeled interactions with Gαq (Figure 1 A–B).
Additionally, there are differences in the binding of RGS2 and RGS4 to AlF4−- activated versus constitutively active RC and QL forms of a few of the helical domain mutants (Table 1 and Figure 4). For example, the mutant Q81A-RC has decreased binding to both RGS proteins as does Q81A activated by AlF4− (Table 1 and Figure 4 A and C); however, Gαq-QL and the Gαq-Q81A-QL mutant bind to RGS2 to a similar level (Figure 4 B). Also, the AlF4− activated Q152A and Gαq-Q152A-QL show decreased binding to RGS2 and RGS4, but Q152A-RC binds RGS2 and RGS4 equally as well as wild-type Gαq-RC does (Table 1 and Figure 4 A, B and C).
Q81A-RC and T187K-RC have reduced sensitivity to RGS2 mediated inhibition of inositol phosphate production
We next wanted to examine the ability of RGS2 and RGS4 to inhibit each of the Gαq point mutants in the RC form. These assays were performed in a manner similar to the inositol phosphate assays with GRK2. The Gβγ subunits were expressed in every sample and increasing amounts of RGS2 were cotransfected with each mutant. We were not able to assess the ability of RGS4 to inhibit the Gαq mutants because transfection of several different RGS4 constructs decreased the expression of Gαq or mutants of Gαq (data not shown). The P185K-RC mutant was not included in these experiments because, like GRK2-K220R, co-expression of RGS2 decreased its expression (data not shown).
Inositol phosphate assays performed to determine the sensitivity of the Gαq point mutants to inhibition by RGS2 agree with the data from the GST-RGS2 pull-down experiments. The two mutants that have very little effect on the binding of RGS2 to Gαq, R92A and Q152A, are also susceptible to RGS2 mediated inhibition of inositol phosphate production (Figure 5 A). In contrast, low amounts of transfected RGS2 DNA do not decrease the inositol phosphate production stimulated by Q81A-RC (Figure 5 A). At the highest amount of RGS2 transfected, 20 ng, Q81A-RC stimulated inositol phosphate production is decreased by about 25 %, while similar levels of RGS2 decrease R183C stimulated inositol phosphate production by 43 % (Figure 5 A). This agrees with the pull-down data in Figure 5 and suggests that this residue is involved in the Gαq-RGS2 interaction. Finally, T182 in Gαi, which corresponds to T187 in Gαq, is found at the center of the Gαi-RGS4 interface and therefore mutation at this position in Gαq should disrupt any interaction with RGS4 and RGS2. Figure 4 shows that very little T187K-RC is pulled-down with GST-RGS2. Figure 5 A also shows that signaling by T187K-RC is not inhibited by co-expression of RGS2. Figure 3 shows that this mutation has no effect on the ability of GRK2-K220R to inhibit Gαq signaling, once again highlighting the difference between GRK2 and RGS2 binding sites on Gαq. As with GRK2, coexpression of RGS2 does not decrease the expression levels of Gαq-RC or any of the mutants (Figure 5 B).
Figure 5.
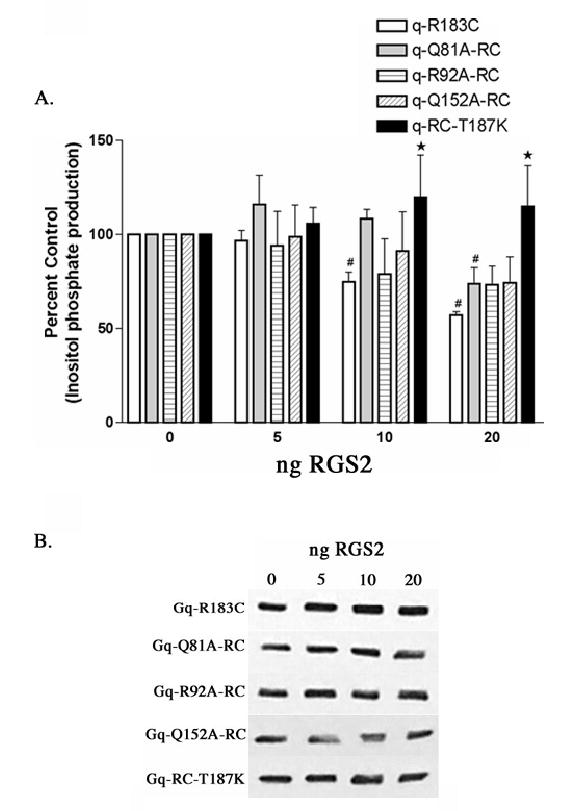
RGS2 inhibition of inositol phosphate production stimulated by Gαq-RC and Gαq-RC mutants. (A) HEK-293 cells were transfected with 0.1 μg of the constitutively active Gαq-R183C, Gαq-Q81A/RC, Gαq-R92A/RC, Gαq-Q152A/RC, or Gαq-RC/T187K and 0.2 μg of myc, His-tagged Gβ and 0.1 μg of Gγ and increasing amounts of RGS2 and empty vector up to a total of 1.0 μg of DNA. 24 hrs after transfection the cells were labeled with 2 μCi/ml myo-[3H]inositol and 16 hours later inositol phosphate production was determined, as described in Experimental Procedures. The results shown are averages from three independent experiments each done in triplicate and displayed as percent control + SD. The control is the inositol phosphate production stimulated by each mutant in the absence of any co-expressed RGS2. The statistical significance of the difference between the indicated bar and Gαq-R183C, in the absence of any additional mutations, transfected with equal amounts of RGS2 is denoted by ⋆ (p < 0.05) by oneway ANOVA followed by a Dunnett post-test. A (#) indicates a statistically significant difference (p < 0.05) by one-way ANOVA followed by a Dunnett post-test, between the indicated bar and the control, either Gαq-RC or a Gαq-RC mutant in the absence of cotransfected RGS2. (B) Western blots of total cellular lysates from a representative inositol phosphate experiment from (A) probed with the EE monoclonal antibody showing that increasing RGS2 expression does not effect expression of Gαq-RC or any of the mutants. We were not able to detect the level of RGS2 overexpression in these experiments; however the decrease in inositol phosphate production suggests that RGS2 expression was increased.
GRK2 L118A does not interact with G αq
While our previous GRK2 mutagenesis studies were driven by an axin/GAIP-based homology model of the GRK2 RH domain (22), the crystal structure and the docking model (Figure 1) used in this study to predict Gαq residues involved in the interface with GRK2 also identified additional GRK2 residues that could be involved in the interface with Gαq. Specifically, the new model predicts that Leu118 of GRK2 is important for the central interaction with Pro185 of Gαq. To test this hypothesis, the L118A mutation was introduced into the GST-GRK2-RH domain (residues 45–178) fusion, and GST pull-down assays were used to assess the ability of the mutant to bind to Gαq/11 from bovine brain extracts in the presence of AlF4−. L118A was markedly impaired in its ability to bind Gαq/11, affirming the importance of the solvent-exposed Leu118 in the Gαq interaction (Figure 6). The Gαq-binding deficiency of L118A is comparable to that of previously identified mutants R106A, D110A and E116A, which also showed severe impairment in Gαq/11 binding (22). To compare the L118A mutation to previously identified mutants, the pull-down assay was modified so as to increase its sensitivity. Some binding (except with the R106A/D110A double mutant) can be detected under these conditions allowing the mutants to be ranked based on their decreasing ability to bind Gαq/11: R106A > L118A > D110A > R106A/D110A (data not shown). Two additional amino acids in the extended α5 helix of GRK2 were also examined; however, neither T111A nor C120A have any effect on the ability of GST-GRK2-(45–178) to bind Gαq/11 (Figure 6).
Figure 6.
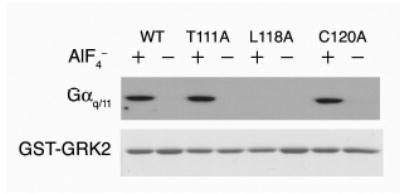
Further mapping of the Gαq/11 binding site on the GRK2 RH domain. Upper Panel. Glutathione-agarose beads bearing GST fusion proteins, either WT GSTGRK2-(45–178) or GST-GRK2-(45–178) substituted as indicated, were incubated with bovine brain extract (as a source of Gαq/11) in the presence (+) or absence (−) of aluminum fluoride (AlF4−). Bound Gαq/11 was visualized by immunoblotting. Lower Panel. Fusion proteins used in the GST-pull-down assay above were separated by SDS-PAGE and visualized by Coomassie staining.
DISCUSSION
Here, Gαq-binding residues of GRK2 identified in a previous study (22) and the crystal structure of GRK2 were both used to construct a model of the Gαq-GRK2 interaction interface. This model was used to identify Gαq residues and additional GRK2 residues that could be involved in the interface (14). While we believe the resulting model is globally correct, there is no way short of determining the crystal structure of the complex to know if it is accurate in detail, especially given that no high-resolution atomic model of Gαq currently exists. Therefore, while the manually docked model described here is consistent with the existing biochemical data, it should not be regarded as more than a conceptual tool to help predict regions of Gαq that are responsible for binding and specificity.
Our results demonstrate that of the Gαq residues tested, Pro185 is the most critical for the Gαq-GRK2 interaction (Figure 2), although mutation of other residues within Switch I, III and the helical domain were also found to influence complex formation. Moreover, we show that mutation of Gαq residues differentially affect interaction with GRK2 and the canonical RGS proteins, RGS2 and RGS4. Specifically, the T187K mutation significantly reduces binding to and inhibition of signaling by RGS2, but does not affect the GRK2-Gαq interaction (compare Figures 2 and 3 to Figures 4 and 5). These results are consistent with the Gαq-GRK2 interface being distinct, but overlapping, with that of Gα-RGS proteins (Figure 1 A–B).
Several lines of evidence are consistent with the proposal that the RH domain of GRK2 interacts with the switch regions of Gαq. First, the activation-dependent nature of the interaction between the GRK2 RH domain and Gαq strongly suggests that the switch regions of Gαq are involved. Secondly, in a study using Gαq-Gα16 chimeras, we have shown that GRK2 binds to a chimeric Gα protein containing the switch regions of Gαq but not a chimeric protein containing the switch regions of Gα16, a member of the Gαq family that does not interact with GRK2 (19). Thirdly, modeling Gαq onto the RH domain of GRK2 predicts that Pro185 of Gαq makes significant contacts with several residues in GRK2, such as Asp110 and Met114, previously identified as being important for the interaction (22). The fact that Pro185 resides at the crux of the interface would also explain why GRK2 selectively interacts with Gαq rather than Gαi, where the corresponding residue is a lysine. Consistent with this idea, the Gαq-P185K mutant does not bind to GRK2 when activated by AlF4−, but it does bind to RGS4 and RGS2 (Table 1 and Figures 2 and 4). P185K-RC not only fails to bind GRK2 but also does not bind RGS2 or RGS4 (Figures 2 and 4). Apparently, in the context of Gαq with the RC mutation, P185K cannot be tolerated. A previous report suggested that Pro185 and Ile190 of Gαq contribute to the higher affinity of RGS2 for Gαq, versus Gαi, by affecting the position of Thr187 relative to the RGS binding pocket (36). If this is true, then it is possible that the combined effects of R183C and P185K change the conformation of Switch I so that it is incompatible with GRK2, RGS2 and RGS4 binding.
The Gαq residues that mediate critical interactions with the A site of RGS proteins are distinct from those that interact with the C site of GRK2. In the model of the Gαq- GRK2 complex, Thr187 is close enough to the interface that mutation to lysine could potentially disrupt the interaction. However, mutation of this residue does not affect the Gαq-GRK2 interaction and therefore does not represent a critical contact site. In contrast, the T187K mutation has a profound effect on the interaction of both RGS2 and RGS4 with Gαq (Figures 4). In addition, RGS2 does not inhibit signaling from the constitutively active, Gαq-T187K-RC, form of this mutant (Figure 5). As mentioned previously, the position of Thr187 in Gαq relative to the binding pocket of RGS2 may determine in part the selectivity of the RGS2-Gαq interaction (36).
Further differences between Gαq binding to GRK2 and RGS2 or RGS4 can be seen by mutation of residues in the helical domain of Gαq. One such mutation, Q152A, located in a loop between the αD and αE helices, disrupted binding to the RH domain of GRK2 and inhibition by full-length GRK2 in cellular inositol phosphate assays (Figures 2 and 3). The Gαq-Q152A-RC mutant interacted with both RGS2 and RGS4 in pull-down assays and its stimulation of PLC-β was inhibited by RGS2 (Figures 4 and 5), indicating that the Q152A mutation selectively disrupts the interaction of Gαq with GRK2. We also identified a mutation in the helical domain of Gαq, Q81A, that has unique effects on RH domain binding specificity. The binding of GST-RGS4, GST-RGS2 and GST-GRK2- (45–178) to AlF4− activated-Q81A is reduced (Table 1 and Figures 2 and 4). In contrast Gαq-Q81A-RC, displays decreased interaction with RGS2 and RGS4 but exhibits no defect in its interaction with GRK2 (Figures 2 and 4). Relative to Gαq-RC, RGS2 inhibition of Gαq-Q81A-RC stimulated inositol phosphate production is decreased (Figure 5), while GRK2 inhibits both Gαq-RC and Gαq-Q81A-RC-stimulated inositol phosphate production to similar levels (data not shown). The finding that the Q81A mutation disrupts the interaction of Gαq with RGS2 and RGS4 is novel. Additionally, this glutamine is conserved only among the Gαq family and therefore may be a residue in the helical domain of Gα that could contribute to the targeting of RGS proteins to specific Gα subunits.
Mutation of other residues in the helical domain of Gαq decreased the interaction with GRK2. Both the K77A and R92A mutations in Gαq decreased binding to GRK2 in the context of Gαq-QL (Figure 2 B). These results likewise suggest that the RH domain of GRK2 interacts with the alpha helical domain of Gαq. There is evidence from other studies that the helical domain imparts some of the Gα selectivity upon interactions with RH domains. Skiba et al. used Gαt/Gαi chimeras to show that the specificity of RGS9 for Gαt resides in the helical region (37). A second study used RGS2/RGS4 chimeras and point mutants to identify residues in RGS2 that confer Gαq selectivity (36). The RGS2 residues identified in this study would interact with residues in the αA helix in the helical domain of Gαq and would be repelled by analogous residues in Gαi (36). In combination with our results, such studies demonstrate that the helical domain of Gα subunits, in particular αA, plays a critical role in the specificity of RH domain-Gα subunit interactions.
Results from several experiments in this study revealed differences in the ability of Gαq to interact with GRK2, depending upon whether the Gα subunit is activated by AlF4−, the R183C mutation or the Q209L mutation. For example, the Q81A mutation disrupts interaction with the RH domain of GRK2 when Gαq is activated by AlF4− but has no effect in the presence of the activating RC and QL mutations (Figure 2). In contrast, Gαq-K77A-QL and Gαq-R92A-QL display greatly decreased interaction with the RH domain of GRK2, but Gαq-K77A-RC, Gαq-R92A-RC, and AlF4−-activated Gαq-K77A and Gαq-R92A efficiently interact with the RH domain of GRK2 (Figure 2). In the cocrystal structure of RGS4 and Gαi, Asn128 of RGS4 projects into the active site of Gαi and contacts the catalytic residue Gln204 (11). Our model predicts that Gln209 in Gαq could also make direct contact with the RH domain of GRK2, by forming hydrogen bonds with the side chain of Asp110 in GRK2. However, in order to do so it would have to adopt a different, more extended conformation than that observed for the analogous Gαi Gln204 residue in the Gαi-RGS4 complex (11). While this extended conformation would be predicted to inhibit GTPase activity, such a conformation of Gαq may be appropriate for binding to GRK2, which exhibits little or no GAP activity (18). The constitutively active RC and QL forms of several Gα subunits have been used extensively and somewhat interchangeably to investigate Gα subunit signaling; however, our results suggest that there are functional differences between these constitutively active mutants that may warrant further investigation.
In conclusion, we have used molecular modeling and mutation studies to identify residues that are important for the interaction between the RH domain of GRK2 and Gαq. This data confirms the unique characteristics of the interaction between Gαq and the C site of the GRK2 RH domain and also identifies new residues in the helical domain of Gα that selectively disrupt the interaction between Gαq and RGS2 or RGS4 but not GRK2. The crystal structure of GRK2 and Gβγ in complex allows for a model in which GRK2 is simultaneously interacting with agonist bound receptor, Gβγ and Gαq (14). It would also be interesting to investigate the possibility that Gαq plays a role in directing the Gβγ-recruited GRK2 to specific, activated Gαq coupled receptors.
Acknowledgments
We thank Chris Fischer for excellent technical assistance. RSM would like to acknowledge Laura A. Tehan, Robyn Pyskadlo and members of the Siena College Spring 2003 Molecular Biology class for contributions to this work. JJGT would also like to acknowledge the UT Austin College of Natural Sciences support for the Center for Structural Biology.
Footnotes
This work was supported by a fellowship from the American Heart Association Pennsylvania-Delaware Affiliate to P.W.D., American Heart Association Scientist Development Grant 0235273N and a Research Corporation Cottrell Scholar grant to J.J.G.T., National Science Foundation Grants MC9728179 and MCB0315888 to R.S.M., National Institute of Health Grant GM44944 to J.L.B. and National Institute of Health Grant GM62884 to P.B.W.
The abbreviations are: RH, RGS homology; GPCR, G protein-coupled receptor; PLCβ, phospholipase Cβ; GRK, GPCR kinase; GAP, GTPase activating protein; AlF4−, aluminum fluoride; RGS, regulator of G protein signaling; GST, glutathione-Stransferase; WT, wild type; DMEM, Dulbecco’s modified Eagle’s medium; GDP, guanosine diphosphate; GTP, guanosine triphosphate.
References
- 1.Bourne HR. Curr Opin Cell Biol. 1997;9:134–142. doi: 10.1016/s0955-0674(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 2.Krupnick JG, Benovic JL. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 3.Hausdorff WP, Caron MG, Lefkowitz RJ. FASEB J. 1990;4:2881–2889. [PubMed] [Google Scholar]
- 4.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson SS, Downey WE, 3rd, Colapietro AM, Barak LS, Menard L, Caron MG. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 6.Gilman AG. Ann Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 7.Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. Nature. 1996;383:172–175. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- 8.Berstein G, Blank JL, Jhon DY, Exton JH, Rhee SG, Ross EM. Cell. 1992;70:411–418. doi: 10.1016/0092-8674(92)90165-9. [DOI] [PubMed] [Google Scholar]
- 9.Ross EM, Wilkie TM. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 10.Hollinger S, Hepler JR. Pharmacol Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 11.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 12.Slep KC, Kercher MA, He W, Cowan CW, Wensel TG, Sigler PB. Nature. 2001;409:1071–1077. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- 13.Spink KE, Polakis P, Weis WI. EMBO J. 2000;19:2270–2279. doi: 10.1093/emboj/19.10.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ. Science. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Wells CD, Sternweis PC, Sprang SR. Nat Struct Biol. 2001;8:805–809. doi: 10.1038/nsb0901-805. [DOI] [PubMed] [Google Scholar]
- 16.Longenecker KL, Lewis ME, Chikumi H, Gutkind JS, Derewenda ZS. Structure (Camb) 2001;9:559–569. doi: 10.1016/s0969-2126(01)00620-7. [DOI] [PubMed] [Google Scholar]
- 17.Zhong H, Neubig RR. J Pharmacol Exp Ther. 2001;297:837–845. [PubMed] [Google Scholar]
- 18.Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T. J Biol Chem. 1999;274:34483–34492. doi: 10.1074/jbc.274.48.34483. [DOI] [PubMed] [Google Scholar]
- 19.Day PW, Carman CV, Sterne-Marr R, Benovic JL, Wedegaertner PB. Biochem. 2003;42:9176–9184. doi: 10.1021/bi034442+. [DOI] [PubMed] [Google Scholar]
- 20.Usui H, Nishiyama M, Moroi K, Shibasaki T, Zhou J, Ishida J, Fukamizu A, Haga T, Sekiya S, Kimura S. Int J Mol Med. 2000;5:335–340. doi: 10.3892/ijmm.5.4.335. [DOI] [PubMed] [Google Scholar]
- 21.Sallese M, Mariggio S, D’Urbano E, Iacovelli L, De Blasi A. Mol Pharmacol. 2000;57:826–831. [PubMed] [Google Scholar]
- 22.Sterne-Marr R, Tesmer JJ, Day PW, Stracquatanio RP, Cilente JA, O’Connor KE, Pronin AN, Benovic JL, Wedegaertner PB. J Biol Chem. 2003;278:6050–6058. doi: 10.1074/jbc.M208787200. [DOI] [PubMed] [Google Scholar]
- 23.DiBello PR, Garrison TR, Apanovitch DM, Hoffman G, Shuey DJ, Mason K, Cockett MI, Dohlman HG. J Biol Chem. 1998;273:5780– 5784. doi: 10.1074/jbc.273.10.5780. [DOI] [PubMed] [Google Scholar]
- 24.Lan KL, Sarvazyan NA, Taussig R, Mackenzie RG, DiBello PR, Dohlman HG, Neubig RR. J Biol Chem. 1998;273:12794–12797. doi: 10.1074/jbc.273.21.12794. [DOI] [PubMed] [Google Scholar]
- 25.Ausubel, F. M., Brent, R. E., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl, K. (1992) Short Protocols in Molecular Biology, second Ed., John Wiley & Sons, New York
- 26.Schwede T, Kopp J, Guex N, Peitsch MC. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colovos C, Yeates TO. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vajda S, Camacho CJ. Trends Biotechnol. 2004;22:110–116. doi: 10.1016/j.tibtech.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Wodak SJ, Mendez R. Curr Opin Struct Biol. 2004;14:242–249. doi: 10.1016/j.sbi.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Skiba NP, Yang CS, Huang T, Bae H, Hamm HE. J Biol Chem. 1999;274:8770–8778. doi: 10.1074/jbc.274.13.8770. [DOI] [PubMed] [Google Scholar]
- 31.Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54 ( Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 35.Kong G, Penn R, Benovic JL. J Biol Chem. 1994;269:13084–13087. [PubMed] [Google Scholar]
- 36.Heximer SP, Srinivasa SP, Bernstein LS, Bernard JL, Linder ME, Hepler JR, Blumer KJ. J Biol Chem. 1999;274:34253–34259. doi: 10.1074/jbc.274.48.34253. [DOI] [PubMed] [Google Scholar]
- 37.Skiba NP, Yang CS, Huang T, Bae H, Hamm HE. J Biol Chem. 1999;274:8770–8778. doi: 10.1074/jbc.274.13.8770. [DOI] [PubMed] [Google Scholar]
- 38.DeLano, W. L. (2002), DeLano Scientific, San Carlos, CA, USA