

Abstract
Protective antigen (PA) is the anthrax toxin protein recognized by capillary morphogenesis gene 2 (CMG2), a transmembrane cellular receptor. Upon activation, seven ligand-receptor units self-assemble into a heptameric ring-like complex that becomes endocytozed by the host cell. A critical step in the subsequent intoxication process is the formation and insertion of a pore into the endosome membrane by PA. The pore conversion requires a change in binding between PA and its receptor in the acidified endosome environment. Molecular dynamics simulations totaling ∼136 ns on systems of over 92,000 atoms were performed. The simulations revealed how the PA-CMG2 complex, stable at neutral conditions, becomes transformed at low pH upon protonation of His-121 and Glu-122, two conserved amino acids of the receptor. The protonation disrupts a salt bridge important for the binding stability and leads to the detachment of PA domain II, which weakens the stability of the PA-CMG2 complex significantly, and subsequently releases a PA segment needed for pore formation. The simulations also explain the great strength of the PA-CMG2 complex achieves through extraordinary coordination of a divalent cation.
INTRODUCTION
Anthrax toxin, secreted by the bacterium Bacillus anthracis, consists of three proteins: protective antigen (PA), lethal factor (LF), and edema factor (EF). PA is responsible for the delivery of the two toxic enzymes, LF and EF, which disrupt the normal immune function of the host and promote bacterial survival, as well as induce pathogenesis (see reviews by Collier and Young (1), Scobie and Young (2), and Moayeri and Leppla (3)). As the first step of the intoxication process, the full-length 83-kDa PA binds to a receptor on the surface of a target cell (4,5). A 20-kDa N-terminal fragment of PA is subsequently cleaved by a furin-like enzyme (6,7). The cleavage activates the remaining 63-kDa receptor-bound PA, which spontaneously assembles into a ring-like heptameric structure (8). The oligomerization generates binding sites for up to three molecules of LF and/or EF at the interfaces of neighboring PA monomers (9). The entire toxin-receptor complex is then internalized into the acidic endosome (10,11). The complex, a prepore structure, undergoes conformational changes and forms a pore inserted into the membrane of the endosome (12,13), as illustrated in Fig. 1 a. Through the pore, the toxic enzymes LF and EF are translocated and released to the cytosol.
FIGURE 1.

Pore-insertion steps of the anthrax intoxication process and the system setup for the MD simulations conducted in this study. (a) Heptameric PA-CMG2 anthrax-receptor complex carrying up to three toxic LF and/or EF enzymes after endocytosis (left). The acidification in the endosome triggers the rearrangement of each PA monomer (center), leading to the formation and insertion of a β-barrel-shaped pore that enables the delivery of the cargo (right). For a clear view, only four PA monomers are shown in the center and left complexes. (b) The crystal structure (PDB code 1T6B) of a PA-CMG2 monomer. The four domains (I–IV) of PA and the I domain of CMG2 are colored in cyan, lime, purple, ochre, and mauve, respectively. The Mg2+ ion at the MIDAS site is shown as an orange sphere. Unless otherwise noted, the same color code is used throughout this report. (c) CMG2 domain I and PAII and PAIV were included in our MD simulations. Eight histidines and one glutamate protonated under acidic conditions are colored in red and blue, respectively. PAIV Asp-683 bound to the MIDAS ion is shown in cyan. The two PAII loops β1-β2 and β2-β3 missing in the original crystal structure were built and are shown in silver. (d) The complex was solvated and equilibrated in an explicit water cell represented as a light blue box.
Despite the identification of the PA residues involved in the pore formation (14–16), the mechanism for how the PA-CMG2 complex regulates conformational changes leading to pore formation (Fig. 1 a) is poorly understood. In the absence of the receptor, the cleaved PA monomers are still capable of self-association, and prepore-to-pore conversion occurs at neutral pH 7 either in artificial membranes (17) or in solution (18), whereas the conversion requires an acidic environment below pH 6 at the receptor-bearing cell surface (17,18). To understand why the pore formation process is pH dependent when receptors are present, additional investigations regarding the interactions between PA and its receptors are necessary.
The anthrax toxin was discovered in the 1950s and the high resolution crystal structures for all three components involved in the primary intoxication process have become available since then (14,19,20); two homologous anthrax receptors recognized by PA have been identified recently as well: tumor endothelial marker 8 (TEM8, or anthrax toxin receptor) (4) and capillary morphogenesis gene 2 (CMG2) (5). Both transmembrane receptors share a highly conserved extracellular I domain with a sequence identity of 60%. The crystal structure of CMG2 I domain alone revealed a so-called Rossman fold (21), structurally homologs to von Willebrand factor A type (vWA) proteins best characterized in integrin αA/I domains (22). The most distinctive feature of these vWA-type domains is a structural motif forming the so-called metal ion-dependent adhesion site (MIDAS), where a divalent metal ion is acquired by the receptor and is recognized by a negatively charged amino acid from its ligand. Asp-683PA from PA domain IV (PAIV) has been pinpointed by genetic analysis and mutagenesis studies to bind the metal ion at the MIDAS site of the CMG2 I domain (23,24). However, PA surprisingly binds CMG2 with an affinity at least 1000-fold higher (25) than that of a typical integrin-ligand complex (26), which suggests that additional interactions are involved in PA-CMG2 binding (21).
Recently, the crystal structure of monomeric PA in complex with CMG2 resolved at 2.5 Å resolution (27) and the structure of the heptameric prepore PA-CMG2 assembly resolved at 4.3 Å resolution (18) have confirmed the suggestion. The structure of the PA-CMG2 complex is shown in Fig. 1 b, where the domains I–IV of PA are highlighted. In addition to the binding of PAIV to the MIDAS region of the receptor, the crystal structures revealed interactions between the receptor and PA domain II (PAII) that are most intriguing because the interactions prevent the structural rearrangement for prepore-to-pore conversion. In light of the new crystal structures, it is hypothesized that protonation of a histidine residue His-121CMG2 located on the edge of the binding pocket regulates dissociation of PAII and triggers the actual pore conversion (18,27). This hypothesis seems to be promising in solving the long-lasting puzzle of why the pore formation process is pH dependent.
We have carried out molecular dynamics (MD) simulations to provide structural atomic insights into the pH-regulated stability of the PA-CMG2 complex. We investigated how the protonation of His-121CMG2 and its neighboring glutamate Glu-122CMG2 (both receptor side groups) affect the stability of the complex. The simulations revealed that the glutamate residue plays an essential role in stabilizing the prepore PA-CMG2 complex by forming a salt bridge with an arginine from PA. But the protonation of His-121CMG2 alone is not enough for disrupting the salt bridge. Protonation of Glu-122CMG2 is also required to disrupt the salt bridge and to trigger the dissociation of PAII necessary for initiating pore formation. We further compare the stability of the equilibrated neutral structure to an acidic structure with PAII partially dissociated by means of so-called steered molecular dynamics (SMD) simulations. SMD is a powerful method for probing the stability of proteins in silico (28) and has been applied previously to the probing of the biotin-avidin complex (29,30), to the study of an integrin-ligand complex (31), to the stretching of the adhesive extracellular matrix protein fibronectin (32,33), as well as to the forced dissociation of C-cadherin (34).
METHODS
System setup
The 2.5 Å resolution crystal structure of anthrax toxin protein PA in complex with its receptor, CMG2 I domain (27), was adopted from the Protein Data Bank (PDB; entry code 1T6B) for modeling. Loops β1−β2 and β2−β3 of PAII were poorly resolved in the original structure. The backbones of 28 missing residues in these two loops (residues 276–287, 304–319) were reconstructed. The side chains of the unresolved residues were subsequently built and the conformation of these two loops was optimized with Swiss DeepView (35). The hydrogen atoms of all protein residues were added using the PSFGEN plug-in of the Visual Molecular Dynamics (VMD) program (36). The resulting β1−β2 and β2−β3 loops were minimized for 3000 steps, whereas all other segments of the complex were fixed.
To reduce the size of the system, only PAII (residues 259–456), PAIV (residues 596–735), and CMG2 domain I (residues 41–210) were included in the MD simulations. There are five histidines (residues 263, 299, 304, 310, 336) in PAII, two (residues 597, 616) in PAIV, and one (residue 121) in CMG2 domain I. To simulate the effect of the acidified endosome environment, we designed four systems by varying the protonation state on these eight histidines and one additional glutamate, Glu-122, neighboring His-121 of CMG2 (see Results). Crystal water molecules within 10 Å distance from the investigated complex were retained for building the starting structure of the simulation. Each system was solvated additionally in a cell of size 87 × 90 × 125 Å3 filled with TIP3 (37) water. The dimensions of the water cell were chosen so that the CMG2 domain can be displaced from the two PA domains by at least 30 Å along the longest axis. The crystal structure contains a Mn2+ ion at the MIDAS site. Since Mg2+ is the physiologically relevant ion, we substitute Mg2+ for the Mn2+ ion. Additional sodium or chloride ions were introduced to neutralize the systems. The final systems, shown in Fig. 1 d, contain ∼92,200 atoms.
MD methods
All simulations ware performed using the program NAMD (38) and CHARMM27 forcefields (39). Periodic boundary conditions were imposed. Each of the three systems was first minimized for two consecutive 2000 conjugate gradient steps, where the protein was held fixed and water molecules were allowed to move; subsequently, all atoms were allowed to move. After minimization, each system was equilibrated for 10 ns under constant pressure and temperature conditions (NPT). The pressure was maintained at 1 atm using the Langevin piston method (40), and the temperature was controlled by using Langevin dynamics at 300 K with a damping coefficient of 5 ps−1. A Langevin piston period of 100 fs and a damping time constant of 50 fs were employed for pressure control.
An integration time step of 1 fs and a uniform dielectric constant of 1 were chosen. A multiple time step integration algorithm was employed to evaluate covalent bond interactions every time step, short range nonbonded interactions every two time steps, and long-range electrostatics every four time steps (41). Full electrostatics were computed using the particle-mesh Ewald (PME) method (42) with a grid size of 96 × 96 × 128 Å3. van der Waals interactions were gradually turned off at a distance between 10 Å and 12 Å.
After equilibration, two sets of constant velocity SMD (cv-SMD) simulations (28,30) were performed to probe the stability of the complexes. In a first set of SMD simulations, the PAII was stretched from its binding pocket, whereas CMG2 was fixed. In the second set of simulations, the CMG2 I domain was pulled away from the binding interface, whereas two domains of PA were held fixed. To avoid unfolding the protein, the external force was distributed over the Cα atoms of the β-strands of PAII in the first case and over the Cα atoms of the central β-sheet of the CMG2 domain in the second case, so that the center of mass of the stretched atoms was harmonically constrained with a force 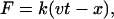 where k is the spring constant, v is the velocity, t is the time, and x denotes the displacement along the pulling direction. Pulling velocities of 0.002 Å/ps, 0.01 Å/ps, and 0.05 Å/ps were chosen. The spring constant k was set to 10 kBT/Å2, where kB denotes the Boltzmann constant and T temperature. When stretching PAII, the force was applied along the vector pointing from MIDAS Mg2+ cation to the Cα atom of Gly-342PA. In the simulations stretching CMG2, the direction of the force was along the vector pointing from the MIDAS Mg2+ cation to the N-terminal Cα atom of the CMG2 domain.
where k is the spring constant, v is the velocity, t is the time, and x denotes the displacement along the pulling direction. Pulling velocities of 0.002 Å/ps, 0.01 Å/ps, and 0.05 Å/ps were chosen. The spring constant k was set to 10 kBT/Å2, where kB denotes the Boltzmann constant and T temperature. When stretching PAII, the force was applied along the vector pointing from MIDAS Mg2+ cation to the Cα atom of Gly-342PA. In the simulations stretching CMG2, the direction of the force was along the vector pointing from the MIDAS Mg2+ cation to the N-terminal Cα atom of the CMG2 domain.
Simulations lasting altogether 136 ns were completed at the National Center for Supercomputing Applications. A 1 ns simulation of the 92,200 atom system required ∼5 h on 128 1.5 GHz Intel Itanium processors.
pKa calculation
The pKa values of the complex and individual CMG2 and PAIV domains were calculated at the H++ server (43) using the program MEAD (see Bashford and Gerwert (44) for details). In the calculations the crystal structure (27) of the complex with missing loops β1−β2 and β2−β3 rebuilt was adopted for modeling the protein, which is treated as a low dielectric ɛin object with embedded charges. The solvent was modeled as a continuum with dielectric constant ɛext. The default dielectric constants ɛin of 4 and ɛext of 80, and a salt concentration 0.15 mol/l were chosen. The finite difference Possion-Boltzmann method was used for electrostatics calculations.
RESULTS
To investigate the stability of the PA-CMG2 complex in neutral and acidic environments, we modeled four systems in which protonation states of all eight histidines of the complex and of one glutamate, Glu-122CMG2, neighboring His-121CMG2 (Fig. 1 c) were varied. A neutral system, where none of these amino acids was protonated, is denoted as the SN structure. The other three acidic systems will be referred to as the SH (all histidines protonated), the SE (Glu-122CMG2 and all histidines but His-121CMG2 protonated), and the SHE (all histidines and Glu-122CMG2 protonated) structures, respectively. Each of the four systems was equilibrated for 10 ns. To further examine the binding strength between PA and CMG2, two sets of cv-SMD simulations (28) were performed after the equilibration for each of the neutral SN and acidic SHE structures. In the first sets of SMD simulations, PAII was stretched from its binding pocket on CMG2. In the second set of SMD simulations, CMG2 domain I was forced to detach from PA. Simulations totaling altogether ∼136 ns were performed for this study on systems of over 92,000 atoms.
The neutral PA-CMG2 complex maintains a stable binding interface
Overall the neutral complex (SN) displayed a stable structure during equilibration, as shown in Fig. 2. The root mean-squared deviation (RMSD) from the original crystal structure for backbone heavy atoms of CMG2 domain I and of PAIV reached 1.25 Å and 1.75 Å, respectively, at 4 ns of the equilibration and subsequently fluctuated around these values during the remainder of the equilibration (Fig. 2 b). The RMSD of PAII fluctuated around a higher value of ∼3 Å, mainly due to the flexible loop segments composed of residues 276−289, 297−327, 396−400, and 422−429. The former two loops, β1−β2 and β2−β3, are implicated in pore formation (14).
FIGURE 2.

Equilibration of the anthrax-receptor complex under neutral condition. (a) Cartoon images of the complex before and after the 10 ns equilibration. In the starting structure, protein residues implicated in the pore formation or located at the binding sites are colored according to the code defined in Fig. 1 b, whereas in the equilibrated structure they are colored in orange. For other protein residues, only their starting structure is shown and they are colored in gray. The initial and final structures were aligned using the coordinates of CMG2. Key residues involved in the binding interactions between PAII and CMG2 and between Arg-344 of PAII and His-121, Glu-122, and Tyr-158 of CMG2 are shown in ball and stick representation. Oxygen, hydrogen, carbon, and nitrogen atoms are colored in red, white, cyan, and blue, respectively. (b) The profiles of backbone heavy atom RMSD for the three domains and the whole complex. The RMSD values were calculated by aligning individual domains to the crystal structure or in the case of the overall RMSD to the whole complex. (c) Changes of the BPSAs between PAII and CMG2 and between PAIV and CMG2 over the course of the equilibration.
During the equilibration, both binding interfaces between the receptor and the ligand were slightly enlarged. As shown in Fig. 2 c, the buried protein surface area (BPSA) between PAII and CMG2 increased during the equilibration from an initial value of 630 Å2 to the average value of 730 Å2, whereas it expanded from 1300 Å2 to the average value of 1410 Å2 between PAIV and CMG2 (Fig. 2 c). The increase at the PAII-CMG2 interface is primarily due to the rearrangement of the PAII β3−β4 loop so that it forms a few new contacts with the receptor (Fig. 3). Other than the two missing PAII loops, β1−β2 and β2−β3, the β3−β4 loop region exhibits the highest mobility in the crystal structure of PAII (average B factor of 28 Å2 for the loop residues 340–351 as compared to an average B factor of 19 Å2 for all resolved PAII residues). Upon the start of the equilibration at 300 K a short α-helix (residue 345–351) of the β3−β4 loop reoriented quickly toward the receptor within 50 ps so that the side chain of Thr-349PA and the backbone hydrogen of Arg-344PA on the loop form a couple of stabilized hydrogen bonds with a polar receptor residue Tyr-158CMG2 (Fig. 3, a–b, and Supplementary Material). In addition, an initially weak salt bridge between Arg-344PA and Glu-122CMG2 became stabilized during the equilibration (Fig. 3, see below). Transitory contacts formed between PAII β3−β4 loop residues (Glu-285PA and Thr-286PA) and the CMG2 α4 helix contributes to the small BPSA peaks observed at 2.8 ns and 8.1 ns for the PAII-CMG2 interface. The increase at the PAIV-CMG2 interface is mainly caused by new contacts formed between the PAIV β3−β4 loop and the CMG2 α2 helix at the edge of the binding interface, including side-chain hydrogen bonds between Asp-648PA and Arg-111CMG2 and between Glu-650PA and Arg-111CMG2.
FIGURE 3.

Key contacts at the PAII and CMG2 binding interface. (a–c) Snapshots of a salt bridge formed between Arg-344 of PAII and Glu-122 of CMG2. Formed hydrogen bonds are represented as dotted black lines. (d) The interaction energy between Arg-344PA and Glu-122CMG2 and the distances of two hydrogen bonds formed between the residues. (e) Number of water molecules within 2.5 Å of the two oxygen atoms on the side chain of Glu-122CMG2 or the unprotonated nitrogen of His-121CMG2.
The neutral PAII-CMG2 interface develops a strong Arg-344PA-Glu-122CMG2 salt bridge
Glu-122 is conserved between CMG2 and TEM8, the two anthrax toxin receptors identified to date. Sitting at the edge of the binding interface between PAII and CMG2, Arg-344PA and Glu-122CMG2 are separated by a distance of at least 3 Å between their side-chain tips, forming only one salt bridge bond in the crystal structure (Fig. 3 a). Arg-344PA is located on the PAII loop β3−β4, the hairpin tip of which is inserted into the CMG2 pocket accommodating PAII. During the equilibration simulation, the guanidinium moiety of Arg-344PA extended slightly to strengthen the salt bridge by forming two hydrogen bonds with Glu-122CMG2 (Fig. 3, a–d). After 5 ns equilibration this bidentate salt bridge became stabilized as the bonding distance decreased to <2.5 Å, counteracting occasional disturbances due to attacks from surrounding water molecules. An analysis of the number of collision events between water molecules and the two oxygen atoms on the carboxylic moiety of the Glu-122CMG2, revealing how frequently the two atoms were contacted by one or two surrounding water molecules, is shown in Fig. 3 e. No water molecule approached the deprotonated nitrogen on the side chain of His-121CMG2, suggesting that this residue cannot be protonated directly through solvent access.
Unbinding of PAII from the PA-CMG2 complex requires strong force
To examine the strength of the binding between PAII and CMG2, we performed cv-SMD simulations (see Methods) in which PAII was pulled out of its binding pocket spanned by helices α3 and α4 of CMG2. Results from a representative cv-SMD simulation at pulling velocity of 0.002 Å/ps are shown in Fig. 4. Under a gradually increasing force applied to the β-strands of PAII, the BPSA of the complex was ∼770 Å2 during the first 2 ns of the simulation, and then decreased to 100 Å2 from 2.4 to 3 ns. Corresponding to this rapid drop, the applied force decreased from 1000 pN to ∼200 pN at 3 ns, and only 2 of 24 contacting residue pairs (defined as two residues having at least one atom within 3.5 Å of each other) found in the equilibrated structure were still maintained at the same time. A closer inspection of specific binding interactions revealed that the Arg-344PA-Glu-122CMG2 salt bridge was disturbed at the beginning stage of the SMD simulation (Fig. 4, d–f). The two original hydrogen bonds formed during equilibration became broken, weakening the interaction between Arg-344PA and Glu-122CMG2 as the interaction energy dropped from −90 kcal/mol to −30 kcal/mol at 1.5 ns (Figs. 4 d and 3 e). Despite their disturbance, the two amino acids maintained considerable attractive interactions that were still four times stronger than any of the two hydrogen bonds formed between Tyr-158CMG2 and Thr-349PA and between Tyr-158CMG2 and the backbone hydrogen of Arg-344PA. These bonds and the salt bridge were concurrently ruptured at 2.4 ns, leading to the detachment of PAII from the binding pocket and the release of the originally restrained β3−β4 loop.
FIGURE 4.

Probing the binding site between PAII and CMG2 of the equilibrated neutral complex in a representative 0.002 Å/ps cv-SMD simulation. (a) Force is applied to the β-strands of PAII along the vector connecting the MIDAS ion (orange sphere) and the Cα atom (cyan sphere) of PAII Gly-342. The force direction is represented by a yellow arrow. CMG2 is shown in surface representation. The color code for positively charged, negatively charged, polar, and hydrophobic amino acids on the surface is blue, red, green, and white, respectively. (b) Profiles of applied force and resulting BPSA between PAII and CMG2 from the SMD simulation. (c) Interaction energy versus time profiles for three pairs of residues involved in binding between PAII and CMG2. (d–f) Snapshots of the PAII-CMG2 binding interface showing the disruption of the Arg-344PA-Glu-122CMG2 salt bridge and a couple of polar interactions between Tyr-158CMG2 and PAII.
Protonation of histidines does not destabilize the PAII-CMG2 binding
To test the hypothesis that protonation of histidines, particularly of His-122CMG2, drives the β3−β4 loop of PAII away from the receptor for the purpose of pore formation, we equilibrated the SH structure in which all histidines of the complex are protonated. After 10 ns of equilibration, the RMSD values for backbone heavy atoms of CMG2 and of PAIV from the crystal structure were <2 Å, slightly higher than that of the neutral structure, whereas the RMSD of PAII gradually grew to 5 Å, ∼2 Å larger than that of the neutral structure. However, the higher RMSD of PAII primarily resulted from a swing-like movement of the loop β2−β3 containing three histidines (Fig. 5 a). Compared to the neutral structure, the SH complex preserves a similar binding interface between PAII and CMG2, as indicated by the close overlap of the starting and equilibrated loop residues involved in the binding. The BPSA of the equilibrated PAII-CMG2 complex is ∼10% smaller than that of the neutral structure (Fig. 5 g). Despite this, the flatness of the BPSA curve does not indicate any tendency of PAII dissociation.
FIGURE 5.

Conformational changes of the anthrax-receptor complex under acidic conditions and interactions of key residues at the PAII-CMG2 binding surface. (a) Cartoon images of the initial and equilibrated SH structures in which all eight histidines are protonated. The color scheme and the alignment method are the same as in Fig. 2 a. (b–c) Despite initial disruption, the Arg-344PA-Glu-122CMG2 salt bridge is stably reformed after 5 ns equilibration. (d) Two overlaid cartoon snapshots of the SE structure where Glu-122CMG2 and all histidines, but His-121CMG2, are protonated. The snapshot taken at 2.2 ns is colored in the same scheme as the starting structure in a. The final structure reached during the equilibration is shown in orange. (e) The Arg-344PA-Glu-122CMG2 salt bridge is no longer formed. However, (f) Arg-344PA stays in the proximity of His-121CMG2. (g) Cartoon representations of the starting and 10-ns-equilibrated SHE structures with Glu-122CMG2 and all eight histidines protonated. In this case (h–i), Arg-344PA separates gradually from Glu-122CMG2. (j) Change of BPSA between PAII and CMG2 over the course of equilibrations of structures SN, SH, SE, or SHE. (k) Profiles of the Arg-344PA-Glu-122CMG2 interaction energy from these four equilibrations.
During the initial stage of the equilibration, the protonated His-121CMG2 attracted Glu-122CMG2 by forming a hydrogen bond and repelled Arg-344PA so that the Arg-344PA-Glu-122CMG2 salt bridge was no longer stabilized (Fig. 5 b). Not retracting from the binding pocket, however, the loop β3−β4 rearranged slightly to allow the reorientation of Arg-344PA, which maintained with Glu-122CMG2 the same bidentate salt bridge formed during the equilibration of the neutral structure (Figs. 5 c and 3 c). The salt bridge broke and reformed several times until it was stabilized after 5 ns equilibration. Correspondingly, the PAII-CMG2 complex was prevented from dissociation and the liberation of the β3−β4 loop became impossible.
Protonation of both His-121CMG2 and Glu-122CMG2 leads to the dissociation of PAII from CMG2
Since a protonated His-122CMG2 does not disrupt the Arg-344PA-Glu-122CMG2 salt bridge, additional regulation is necessary to induce the dissociation of PAII from the PA-CMG2 complex. Glu-122CMG2 is a straightforward candidate for playing such a role in a low pH environment. Indeed, we found that the protonation of both Glu-122CMG2 and His-121CMG2 triggered the dissociation of PAII from CMG2, as revealed in the equilibration simulations of the SE (Fig. 5, d–f) and SHE structures (Fig. 5, g–i). During the equilibration of the SE structure, where Glu-122CMG2 and all histidines other than His-121CMG2 were protonated, Arg-344PA was repelled from Glu-122CMG2 immediately after the equilibration started. Moreover, Thr-158CMG2, which was embedded in a pocket formed by PAII β3−β4 loop residues during the equilibration of the neutral structure (Fig. 2 e), slipped out of the pocket and resulted in the decrease of the BPSA of the PAII-CMG2 interface from an initial value of 600 Å2 to a value of 400 Å2 at 2.2 ns (Fig. 5 e). However, further dissociation of PAII was not observed within 10 ns equilibration as Arg-344PA remained in the proximity of His-121CMG2 (Fig. 5 f). Instead, Thr-158CMG2 flipped back toward its original position, restoring partial contacts with the PAII β3−β4 loop. The movement corresponds to an increase of PAII-CMG2 BPSA to ∼550 Å2 at the end of the equilibration. By comparison, further protonation of His-121CMG2 drove away Arg-344PA in the equilibration of the SHE structure. After a 5 ns fluctuation around 700 Å2, the BPSA of the PAII-CMG2 complex decreased as the dissociation started spontaneously. At the end of the 10 ns equilibration, the BPSA value dropped to 250 Å2, i.e., 65% lower than that of the equilibrated neutral structure. Accompanying this change, the tip of the PAII loop β3−β4 retreated 4 Å from the binding pocket on CMG2 for PAII (Fig. 5 g, and Supplementary Material). Arg-344PA interacted weakly with Glu-122 during the first half of the equilibration, but it was driven 10 Å away at the end of the equilibration (Fig. 5, e–f). In contrast, the PAIV-CMG2 binding interface was well preserved after the equilibration of all three acidic structures (SH, SE, and SHE). The BPSA of PAIV-CMG2 is similar to that of the equilibrated SH structure, only ∼5% lower than the BPSA of the neutral structure.
Glu-122CMG2 has a higher pKa value than His-121CMG2
To examine the likelihood of the protonation of His-121CMG2, of the neighboring Glu-122CMG2, and of other histidines of the complex, we calculated the pKa values for these amino acids using the crystal structure. The resulting pKa values are shown in Table 1. Whereas histidines on PAII and PAIV generally have pKa values >4, except His-616PA which is buried in the hydrophobic core of PAIV, His-121CMG2 exhibits a negative pKa value either in the absence or in the presence of PA. The primary reason for the low pKa of His-121CMG2 is that it is partially buried in a valley surrounded by two ridges comprised of outward-pointing residues on helices α3 and α4 of the receptor, respectively. The Arg-344PA-Glu-122CMG2 salt bridge formed between the ligand and receptor flanks His-121CMG2 farther from the top and virtually eliminates solvent accessibility of the side chain of this residue. His-121CMG2 therefore has an even lower pKa value in the complex form compared to that of the receptor-only form. By comparison, Glu-122CMG2 has a higher pKa of 5 in the absence of PA. However, the formation of the salt bridge with Arg-344PA decreases the pKa of Glu-122CMG2 to 2.3. These values are below the expected pH threshold between pH 5 to 6 for two reasons: First, interaction between the large number (over 100) of titratable side chains of the complex complicates the calculations and likely introduces an error; second, conformational changes occur in acidic condition although the crystal structure was determined at basic pH 8 and was used for the pKa calculations. Nevertheless, the results suggest that the protonation of Glu-122CMG2 likely happens before that of His-121CMG2.
TABLE 1.
pKa values of seven histidines and one glutamate of the PA-CMG2 complex
| CMG2
|
PAII
|
PAIV
|
Complex
|
|||||
|---|---|---|---|---|---|---|---|---|
| Titratable residue | pKintr | pKa | pKintr | pKa | pKintr | pKa | pKintr | pKa |
| His-121CMG2 | −1.7 | −1.7 | — | — | — | — | −4.4 | −6.3 |
| Glu-122CMG2 | 5.6 | 5.0 | — | — | — | — | 7.4 | 2.1 |
| His-263PA | — | — | 5.0 | 5.7 | — | — | 5.1 | 5.6 |
| His-299PA | — | — | 5.3 | 4.6 | — | — | 4.9 | 4.0 |
| His-304PA | — | — | 4.1 | 4.2 | — | — | 4.2 | 3.8 |
| His-310PA | — | — | 6.3 | 6.3 | — | — | 6.3 | 6.3 |
| His-336PA | — | — | 6.3 | 6.9 | — | — | 5.6 | 5.2 |
| His-597PA | — | — | — | — | 4.2 | 8.8 | 4.2 | 8.8 |
| His-616PA | — | — | — | — | 3.1 | 1.1 | 3.2 | 0.8 |
The calculation was performed for separated individual domains CMG2, PAII, and PAIV, as well as for the whole complex. The intrinsic pK, denoted as pKintr, is defined as the pKa of a titratable group when all other titratable groups of the protein were neutral.
Separating the PAII β3−β4 loop from CMG2 requires weaker force in the SHE structure compared to the neutral structure
To accelerate the separation of the PAII loop β3−β4 from CMG2, we carried out cv-SMD simulations (see Methods) in which the PAII-CMG2 binding was completely eliminated by pulling PAII along the same direction as in the previous set of SMD simulations. Compared to detaching PAII of the neutral structure, no major peak force region was encountered in the case of the SHE structure (Fig. 6 a). The maximum force required for detaching PAII from CMG2 is ∼40–50% lower than that arising in cv-SMD simulations of the SN structure at the same pulling velocity ranging from 0.05 Å/ps to 0.002 Å/ps (Fig. 6 b). During the equilibration of the SHE structure, a transient contact was formed between Tyr-158CMG2 and Gly-342PA when loop β3−β4 retreated from the CMG2 binding pocket (Fig. 6 c). This hydrogen bond was easily disrupted at 2.65 ns as shown in one representative SMD simulation (Fig. 6 d).
FIGURE 6.

Forced detachment of PAII from the CMG2 binding pocket in SMD simulations of the SHE structure. (a) Profiles of applied force and BPSA between PAII and CMG2 from a representative 0.002 Å/ps cv-SMD simulation. (b) Comparison of peak forces required to unbind PAII from CMG2 between the SN and the SHE structures in cv-SMD stretching simulations with various constant velocities. Significantly lower force is required to detach PAII when the Arg-122PA-Glu-122CMG2 salt bridge is broken under low pH conditions. (c–d) Snapshots of the PAII-CMG2 binding interface obtained from the representative SMD simulation.
The binding of PA to CMG2 establishes an optimal MIDAS cation coordination sphere
Having characterized the PAII-CMG2 binding interactions, we next examine structural changes at the MIDAS site, the primary binding site between PAIV and CMG2. As shown in Fig. 7, the upper surface of CMG2 of the MIDAS site is encompassed by three loops: α1−β1, α2−α3, and β4−α4. The MIDAS motif includes Asp-50CMG2, Ser-52CMG2, and Ser-54CMG2 located on α1−β1, Thr-118CMG2 located on α2−α3, and Asp-148CMG2 located on β4−α4. The complete coordination sphere of the MIDAS ion includes Ser-52CMG2, Ser-54CMG2, Thr-118CMG2, two water molecules W1 and W2, and Asp-683PA from PAIV (see Fig. 7). Although Asp-50CMG2 and Asp-148CMG2 do not contact the metal ion directly, their side-chain carboxylic fingers act together to firmly grip water molecule W1. Both water molecules W1 and W2, taken from the crystal structure, are tightly bound to the ion throughout all three equilibrations.
FIGURE 7.

Conformational changes of the MIDAS coordination sphere region upon the dissociation of PAII from CMG2. The equilibrated SN structure is shown in gray. The equilibrated SHE structure, where PAII becomes partially dissociated, is colored following the code defined in Fig. 1 b. Residues not belonging to the binding sites on CMG2 are dimmed. The SN and SHE structures were aligned using the coordinates of CMG2. The amino acids and water molecules W1 and W2 coordinating the MIDAS cation are shown in licorice representation. The coordination of water W1 by Asp-148CMG2 is disturbed due to the dissociation of PAII in the SHE structure. Shown in the inset are the profiles for the distance between MIDAS water W1 and two side-chain oxygens of Asp-148CMG2 over the course of the equilibration of SN and SHE structures.
Loop α2−α3 inserts into a hydrophobic pocket flanked by two loops of PAIV; loop α1−β1 contains residues interacting with PAIV (Fig. 7). Comparing the SHE structure to the SN structure, the movement of loops α1−β1 and α2−α3 is small because PAIV-CMG2 binding interactions stabilizing the conformation of these two loops were essentially not disturbed by the protonation of histidines and Glu-122PA. A backbone RMSD of <1 Å between the equilibrated SN and SHE structures was measured for the two loops, which contain all amino acids of the MIDAS motif except Asp-148CMG2. In contrast, the dissociation of PAII from the receptor releases the restraint on the β4−α4 loop emanating from helix α4 that shapes one edge of the binding pocket for PAII (Fig. 7). The loop displays a backbone RMSD of 2.7 Å between the SN and SHE structures. In the latter structure the conformational changes upon protonation cascaded from the end at the CMG2 binding pocket to the other end at the MIDAS region, leading to a 2 Å shift of the Asp-148CMG2 Cα atom (Fig. 7). Accompanying this change is a perturbation of the tight binding of water W1 to Asp-148CMG2, which flipped its two side-chain carboxylic oxygen atoms to maintain the water coordination (Fig. 7, inset). By comparison, such flip behavior was not observed in the equilibration of the neutral structure, suggesting that the SN structure maintains a more stabilized MIDAS configuration than does the SHE structure where PAII become partially dissociated.
Dissociation of PAII weakens the PA-CMG2 complex
To compare the overall stability of the equilibrated SN and SHE complexes, the latter with PAII partially dissociated, cv-SMD simulations were conducted for each complex separating PA from CMG2, as sketched in Fig. 8 a. In both cases, i.e., SN and SHE, a prominent force peak was observed consistently in the force versus time profiles obtained from 15 independent cv-SMD simulations at various stretching velocities (Fig. 8, b and c). The major force peaks correspond to destabilizing the coordination through the MIDAS ion, as shown in two series of snapshots from two representative 0.002 Å/ps cv-SMD simulations (Fig. 8, d–m, and Supplementary Material). However, the scenarios of the destabilization are different. Disrupting the SN complex requires significantly higher force than disrupting the SHE structure, due to the tightness of the binding between PAII and the receptor.
FIGURE 8.

Forced dissociation of the receptor CMG2 from the ligand PA in cv-SMD simulations. (a) The force is applied to the central β-sheet of CMG2 along the direction pointing from Mg2+ to the Cα atom of the CMG2 N-terminus, as indicated by a blue arrow. (b) Force versus time profiles from one representative 0.002 Å/ps cv-SMD simulation of the SN and SHE structures. (c) Comparison of the peak unbinding force calculated in 30 SMD simulations of the SN (⋄) and the SHE (○) structure. Each symbol represents an independent simulation. Note that two close symbols may overlap. Snapshots obtained from the representative cv-SMD simulations (d–h) of the SN structure and (i–m) of the SHE structure. The trajectories of these two simulations are provided as movies in the Supplementary Material. Water molecules coordinating the MIDAS ion are shown in light blue.
In the representative SMD simulation of the neutral complex (Fig. 8, d–h), the contacts between the receptor and the MIDAS ion were greatly weakened at 4.46 ns, coinciding with a precipitous decrease of the pulling force from over 3000 pN to <500 pN (Fig. 8 b). Immediately after the force peak, the MIDAS coordination residues Ser-52CMG2 and Thr-118CMG2 were replaced by two water molecules coordinating the MIDAS ion (Fig. 8 f). The BPSA between PAIV and CMG2 decreased from 1000 Å2 to 300 Å2 at 4.6 ns. However, the Arg-344PA-Glu-122CMG2 salt bridge and other interactions between PAII and CMG2 were still maintained. At 5 ns, CMG2 was completely detached from the binding interface of PAIV, tilting ∼60° toward PAII (Fig. 8 g). The remaining contacts were ruptured sequentially by a rather weak force.
PAII is largely dissociated from CMG2 in the equilibrated SHE structure. The SMD force easily disrupted a transient contact formed between Tyr-158CMG2 and Gly-342PA, the rupture occurring at 2.47 ns in one representative SMD simulation of the acidic structure (Fig. 8, b and j). By the time of occurrence of the peak force at 3.5 ns, the stretching had diminished the BPSA between CMG2 and PAII to 80 Å2. The peak force of 2000 pN coincides with the breakage of the contacts between the MIDAS water W1 and receptor residues Asp-50CMG2 and Asp-148CMG2. The configuration of the latter had already been disturbed during the equilibration of the acidic complex (see Fig. 7). The release of water W1 allowed the molecule to attack the remaining MIDAS ion coordination. W1 subsequently facilitated the separation of MIDAS coordination residue Ser-52CMG2 from the ion by replacing the coordination of Ser-52CMG2 (Fig. 8 l). At 4.02 ns, the interactions between the MIDAS ion and two surrounding serines were ruptured. At this point, the BPSA between PAIV and CMG2 started a rapid decrease from 900 Å2 to 300 Å2 at 4.5 ns, and the pulling force dropped from 2000 pN to below 500 pN. As the stretching continued, the remaining receptor-ligand contacts were easily abolished without encountering a further major force peak. Based on the average from 30 independent simulations pulling SN and SHE structures at velocities of 0.05 Å/ps, 0.01 Å/ps, and 0.002 Å/ps, the peak unbinding force was consistently found ∼20% lower for the acidic complex compared to that of the neutral complex (Fig. 8 c). Decreasing the pulling velocity 25-fold reduced the peak force by ∼30% in each case, leaving the key features of the scenarios described unchanged.
DISCUSSION
One key question regarding the anthrax intoxication mechanism is how the heptameric PA-CMG2 complex initiates formation of a β-barrel-like pore for ferrying its toxic cargo. It was suggested that the PA segment lining the eventual 14-β-stranded barrel, i.e., loops β1-β2, β2-β3, and β3-β4 and β-strands β2 and β3, becomes dislodged from the PAII domain upon acidification in the endosome environment (14). However, the new crystal structures exhibit an unanticipated PAII-CMG2 binding that inhibits the rearrangement and dislodging of these secondary structures (18,27). On the other hand, it was known that low acidic conditions are required for pore formation. Early studies suggest that prepore initiation proceeds under acidic condition between pH 5 and 6 (8,17). Recent experiments indicate that the threshold for converting the PA-CMG2 prepore complex to the pore is between pH 5.2 and 5.5 (45). Therefore, the apparent contradiction caused by the PAII-CMG2 binding can be reconciled if PAII dissociates from CMG2 under low pH conditions (18,27). Addressing this important issue, our MD simulations provide structural insights into the mechanism for the pH-regulated PA-CMG2 binding and conformational change relevant to pore formation.
One consistent finding is that PAII orients its β3−β4 loop under neutral condition so that Arg-344PA stabilizes a salt bridge formed with the conserved receptor amino acid Glu-122CMG2. The breakage of this salt bridge was detected as the key event during both mechanical unbinding via applying external force and chemical unbinding through lowering pH. The peak pulling force for detaching PAII from the complex corresponds to abolishing the salt bridge. In simulations of low pH structures, the salt bridge became disrupted before PAII dissociated from the binding site. Therefore, the Arg-344PA-Glu-122CMG2 salt bridge is identified as the key contributor to the functionally relevant PAII-CMG2 binding interaction.
Our simulations also suggest that the protonation of His-121CMG2 alone only slightly alters the configuration of Arg-344PA, which remains capable of forming a salt bridge with Glu-122CMG2. The observation that the Arg-344PA-Glu-122CMG2 salt bridge is stably reformed after the initial interruption upon protonating His-121CMG2 implies that additional structural changes related to the low pH condition are required for releasing PAII. However, since the accessible timescale of MD simulations is limited to tens of nanoseconds, we cannot rule out the possibility that PAII dissociates from CMG2 on a much longer timescale upon the protonation of only His-121CMG2. As an alternative, we propose that Glu-122CMG2 also plays a role in triggering the dissociation of PAII under low pH conditions. First, the protonation of the Glu-122CMG2 carboxylate side chain eliminates the salt bridge, greatly weakening the interactions between PAII and CMG2. Second, the protonation of Glu-122CMG2 creates the solvent accessibility for protonating His-121CMG2, the titratable nitrogen on the tip of His-121CMG2 being buried by the salt bridge and inaccessible to solvent as long as the salt bridge is formed. The protonation of both His-121CMG2 and Glu-122CMG2 initiates rapid dissociation of PAII.
Although the anthrax receptor CMG2 I domain resembles integrin αA/I domains adopting a homologous MIDAS binding motif (23), binding assays have shown that the affinity constant KD associated with the binding between anthrax toxin PA and its receptor CMG2 is ∼200 pM (25), at least 1000-fold higher than KD for integrin-ligand complexes (26). To explain the extremely high affinity of the anthrax-receptor complex, additional binding interactions between PA and CMG2 have been postulated. Indeed, along with the MIDAS interactions between PAIV and the receptor, a second binding site between PAII and CMG2 was unveiled in the crystal structures of the complex (18,27). Although the configuration of the MIDAS motif on CMG2 exhibits the same “open” form as typical integrin αA/I domains (26,46,47), the second binding site is conserved only between PA receptors but not seen in integrin αA/I domains. Consistent with previous biochemical and crystallography data, our SMD study demonstrates that PA-CMG2 is a very stable complex as a strong force is required to rupture it. In particular, the PAII-CMG2 binding contributes significantly to the stability of the complex. Forced separation of PAII in the equilibrated neutral complex results in a force peak twice that of detaching PAII in the acidic SHE complex where PAII became partially dissociated before the stretching. The force peak detected in the latter case is similar to the maximum force required for destabilizing an integrin αVβ3-ligand complex investigated previously (31). However, we note that a different type of cation (Mn2+ instead of Mg2+) was adopted in the previous SMD study and that integrin αVβ3 belongs to a subset of integrins where the αA/I domain is absent and the binding to its ligand is achieved via a homologous MIDAS ion on its βA/I domain.
The MIDAS interactions play a major role in stabilizing the whole complex because the peak force for separating CMG2 from the ligand consistently correlates with the disruption of the MIDAS contacts. The SMD simulations further revealed a mechanism by which the PA-CMG2 complex achieves a high affinity state through maintaining the configuration of the MIDAS coordination sphere. On CMG2 the MIDAS site is linked to the PAII binding site through the loop β4−α4. The PAII-CMG2 interactions restrict the movement of the β4−α4 loop bearing Asp-148CMG2, which acts with Asp-50CMG2 to firmly seize one of two water molecules coordinating the MIDAS cation. The coordination is important for stabilizing the PA-CMG2 complex as mutating Asp-50CMG2 to alanine eliminates PA binding (23). When the PAII-CMG2 binding was weakened in the SHE structure, loop β4−α4 became more flexible, and the coordination to the water molecule was easily disrupted as Asp-148CMG2 was stretched away, followed by the breakage of the Asp-50CMG2-water contact. The water molecule W1 subsequently attacked one of the remaining receptor-MIDAS ion contacts, resulting in a much lower peak force than that required for disrupting the intact MIDAS coordination sphere exhibited in the neutral structure. It has been shown that the tight coordination of water molecules by the divalent MIDAS cation shields the attack of free water molecules to the motif residues, thus stabilizing the binding interactions (31). Extending this conclusion, our study now provides an illustration of how the tight coordination of a MIDAS water molecule by both the MIDAS ion and the receptor dramatically enhances the MIDAS interactions.
Many toxin proteins secreted by bacteria initiate their invasion through a pore inserted into the membrane of a host cell (48). The pH-regulated tight binding between the anthrax toxin and its receptor delays pore formation until the complex has been taken up by the host cell and translocated to a destination close to its targets. The MD simulations conducted by us provide structural insights into how the anthrax-receptor complex stabilizes itself and how the stability is weakened through protonation of a few amino acids. We have shown that the binding between PAII and CMG2 contributes significantly to the overall stability of the complex. The PAII-CMG2 binding is regulated by His-121CMG2 and Glu-122CMG2, two conserved amino acids at the binding pocket of the receptor. The protonation of both residues disrupts a salt bridge characterizing the binding stability, which is necessary for releasing PAII for the purpose of pore formation.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
This work was supported by the National Institutes of Health grant PHS5P41RR05969 and National Science Foundation supercomputer time grant NRAC MCA93S028.
References
- 1.Collier, R. J., and J. A. T. Young. 2003. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 19:45–70. [DOI] [PubMed] [Google Scholar]
- 2.Scobie, H. M., and J. A. T. Young. 2005. Interactions between anthrax toxin receptors and protective antigen. Curr. Opin. Microbiol. 8:106–112. [DOI] [PubMed] [Google Scholar]
- 3.Moayeri, M., and S. H. Leppla. 2004. The roles of anthrax toxin in pathogenesis. Curr. Opin. Microbiol. 7:19–24. [DOI] [PubMed] [Google Scholar]
- 4.Bradley, K. A., J. Mogridge, M. Mourez, R. J. Collier, and J. A. T. Young. 2001. Identification of the cellular receptor for anthrax toxin. Nature. 414:225–229. [DOI] [PubMed] [Google Scholar]
- 5.Scobie, H. M., G. J. A. Rainey, K. A. Bradley, and J. A. T. Young. 2003. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA. 100:5170–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beauregard, K. E., R. J. Collier, and J. A. Swanson. 2000. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell. Microbiol. 2:251–258. [DOI] [PubMed] [Google Scholar]
- 7.Klimpel, K. R., S. S. Molloy, G. Thomas, and S. H. Leppla. 1992. Anthrax toxin protective antigen is activated by a cell-surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA. 89:10277–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milne, J. C., D. Furlong, P. C. Hanna, J. S. Wall, and R. J. Collier. 1994. Anthrax protective antigen forms oligomers during intoxication of mammalian-cells. J. Biol. Chem. 269:20607–20612. [PubMed] [Google Scholar]
- 9.Mogridge, J., K. Cunningham, and R. J. Collier. 2002. Stoichiometry of anthrax toxin complexes. Biochemistry. 41:1079–1082. [DOI] [PubMed] [Google Scholar]
- 10.Abrami, L., S. H. Liu, P. Cosson, S. H. Leppla, and F. G. van der Goot. 2003. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 160:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon, V. M., S. H. Leppla, and E. L. Hewlett. 1988. Inhibitors of receptor-mediated endocytosis block the entry of Bacillus-anthracis adenylate-cyclase toxin but not that of bordetella-pertussis adenylate-cyclase toxin. Infect. Immun. 56:1066–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blaustein, R. O., T. M. Koehler, R. J. Collier, and A. Finkelstein. 1989. Anthrax toxin—channel-forming activity of protective antigen in planar phospholipid-bilayers. Proc. Natl. Acad. Sci. USA. 86:2209–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milne, J. C., and R. J. Collier. 1993. pH-dependent permeabilization of the plasma-membrane of mammalian-cells by anthrax protective antigen. Mol. Microbiol. 10:647–653. [DOI] [PubMed] [Google Scholar]
- 14.Petosa, C., R. J. Collier, K. R. Klimpel, S. H. Leppla, and R. C. Liddington. 1997. Crystal structure of the anthrax toxin protective antigen. Nature. 385:833–838. [DOI] [PubMed] [Google Scholar]
- 15.Benson, E. L., P. D. Huynh, A. Finkelstein, and R. J. Collier. 1998. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 37:3941–3948. [DOI] [PubMed] [Google Scholar]
- 16.Nassi, S., R. J. Collier, and A. Finkelstein. 2002. PA(63) channel of anthrax toxin: an extended beta-barrel. Biochemistry. 41:1445–1450. [DOI] [PubMed] [Google Scholar]
- 17.Miller, C. J., J. L. Elliott, and R. J. Collier. 1999. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 38:10432–10441. [DOI] [PubMed] [Google Scholar]
- 18.Lacy, D. B., D. J. Wigelsworth, R. A. Melnyk, S. C. Harrison, and R. J. Collier. 2004. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. USA. 101:13147–13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drum, C. L., S. Z. Yan, J. Bard, Y. Q. Shen, D. Lu, S. Soelalman, Z. Grabarek, A. Bohm, and W. J. Tang. 2002. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature. 415:396–402. [DOI] [PubMed] [Google Scholar]
- 20.Pannifer, A. D., T. Y. Wong, R. Schwarzenbacher, M. Renatus, C. Petosa, J. Bienkowska, D. B. Lacy, R. J. Collier, S. Park, S. H. Leppla, P. Hanna, and R. C. Liddington. 2001. Crystal structure of the anthrax lethal factor. Nature. 414:229–233. [DOI] [PubMed] [Google Scholar]
- 21.Lacy, D. B., D. J. Wigelsworth, H. M. Scobie, J. A. T. Young, and R. J. Collier. 2004. Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA. 101:6367–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimaoka, M., J. Takagi, and T. A. Springer. 2002. Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 31:485–516. [DOI] [PubMed] [Google Scholar]
- 23.Bradley, K. A., J. Mogridge, G. J. A. Rainey, S. Batty, and J. A. T. Young. 2003. Binding of anthrax toxin to its receptor is similar to alpha integrin-ligand interactions. J. Biol. Chem. 278:49342–49347. [DOI] [PubMed] [Google Scholar]
- 24.Rosovitz, M. J., P. Schuck, M. Varughese, A. P. Chopra, V. Mehra, Y. Singh, L. M. McGinnis, and S. H. Leppla. 2003. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J. Biol. Chem. 278:30936–30944. [DOI] [PubMed] [Google Scholar]
- 25.Wigelsworth, D. J., B. A. Krantz, K. A. Christensen, D. B. Lacy, S. J. Juris, and R. J. Collier. 2004. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J. Biol. Chem. 279:23349–23356. [DOI] [PubMed] [Google Scholar]
- 26.Shimaoka, M., T. Xiao, J. H. Liu, Y. T. Yang, Y. C. Dong, C. D. Jun, A. McCormack, R. G. Zhang, A. Joachimiak, J. Takagi, J. H. Wang, and T. A. Springer. 2003. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 112:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santelli, E., L. A. Bankston, S. H. Leppla, and R. C. Liddington. 2004. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 430:905–908. [DOI] [PubMed] [Google Scholar]
- 28.Isralewitz, B., M. Gao, and K. Schulten. 2001. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11:224–230. [DOI] [PubMed] [Google Scholar]
- 29.Grubmuller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- 30.Izrailev, S., S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten. 1997. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72:1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Craig, D., M. Gao, K. Schulten, and V. Vogel. 2004. Structural insights into how the MIDAS ion stabilizes integrin binding to an RGD peptide under force. Structure. 12:2049–2058. [DOI] [PubMed] [Google Scholar]
- 32.Craig, D., M. Gao, K. Schulten, and V. Vogel. 2004. Tuning the mechanical stability of fibronectin type III modules through sequence variations. Structure. 12:21–30. [DOI] [PubMed] [Google Scholar]
- 33.Gao, M., D. Craig, O. Lequin, I. D. Campbell, V. Vogel, and K. Schulten. 2003. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc. Natl. Acad. Sci. USA. 100:14784–14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bayas, M. V., K. Schulten, and D. Leckband. 2004. Forced dissociation of the strand dimer interface between C-cadherin ectodomains. Mech. Chem. Biosyst.. 1:101–111. [PubMed] [Google Scholar]
- 35.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 37.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 38.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacKerell, A. D., D. Bashford, M. Bellot, R. L. Dunbrack Jr., J. Evansec, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kucknir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrick, J. Smith, R. Sote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-hydrogen empirical potential for molecular modeling and dynamics studies of proteins using the CHARMM22 force field. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 40.Feller, S. E., Y. H. Zhang, R. W. Pastor, and B. R. Brooks. 1995. Constant-pressure molecular-dynamics simulation—the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- 41.Schlick, T., R. D. Skeel, A. T. Brunger, L. V. Kale, J. A. Board, J. Hermans, and K. Schulten. 1999. Algorithmic challenges in computational molecular biophysics. J. Comput. Phys. 151:9–48. [Google Scholar]
- 42.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald—an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 43.Gordon, J. C., J. B. Myers, T. Folta, V. Shoja, L. S. Heath, and A. Onufriev. 2005. H++: a server for estimating pK(a)s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33:W368–W371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bashford, D., and K. Gerwert. 1992. Electrostatic calculations of the Pka values of ionizable groups in bacteriorhodopsin. J. Mol. Biol. 224:473–486. [DOI] [PubMed] [Google Scholar]
- 45.Rainey, G. J. A., D. J. Wigelsworth, P. L. Ryan, H. M. Scobie, R. J. Collier, and J. A. T. Young. 2005. Receptor-specific requirements for anthrax toxin delivery into cells. Proc. Natl. Acad. Sci. USA. 102:13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley, J., C. G. Knight, R. W. Farndale, M. J. Barnes, and R. C. Liddington. 2000. Structural basis of collagen recognition by integrin alpha 2 beta 1. Cell. 101:47–56. [DOI] [PubMed] [Google Scholar]
- 47.Lee, J. O., L. A. Bankston, M. A. Arnaout, and R. C. Liddington. 1995. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure. 3:1333–1340. [DOI] [PubMed] [Google Scholar]
- 48.Parker, M. W., and S. C. Feil. 2005. Pore-forming protein toxins: from structure to function. Prog. Biophys. Mol. Biol. 88:91–142. [DOI] [PubMed] [Google Scholar]