

Abstract
Fluorocarbon gases (gFCs) were found to inhibit the liquid-expanded (LE)/liquid-condensed (LC) phase transition of dipalmitoyl phosphatidylcholine (DPPC) Langmuir monolayers. The formation of domains of an LC phase, which typically occurs in the LE/LC coexistence region upon compression of DPPC, is prevented when the atmosphere above the DPPC monolayer is saturated with a gFC. When contacted with gFC, the DPPC monolayer remains in the LE phase for surface pressures lower than 38 mN m−1, as assessed by compression isotherms and fluorescence microscopy (FM). Moreover, gFCs can induce the dissolution of preexisting LC phase domains and facilitate the respreading of the DPPC molecules on the water surface, as shown by FM and grazing incidence x-ray diffraction. gFCs have thus a highly effective fluidizing effect on the DPPC monolayer. This gFC-induced fluidizing effect was compared with the fluidizing effect brought about by a mixture of unsaturated lipids and proteins, namely the two commercially available lung surfactant substitutes, Curosurf and Survanta, which are derived from porcine and bovine lung extracts, respectively. The candidate FCs were chosen among those already investigated for biomedical applications, and in particular for intravascular oxygen transport, i.e., perfluorooctyl bromide, perfluorooctylethane, bis(perfluorobutyl)ethene, perfluorodecalin, and perfluorooctane. The fluidizing effect is most effective with the linear FCs. This study suggests that FCs, whose biocompatibility is well documented, may be useful in lung surfactant substitute compositions.
INTRODUCTION
This work reports an investigation of the effects of the adsorption of fluorocarbon gases (gFCs) on the structure and behavior upon compression and expansion of a dipalmitoyl phosphatidylcholine (DPPC) monolayer, taken as a simplified model of lung surfactant (LS), with the objective of determining the potential of fluorocarbons (FCs) for new LS compositions. Due to their high biological inertness, remarkable ability to solubilize oxygen, and extremely low solubility in water, FCs have been investigated for various biological applications (1), in particular for intravascular oxygen transport (2) and for the stabilization of gaseous microbubbles used as contrast agent in ultrasound imaging (3). Partial liquid ventilation (PLV) with FCs has been explored as a treatment of acute respiratory distress syndrome (ARDS). Improved oxygenation and lung compliance were achieved in preterm animal models (4) as well as in premature infants (5,6). FC-based PLV was also reported to have an antiinflammatory effect in the alveolar environment of trauma patients (7). The fact that FCs attenuate the proinflammatory and procoagulatory responses of activated monocytes and of alveolar macrophages may contribute to the protective role of FCs in injuries associated with local activation of inflammatory processes (8). Delivery of vaporized FCs to oleic acid-injured ARDS sheep resulted in significant and sustained improvements of gas exchange and of lung compliance (9,10). Although these results suggest that FCs may be useful in pulmonary disease therapy, no study aiming at determining the influence of FCs on LS or LS models appears to have been reported so far, to our knowledge.
The native LS is a complex mixture of lipids and proteins that forms a monolayer at the alveolus/air interface of mammalian species (11,12). LS is secreted into the alveolar space by epithelial type II pneumocytes via exocytosis (13,14). It contains several lipids, primarily DPPC, small fractions of polyunsaturated fatty-acid-containing phospholipids (PUFA-PL), anionic PLs such as phosphatidylglycerols (PGs), neutral lipids such as diacyl- and triacylglycerols, free fatty acids such as palmitic acid (PA), plasmalogens, and cholesterol (15). LS also contains four specific proteins (SP-A, SP-B, SP-C, and SP-D). One key role of LS is to form a monolayer that lowers the air/alveoli surface tension upon compression (i.e., during expiration), reduces the work of breathing, and respreads easily on expansion (i.e., during inspiration) (16,17).
The LS function has been described by the “squeeze out” model (18). Upon compression, the minor components (predominantly nonphosphatidylcholine compounds) are squeezed out from the monolayer, which results in an enrichment of the monolayer in phosphatidylcholine compounds. Surface tension is then reduced and alveolar collapse is prevented. However, the physicochemical properties of the major surfactant-PL fraction were not in accordance with the predicted requirements of this model (19). A new concept involving a “surface-associated reservoir” was suggested as an improved model (20,21). In this model of surfactant function, layers of LS are folded in the subphase and remain in contact with the monolayer at the air/liquid interface. One key role of the minor components of LS is to decrease the surface viscosity of the monolayer (19). SP-B and SP-C are small amphiphilic proteins that were recently shown to play an important role in the surface activity of the LS (22,23). Recent work has focused on designing lipid mixtures that combine the surface activity of PLs (and neutral lipids) and the fluidizing properties of plasmalogens, PUFA-PL, and SP-B (15).
Although DPPC can generate near-zero surface tension at the air/water interface during compression, it is a poor LS when used alone. This is because it tends to form rigid, multilamellar structures in solution and does not adsorb efficiently at the air/water interface. When present at the interface, DPPC forms a monolayer that is in the semicrystalline liquid-condensed (LC) state (24). Moreover, DPPC does not respread upon expansion because of the formation of stable, two-dimensional crystalline domains at the air/water interface.
Several LS substitutes (for example, Curosurf (Chiesi Pharmaceutici, Parma, Italy) and Survanta (Ross Laboratories, Columbus, OH)) have been developed and are being clinically used in the treatment of neonatal respiratory distress syndrome (NRDS). These replacement LS consist of purified preparations of bovine (Survanta) or porcine (Curosurf) LS. Being natural extracts, these preparations are, however, not devoid of potential viral contamination and inherent immunological risks. Other drawbacks include a costly purification procedure and the difficulty of achieving batch-to-batch consistency. Therefore, there is a clear need for alternative synthetic LS substitutes (25).
We found that FC gases have a strong effect on the physical state of DPPC Langmuir monolayers upon compression. Langmuir monolayers provide useful model systems for studying LS. Great care is necessary when extrapolating Langmuir monolayer behavior to LS behavior in vivo. General correlations between in vitro and in vivo behavior start, however, to emerge (26). The behavior of surfactants in monolayers is characterized by surface pressure – molecular area (π – A) compression isotherms. Compression isotherms of DPPC monolayers were measured under an atmosphere saturated with various gFCs. Fluorescence microscopy (FM) and grazing incidence x-ray diffraction (GIXD) were used to determine the presence, morphology, and degree of order of the organized domains within the monolayers. Because FCs have low intermolecular cohesiveness, their vapor pressures are high with regard to their molecular weights (27). The FCs selected for this study, perfluorooctyl bromide (PFOB), perfluorooctylethane (PFOE), bis(perfluorobutyl)ethene (F-44E), perfluorodecalin (FDC), and perfluorooctane (PFO), were chosen among those most thoroughly investigated for biomedical applications, in particular for intravascular oxygen transport (2). Both the ability of gFCs to prevent the formation of crystalline DPPC domains and their ability to dissolve such domains once formed were determined. We have also compared the behavior of DPPC monolayers contacted with gFCs with that of monolayers of the commercial LS substitutes Curosurf and Survanta.
MATERIALS AND METHODS
Materials
PFOB and PFOE were kindly provided by Alliance Pharmaceutical (San Diego, CA), FDC by Air Products (Allentown, PA), and F-44E by DuPont (Wilmington, DE). PFO and l-α-1,2-dipalmitoyl-sn-3-glycero-phosphatidylcholine (DPPC, purity >99%) were purchased from Sigma (St. Louis, MO). Samples of Curosurf and Survanta were a gift from Prof. J. Messer (Service de Néonatalogie, Hôpital de Hautepierre, Strasbourg, France). Water was purified using a Millipore system (pH = 5.5; surface tension: 72.1 mN m−1 at 20°C, resistivity: 18 MΩ cm). The fluorescent dye (2-[6-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino] hexanoyl-1-hexadecanoyl-sn-glycero-3-phosphocholine, NBDC6-HPC) was purchased from Molecular Probes (Eugene, OR).
Methods
Compression of DPPC monolayers under FC gases
Surface pressure, π, versus molecular area, A, isotherms were recorded on a Langmuir minitrough (Riegler & Kirstein, Potsdam, Germany) equipped with two movable barriers (speed: 2.0 mm min−1, which corresponds to a reduction of the total area of 3% min−1). The surface pressure was measured using the Wilhelmy plate method. The trough was enclosed in a gas-tight box (volume = 9 L). The gas-tight box was flushed either with pure N2 or with N2 saturated with the chosen FC. In the latter case, N2 was allowed to bubble at room temperature through the liquid FC before being flushed into the gas-tight box. The flow rate of the gas phase (N2 or N2 saturated with the FC) was set to 1.2 L min−1 for PFOB, PFOE, F-44E, and FDC and 0.5 L min−1 for PFO. The evaporation rate of these FCs was then ∼6 mL h−1. The temperature measured inside the gas-tight box was 26°C ± 0.5°C. The errors on π and A were estimated to be ± 1 mN m−1 and ± 1 Å2, respectively. The molecular formula and physical characteristics of the FCs investigated are collected in Table 1. Spreading solutions of DPPC (1.0 mmol L−1 for minitrough experiments and 2.0 mmol L−1 for GIXD experiments) were prepared in chloroform (analytical grade). A total of 15 μL of DPPC solution was spread on the water surface.
TABLE 1.
Structural formula and physical characteristics of the FCs investigated (from Riess (2))
| Structural formula | Code name (MW) | Boiling point (°C) | Vapor pressure (torr, 37°C) | *CST in hexane (°C) |
|---|---|---|---|---|
| CF3(CF2)7Br | PFOB (499) | 143 | 10.5 | −20 |
| CF3(CF2)7CH2CH3 | PFOE (448) | 164 | 11.5 | −8 |
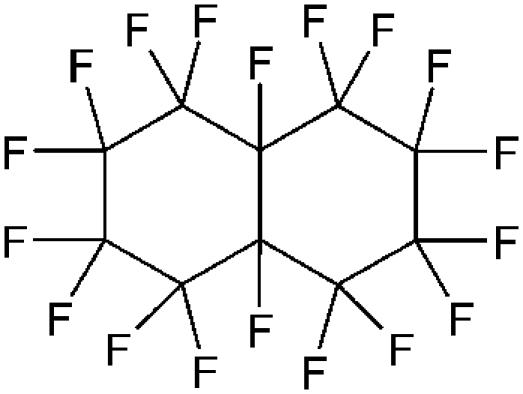 |
FDC (462) | 142 | 13.5 | 22 |
| CF3(CF2)3CH=CH(CF2)3CF3 | F-44E (464) | 150 | 12.5 | 19.5 |
| CF3(CF2)6CF3 | PFO (438) | 102 | 52–64 | † |
Critical solution temperature (CST).
Not determined.
Compression of monolayers of Curosurf and Survanta
Curosurf and Survanta are available in the form of aqueous dispersions. The samples were frozen to −80°C, transferred to the lyophilizer (Flexi-Dry, FTS Systems Inc., Stone Ridge, NY) and lyophilized at −30°C and 20 Pa for 18 h. The lyophilized samples were then solubilized in chloroform at the concentration of 1 mg mL−1. No correction was made to take into account the amount of salts present in Curosurf and Survanta. In addition to the lipids and proteins (Table 2), which have a minimal solubility in water, Curosurf and Survanta contain several components that are soluble in the subphase. As a consequence, the values of the extrapolated areas of the isotherms depend on the amount deposited on the water surface. A total of 12 μL of each surfactant solution was spread on the surface.
TABLE 2.
Compositions of Curosurf and Survanta
Fluorescence microscopy
FM was achieved with the above balance equipped with an Olympus (Tokyo, Japan) fluorescence microscope (20× power objective) mounted on an x, y translation stage to allow scanning of the trough over different regions. An Olympus 100 W high-pressure mercury lamp was used for excitation. A dichroic mirror/barrier filter assembly was used to filter and direct the excitation light onto the monolayer (450–490 nm) and to filter out the emitted fluorescence (520 nm). The emitted fluorescence was collected by the objective and detected via a Hamamatsu (Hamamatsu City, Japan) intensified camera. The microscope was linked to the gas-tight box of the trough through an extensible gusset, allowing easy control of the partial pressure of the FC. The surface pressure was kept constant during FM experiments. The fluorescent dye NBDC6-HPC was used at a lipid mole ratio of 1% (1 × 10−5 mol L−1).
Grazing incidence x-ray diffraction
GIXD experiments were achieved at the D41B beamline of the LURE-DCI synchrotron source (Orsay, France). The x-ray wavelength λ = 1.646 Å of the incoming x-ray beam was selected using a focusing Ge (111) monochromator. The monocrystalline surface of a Ge plate (〈111〉) is used to select a monochromatic beam of wavelength 1.646 Å from the white synchrotron beam (28). The grazing angle of incidence θi = 2.08 mrad was set slightly below the critical angle for total external reflection of the x-rays at the air/water interface (∼2.8 mrad at 1.646 Å). For the acquisition of the diffracted intensity, we used a new setup composed of a two-dimensional detector and a single vertical slit positioned between the sample and the detector (28). The resulting qxy resolution was 0.007 Å−1 for the q-range explored here. The shape of the Bragg rods gave information about the tilt angle t and tilt azimuth φ (29). In the following, the rectangular description of the chain lattice will be used (30). The diffraction pattern exhibits two peaks. The peak located at low qxy corresponds to the degenerate [11] and [ī1] out-of-plane reflections, and the other peak to the [02] in-plane reflection. Since the maximum of intensity along the Bragg rods [11] and [ī1] is located out of the plane and in the scattering plane along the Bragg rod [02], the chains are tilted to the nearest neighbor. Thus, the observed phase is L2d (t ≠ 0, φ = 0), according to the nomenclature introduced in Kaganer et al. (30).
The Teflon Langmuir trough mounted on the diffractometer was equipped with a movable single barrier. The surface pressure, π, measured using the Wilhelmy plate method, was kept constant throughout a given scan. The trough was enclosed in a gas-tight box with Kapton windows flushed with water-saturated helium. Helium is the only gas that exerts sufficiently low absorption and scattering of the incident and diffracted x-ray beam to allow measurement of the diffraction of the monolayer at the interface. It is noteworthy that the diffraction and scattering level by N2 is too high to enable the diffraction measurement. When appropriate, helium was saturated with the chosen FC. With this chamber, the partial pressure of the FC was readily controlled. The temperature was regulated to 20°C ± 0.5°C. A total of ∼ 50 μL of DPPC solution were spread on the water surface. The film was compressed stepwise, and Bragg peaks were recorded at each set pressure step. The total duration of a scan was typically 10 min.
RESULTS
Compressing a Langmuir monolayer of DPPC under an atmosphere saturated with gFCs
The isotherms of the DPPC monolayer compressed on a pure water subphase in an atmosphere of air or N2 or in an atmosphere of N2 saturated with PFOB, PFOE, PFO, F-44E, or FDC are shown in Fig. 1. The whole range of surface pressures, from 0 to 72 mN m−1, was investigated (see inset in Fig. 1). This corresponds to surface tensions from 72 to 0 mN m−1 and includes the physiological surface tensions.
FIGURE 1.

Compression isotherms of DPPC on water under an atmosphere of (1) air (25°C), (2) N2 (26°C), and (3) N2 saturated with various FCs at 26°C. No significant difference was observed on the isotherms of the FCs investigated (see Table 1). The letters (a, b, c, a′, b′, and c′) correspond to the surface pressures at which the monolayers have been imaged with FM (see Fig. 2). (Inset) Compression isotherms of DPPC under an atmosphere of air (dashed line) and under an atmosphere of N2 saturated with gPFOB (solid line). Near zero surface tensions can be achieved with the DPPC monolayer contacted with gPFOB.
When compressed under ambient air (without the gas-tight box) at 25°C ± 0.5°C, the DPPC isotherm presented the characteristic first-order transition from the disordered LE phase to the ordered LC phase (31,32). The phase transition surface pressure, πeq, at a given temperature is a characteristic quantity for a given lipid in a monolayer on a given subphase. The πeq value determined for DPPC from the isotherm was ∼10 mN m−1. This value is in agreement with the literature, which reports for DPPC on pure water, a πeq value of ∼4 mN m−1 at 20°C shifted by ∂πeq/∂T ∼ + 1.5 mN m−1 K−1, until a tricritical point is reached at Tt ∼43°C (32). Enclosing the trough in the gas-tight box resulted in an increase of temperature of ∼1°C (26°C ± 0.5°C), which results in an increase of πeq to ∼13 mN m−1. The LE/LC coexistence region is identified by the observation, by FM, of discrete domains of LC phase within a continuous LE phase (Fig. 2). When π increases, the LC domains increase in size, become more numerous, and progressively merge into a continuous LC phase.
FIGURE 2.

Fluorescence micrographs of DPPC under pure N2 (left panel) and of DPPC compressed under an N2 atmosphere saturated with gPFOB in a gas-tight box (right panel). The images were obtained using the fluorescent dye NBDC6-HPC, which is preferentially soluble in the disordered regions of the monolayer (39). As a result, LE regions appear bright, whereas the LC domains appear dark. The temperature was 26°C, and the subphase was pure water. The lateral pressures were (a) 2 mN m−1, (b) 13 mN m−1, (c) 20 mN m−1, (a′) 13 mN m−1, (b′) 20 mN m−1, and (c′) 35 mN m−1. The gaseous FC present above the monolayer prevents the formation of LC domains.
Compressing the DPPC monolayer under a N2 atmosphere saturated with a gFC changes the phase behavior drastically. The LE/LC transition at ∼13 mN m−1 (Fig. 1) disappears. The compression isotherm is now characterized by two kinks at ∼28 and ∼38 mN m−1. Below ∼38 mN m−1, the isotherm is shifted toward larger molecular areas, which indicates that FC molecules are incorporated into the DPPC monolayer. It is also noteworthy that, even at the beginning of the compression, the surface pressure is not zero as in the case of pure DPPC, but 2–4 mN m−1, which also shows that FC molecules are inserted into the DPPC monolayers.
The transition observed at ∼28 mN m−1 is no longer of the LE/LC type, as assessed by the fluorescence images that are bright and featureless (Fig. 2), not only at ∼28 mN m−1, but up to ∼35 mN m−1 (Fig. 2 c′). This suggests a conformational change of the FC molecules inserted in the DPPC monolayer. For π higher than ∼38 mN m−1, the isotherm becomes steeper, and the limiting area (∼50 Å2) is then very comparable to the limiting area of the DPPC monolayer compressed in the absence of gFC. This indicates that the FC molecules are progressively squeezed out on the top of the DPPC monolayer. At such high surface pressures, FM images show the presence of very small crystalline domains, suggesting that the LE/LC transition occurred at ∼38 mN m−1. However, for these π values, the images are not focused, probably due to the presence of PFOB molecules expelled from the DPPC monolayer, which likely form a thin liquid film with small droplets on top of the monolayer. It is noteworthy that for π higher than 38 mN m−1, i.e., after squeeze out of the FC molecules, only small LC domains are seen as compared to the essentially continuous LC phase observed for DPPC under pure N2 at the same π values. It is likely that the presence of a thin liquid FC film on top of the DPPC monolayer also disorganizes the DPPC molecules and contributes to the fluidization process at high surface pressures. Finally, it should be noticed that the presence of the gFC above the DPPC monolayer does not destabilize it.
The collapse surface pressure of the DPPC monolayer in contact with gFC is ∼71 mN m−1 (see inset in Fig. 1), i.e., is identical to the collapse surface pressure of the DPPC monolayer in the absence of gFC. This means that combinations of DPPC and gFC allow reaching very low surface tension values, which is necessary if gFCs are to be used in exogenous LS compositions.
These experiments demonstrate that gFC molecules interact with DPPC molecules and prevent the formation of the LC phase until high values of lateral pressure and hence induce a fluidizing effect in the monolayer.
An important issue with regard to the potential LS replacement application was to ensure that there is no delipidating effect of gPFOB on the DPPC monolayer. Fig. 3 represents the compression/expansion isotherms of DPPC in an atmosphere of N2 saturated with gPFOB. It shows clearly that very few DPPC molecules have desorbed from the air/water interface during the compression/expansion cycle. However, the shape of the expansion isotherm has significantly changed. Although the compression isotherm was characterized by two kinks at 28 and 38 mN m−1, only the latter kink was observed during expansion. The plateau observed around 38 mN m−1 is more clearly visible than during compression, and the expansion isotherm is much more expanded than the compression isotherm. FM images clearly show the presence of very small crystalline domains for pressures higher than 38 mN m−1 (data not shown). For π values lower than 38 mN m−1, the images were bright and featureless, demonstrating that the monolayer was in a homogeneous LE phase (Fig. 2). This confirms that the LE/LC phase transition occurs at ∼38 mN m−1 for the DPPC monolayer in contact with gPFOB.
FIGURE 3.

Compression isotherms of DPPC (26°C) on a pure water subphase. The first compression (solid line) and subsequent expansion (dotted line) were achieved under an atmosphere of N2 saturated with gPFOB. gPFOB was then evacuated for 2 h by flushing with N2. The second compression (dashed line) was obtained under an atmosphere of pure N2.
The fact that the kink at 38 mN m−1 is much more visible during expansion than during compression as well as the fact that the expansion isotherm is much more expanded than the compression isotherm clearly indicate that PFOB molecules that were squeezed out to the top of the DPPC monolayer during the first compression are reincorporated into the monolayer during expansion. It is not surprising that the change in orientation of the FC molecules that is evidenced upon compression by the kink at ∼28 mN m−1 is no longer visible during expansion.
We have designed another experiment aimed at detecting any delipidating effect of the gFC in which the gas-tight box that had contained gPFOB-saturated N2 was flushed with pure N2 after the first compression/expansion cycle. This allowed us to thoroughly remove gPFOB (Fig. 3). It was observed that the compression isotherm of the DPPC monolayer after removal of gPFOB is similar to that obtained when compression was achieved in the absence of gPFOB. This indicates that the PFOB molecules are easily removed from the DPPC monolayer by evaporation. It also establishes that the fluidizing effect of gPFOB is a reversible phenomenon. The fact that, during the second compression, the limiting area is nearly identical to that observed during the first compression further indicates that the presence of gPFOB molecules does not provoke any significant loss of DPPC molecules to the subphase.
Similar results were obtained with the other FCs investigated. All gFCs interacted with DPPC, thus preventing the formation of the LC phase and inducing a fluidizing effect in the monolayer. This fluidizing effect was always reversible; removal of the gFC by N2 flushing of the gas-tight box resulted in the restoration of the isotherm of a pure DPPC monolayer, with the same limiting molecular area, which further demonstrates that no significant amount of DPPC molecules had been desorbed by the gFC.
Effect of gFCs on preformed LC domains
To assess the effect of gFCs on the DPPC semicrystalline domains that are already formed, DPPC monolayers were first compressed to 13 mN m−1 and gFC-saturated N2 was then allowed to flow into the gas-tight box that encloses the trough. The fluorescence images of Fig. 4 clearly show that, 3 min after the introduction of gPFOB, the LC domains have become significantly smaller. After 7 min, these domains have totally disappeared, indicating that the DPPC monolayer has become totally fluid. When the DPPC monolayer was contacted with gPFOE at 13 mN m−1, total fluidization of the monolayer was observed after 5 min (Fig. 5).
FIGURE 4.

Fluorescence images of a DPPC monolayer compressed at 13 mN m−1 under N2 (upper left). At time t, the atmosphere of N2 above the monolayer was saturated with gPFOB. One can see that the LC domains progressively disappear over time. After 7 min, the monolayer is totally fluidized.
FIGURE 5.

Fluorescence images of a DPPC monolayer compressed at 13 mN m−1 under N2 (upper left). At time t, the atmosphere of N2 above the monolayer was saturated with gPFOE. The LC domains rapidly disappear. After 5 min, the monolayer is totally fluidized.
The fluidizing capacity of gFCs depends somewhat on the structure of the FC. PFO and PFOE were the most efficient in this respect, inducing total fluidization within ∼5 min. The effect of F-44 E was comparable to that of PFOB (total fluidization in ∼7 min). The effect was slower with FDC (fluidization required ∼10 min). The lesser fluidizing capacity of the bicyclic FDC may be assigned to lesser affinity for hydrocarbon oils, as indicated by a higher critical solubility temperature (CST) in hexane (Table 1) and to its globular, bulky molecular shape.
FM provides information on a local scale. Small crystalline domains present at the interface may escape from the investigation field and not be taken into account. To have a definitive, independent assessment of any possible crystalline regions present in the monolayer, we performed GIXD using synchrotron radiation. An x-ray beam that hits the surface of water at an incidence lower than the critical angle of water (∼2.5 mrad) is totally reflected. Under these conditions, an evanescent wave is formed that is scattered by the monolayer. If the monolayer is ordered (i.e., crystalline), the wave is diffracted and Bragg peaks are obtained. GIXD thus provides global information on the zones of the monolayer that are ordered.
Fig. 6 a shows the integrated diffracted intensity (I/I0) as a function of the in-plane component of the scattering wave vector (qxy) of a DPPC monolayer compressed at 20 mN m−1. At this surface pressure, two Bragg peaks were obtained, indicating a rectangular unit cell (NN tilted, L2d phase). The calculated area per chain of ∼23 Å2 and the tilt angle obtained from Bragg rod analysis are in agreement with published data for DPPC. When the He atmosphere was saturated with gPFOB, the diffraction peaks disappeared within a few minutes (Fig. 6 b), establishing the dissolution of the LC domains and the rapid respreading of the DPPC molecules.
FIGURE 6.

Intensity of the diffracted grazing x-rays, I/I0, as a function of the in-plane vector, qxy. For GIXD experiments, the DPPC monolayer was compressed under a helium atmosphere to reduce x-ray scattering. When compressed at 20 mN m−1, the DPPC monolayer shows the two Bragg peaks characteristic of the tilted LC phase of DPPC at 1.42 and 1.51 Å−1 (curve a). When the He atmosphere is saturated with gPFOB (curve b), the peaks disappear rapidly, establishing the dissolution of the crystalline LC domains and the rapid uniform respreading of the DPPC molecules.
When pure He was subsequently allowed to flow again into the gas-tight box, the compression of the DPPC monolayer being maintained at 20 mN m−1, the two diffraction peaks characteristics of DPPC slowly reappeared, establishing the reformation of the LC domains. This indicates that PFOB molecules are adsorbed at the DPPC monolayer and inhibit the crystallization of these DPPC molecules. When the flow of gPFOB is stopped, the PFOB molecules adsorbed at the interface are removed by evaporation and are then evacuated with the He flow.
It is noteworthy that total fluidization has not been obtained in the case of FDC (Fig. 7). The DPPC peak at qxy = 1.51 Å−1 is less intense but still visible even if the gas-tight box of the trough has been saturated with gFDC for over 1 h. This indicates that parts of the monolayer still retain some weak organization. The fact that FDC is not as efficient as the other FCs with respect to the DPPC monolayer fluidization may be ascribed to the globular shape of the FDC molecule, which is expected to be less favorable to insertion between the hydrophobic fatty chains of DPPC than the linear shape of the other FCs investigated.
FIGURE 7.

In contrast with Fig. 6, the Bragg peaks characteristic of the LC domains of DPPC (a) did not disappear totally when gaseous FDC was introduced in the gas-tight box instead of PFOB (b).
Comparison with Curosurf and Survanta
The compression isotherms of monolayers of the replacement LS Curosurf and Survanta are shown on Fig. 8. The Curosurf and Survanta formulations contain components that are soluble in the subphase. The nature and exact amounts of these soluble compounds being unknown, no relationship can be established between the deposited amount and the apparent area per molecule. However, the shapes of the isotherms (steep versus expanded) are characteristic of the degree of organization and fluidity of the monolayer. It is clearly seen that the compression isotherm of Survanta is steeper than that of Curosurf, in agreement with Ivanova et al. (33), demonstrating a lower compressibility (comparable to that of the LC phase of DPPC). A slight kink is observed for Curosurf at ∼28 mN m−1, and the isotherm becomes steeper at higher surface pressures. FM images (Fig. 9, left panel) indeed show that the Curosurf monolayer is in the fluid LE state, except at the highest lateral pressures (i.e., 45 mN m−1), for which some small crystalline domains can be observed (Fig. 9 c). By contrast, definite crystalline domains are already seen in the Survanta monolayer at low surface pressures, actually even at zero surface pressure for A = 80 Å2 (Fig. 9 a′). The number of these domains increases rapidly with π, until they practically form a continuous LC phase at 45 mN m−1 (Fig. 9 c′).
FIGURE 8.

Compression isotherms of (1) Curosurf and (2) Survanta as compared to those of pure DPPC compressed under N2 (3) and DPPC compressed under N2 + gPFOB (4) (at 26°C).
FIGURE 9.

Fluorescence images of a Curosurf monolayer (left panel) compressed at (a) 10 mN m−1, (b) 20 mN m−1, and (c) 45 mN m−1, and of a Survanta monolayer (right panel) at (a′) 0 mN m−1 (80 Å2), (b′) 10 mN m−1, and (c′) 45 mN m−1.
The difference in fluidity between Survanta and Curosurf may be related to the fact that the former has the lowest content in components that decrease the viscosity of the monolayer, such as plasmalogens, PUFA-PL, and SP-B (Table 2). In addition, Survanta has the highest concentration in disaturated PLs that tend to form crystalline islets and in cholesterol that tends to increase the monolayer's viscosity (15).
DISCUSSION
To our knowledge, this is the first time that such a drastic change in the physical state of PL monolayers, when contacted with a volatile oil, has been reported. This effect has been observed with several FCs. Liquid FCs have, among liquid oils, the highest vapor pressures relative to their molecular weight. This is a consequence of the liquid's low cohesiveness, consequent to the weakness of the van der Waals interactions between fluorinated chains. The observed fluidization of the DPPC monolayer may result from the progressive insertion of FC molecules into the DPPC monolayer. This may involve a mechanism comparable to that proposed for hydrocarbon oils contacting PL monolayers (34). It is likely that the FC condenses upon compression and forms a thin liquid film on top of the DPPC molecules. The FC molecules are reincorporated into the monolayer upon expansion. The fluidizing effect of gFCs that is established here results from the inhibition by the gFCs of the formation of an LC phase and from the fact that an LE phase is more fluid than a crystalline LC phase.
Improving our understanding of the interactions between FCs and PL interfaces is a key issue for the medical use of FCs since several FC-based systems are being developed (1). One can note the growing interest for gaseous microbubbles as contrast agents for ultrasound diagnosis (echosonography) and therapy (3,35). The most recent commercial microbubbles have a shell made of PLs and are osmotically stabilized by an FC gas (3). A synergistic effect between shell and internal gas components has recently been demonstrated (36).
We believe that the observations reported here are important because they suggest that a gFC/PLs combination could be the basis for the development of new synthetic LS replacement compositions. One advantage of this new approach is that the acting components (FC + DPPC) are synthetic. Another is that the fluidizing effect induced by gFCs is highly reproducible. During expansion (inspiration) the gFC molecules are likely inserted into the monolayer, thus preventing the formation of crystalline domains of DPPC and facilitating DPPC respreading. During compression (expiration), the FC molecules are expelled, leaving a DPPC-rich monolayer capable of producing near zero surface tension, but remain readily available, probably as a condensed thin liquid film on top of the DPPC monolayer. We have also established that the contact of the DPPC monolayer with gFC does not induce any detectable desorption of the PLs to the subphase. Such a mechanism of action would not require a “reservoir” effect provided by folded layers of PLs and proteins.
Replacing the air/water interface by a gFC-saturated air/water interface appears to have a significant impact on the adsorption of DPPC molecules at the interface. We have recently shown that the interfacial tension was much lower at a gPFOB/water interface than at the air/water interface and that the kinetics of adsorption of DPPC molecules at the interface were strongly accelerated (F. Gerber, M. Sanchez-Dominguez, T. F. Vandamme, and M. P. Krafft, unpublished). We can thus infer that, in vivo, the presence of gFC in the lung will help recruit DPPC molecules at the alveolar interface, thus decreasing the interfacial tension and improving the pulmonary function. This function would be combined and develop a synergy with the LS function, which will provide a further advantage over the commercial LS compositions.
CONCLUSIONS AND PERSPECTIVES
Fluorescence images and GIXD experiments provide definite evidence that contacting a DPPC monolayer with certain FC gases results in a very effective fluidization of the monolayer. FC gases prevent the formation of LC domains upon compression of the DPPC monolayer. FC gases are also able to induce the dissolution of LC domains when such domains are already constituted and thus facilitate the respreading of the DPPC molecules in an LE phase. This fluidizing effect is fully reversible. These results suggest that FC gases/lipid combinations may provide a useful basis for the design of novel and synthetic LS compositions. The administration of combinations of DPPC (or mixtures of lipids) and a gFC could be conveniently achieved using pressurized meter dose inhalers.
Acknowledgments
The authors thank Prof. J. Messer (Service de Néonatalogie, Hôpital Hautepierre, Strasbourg) for the gift of samples of Survanta and Curosurf, and Alliance Pharmaceutical (San Diego, CA) and DuPont (Wilmington, DE) for the gift of FCs.
References
- 1.Riess, J. G. 2004. Fluorous materials for biomedical uses. In Handbook of Fluorous Chemistry. J. A. Gladysz, I. Horváth, and D. P. Curran, editors. Wiley-VCH, Weinheim, Germany. 521–573.
- 2.Riess, J. G. 2001. Oxygen carriers (“Blood substitutes”)—Raison d'être, chemistry, and some physiology. Chem. Rev. 101:2797–2920. [DOI] [PubMed] [Google Scholar]
- 3.Schutt, E. G., D. H. Klein, R. M. Mattrey, and J. G. Riess. 2003. Injectable microbubbles as contrast agents for diagnostic ultrasound imaging: the key role of perfluorochemicals. Angew. Chem. Int. Ed. Engl. 42:3218–3235. [DOI] [PubMed] [Google Scholar]
- 4.Wolfson, M. R., N. E. Kechner, R. F. Roache, J. DeChadarevian, H. E. Friss, S. D. Rubenstein, and T. H. Shaffer. 1998. Perfluorochemical rescue after surfactant treatment: effect of perflubron dose and ventilatory frequency. J. Appl. Physiol. 84:624–640. [DOI] [PubMed] [Google Scholar]
- 5.Leach, C. L., J. S. Greenspan, D. Rubenstein, T. H. Shaffer, M. R. Wolfson, J. C. Jackson, R. DeLemos, and B. P. Fuhrman. 1996. Partial liquid ventilation with perflubron in premature infants with severe respiratory distress syndrome. N. Engl. J. Med. 335:761–766. [DOI] [PubMed] [Google Scholar]
- 6.Sukumar, M., M. Bommaraju, J. E. Fisher, F. C. Morin, M. C. Papo, B. P. Fuhrman, L. J. Hernan, and C. L. Leach. 1998. High-frequency partial liquid ventilation in respiratory distress syndrome: hemodynamics and gas exchange. J. Appl. Physiol. 84:327–334. [DOI] [PubMed] [Google Scholar]
- 7.Croce, M. A., T. C. Fabian, J. H. J. Patton, S. M. Melton, M. Moore, and L. Trenthem. 1998. Partial liquid ventilation decreases the inflammatory response in the alveolar environment of trauma patients. J. Trauma. 45:273–282. [DOI] [PubMed] [Google Scholar]
- 8.Koch, T., M. Ragaller, D. Haufe, A. Hofer, M. Grosser, D. M. Albrecht, M. Kotzsch, and T. Luther. 2001. Perfluorohexane attenuates proinflammatory and procoagulatory response of activated monocytes and alveolar macrophages. Anesthesiology. 94:101–109. [DOI] [PubMed] [Google Scholar]
- 9.Bleyl, J. U., M. Ragaller, U. Tschö, M. Regner, M. Kanzow, M. Hübler, S. Rasche, and M. Albrecht. 1999. Vaporized perfluorocarbon improves oxygenation and pulmonary function in an ovine model of acute respiratory distress syndrome. Anesthesiology. 91:461–469. [DOI] [PubMed] [Google Scholar]
- 10.Bleyl, J. U., M. Ragaller, U. Tschö, M. Regner, M. Hübler, M. Kanzow, O. Vincent, and M. Albrecht. 2002. Changes in pulmonary function and oxygenation during application of perfluorocarbon vapor in healthy and oleic acid-injured animals. Crit. Care Med. 30:1340–1347. [DOI] [PubMed] [Google Scholar]
- 11.Zasadzinski, J. A., J. Ding, H. E. Warriner, F. Bringezu, and A. J. Waring. 2001. The physics and physiology of lung surfactants. Curr. Opin. Colloid Interf. Sci. 6:506–513. [Google Scholar]
- 12.Hills, B. A. 1988. The Biology of Surfactant. Cambridge University Press, Cambridge.
- 13.Wright, J. R., and S. Hawgood. 1989. Pulmonary surfactant metabolism. Clin. Chest Med. 10:83–93. [PubMed] [Google Scholar]
- 14.Rooney, S. A. 2001. Regulation of surfactant secretion. Comp. Biochem. Physiol. A. 129:233–243. [DOI] [PubMed] [Google Scholar]
- 15.Rüdiger, M., A. Tölle, W. Meier, and B. Rüstow. 2005. Naturally derived commercial surfactants differ in composition of surfactant lipids and in surface viscosity. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L379–L383. [DOI] [PubMed] [Google Scholar]
- 16.Possmayer, F., S. H. Yu, J. M. Weber, and P. G. Harding. 1984. Pulmonary surfactant. Can. J. Biochem. Cell Biol. 62:1121–1133. [DOI] [PubMed] [Google Scholar]
- 17.Goerke, J. 1998. Pulmonary surfactant: functions and molecular composition. Biochim. Biophys. Acta. 1408:79–89. [DOI] [PubMed] [Google Scholar]
- 18.Clements, J. A., R. F. Hustead, R. P. Johnson, and I. Gribetz. 1961. Pulmonary surface tension and alveolar stability. J. Appl. Physiol. 16:444–450. [DOI] [PubMed] [Google Scholar]
- 19.Rüdiger, M., A. Tölle, W. Meier, and B. Rüstow. 2003. Effects of minor components of alveolar surfactant lipids on surface properties. In Recent Research Developments in Chemistry and Physics of Lipids. Transworld Research Network, Trivandrum, India. 1–14.
- 20.Post, A., A. V. Nahmen, M. Schmitt, J. Ruths, H. Riegler, M. Sieber, and H. J. Galla. 1995. Pulmonary surfactant protein C-containing lipid films at the air-water interface as a model for the surface of lung alveoli. Mol. Membr. Biol. 12:93–99. [DOI] [PubMed] [Google Scholar]
- 21.Schürch, S., R. Qanbar, H. Bachofen, and F. Possmayer. 1995. The surface-associated surfactant reservoir in the alveolar lining. Biol. Neonate. 67:61–76. [DOI] [PubMed] [Google Scholar]
- 22.Ding, J., D. Y. Takamoto, A. von Nahmen, M. M. Lipp, K. Y. C. Lee, A. J. Waring, and J. A. Zasadzinski. 2001. Effects of lung surfactant proteins, SP-B and SP-C, and palmitic acid on monolayer stability. Biophys. J. 80:2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takamoto, D. Y., M. M. Lipp, A. van Nahmen, K. Y. C. Lee, A. J. Waring, and J. A. Zasadzinski. 2001. Interaction of lung surfactant proteins with anionic phospholipids. Biophys. J. 81:153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding, J., I. Doudevski, H. E. Warriner, T. Alig, and J. A. Zasadzinski. 2003. Nanostructure changes in lung surfactant monolayers induced by interactions between palmitoyloleoylphosphatidylcholine and surfactant protein B. Langmuir. 19:1539–1550. [Google Scholar]
- 25.Spragg, R. G., P. Richman, N. Gilliard, and T. A. Merritt. 1987. The future for surfactant therapy of the adult respiratory distress syndrome. In International Symposium on Surfactant Replacement Therapy. B. Lachmann, editor. Springer-Verlag, Rotterdam, The Netherlands. 203–211.
- 26.Banerjee, R. 2002. Surface chemistry of the lung surfactant system: techniques for in vitro evaluation. Curr. Sci. 82:420–428. [Google Scholar]
- 27.Riess, J. G. 2002. Fluorous micro- and nanophases with a biomedical perspective. Tetrahedron. 58:4113–4131. [Google Scholar]
- 28.Fontaine, P., M. Goldmann, M. Bordessoule, and A. Jucha. 2004. Fast and adjustable-resolution grazing-incidence x-ray liquid surface diffraction. Rev. Sci. Instrum. 75:3097–3106. [Google Scholar]
- 29.Als-Nielsen, J., D. Jacquemain, K. Kjaer, F. Leveiller, M. Lahav, and L. Leiserowitz. 1994. Principles and applications of grazing incidence x-ray and neutron scattering from ordered molecular monolayers at the air-water interface. Phys. Rep. 246:251–313. [Google Scholar]
- 30.Kaganer, V. M., H. Möhwald, and P. Dutta. 1999. Structure and phase transitions in Langmuir monolayers. Rev. Mod. Phys. 71:779–819. [Google Scholar]
- 31.Lösche, M., E. Sackmann, and H. Möhwald. 1983. Fluorescence microscopic study concerning the phase diagram of phospholipids. Ber. Bunsenges. Phys. Chem. Chem. Phys. 87:848–852. [Google Scholar]
- 32.Albrecht, O., H. Gruler, and E. Sacknann. 1978. Polymorphism of phospholipid monolayers. J. Phys. (Paris). 39:301–313. [Google Scholar]
- 33.Ivanova, T., I. Minkov, I. Panaiotov, P. Saulnier, and J. E. Proust. 2004. Dilatational properties and morphology of surface films spread from clinically used lung surfactants. Colloid Polym. Sci. 282:1258–1267. [Google Scholar]
- 34.Brezesinski, G., M. Thoma, B. Struth, and H. Möhwald. 1996. Structural changes of monolayers at the air/water interface contacted with n-alkanes. J. Phys. Chem. 100:3126–3130. [Google Scholar]
- 35.Unger, E. C., T. Porter, W. Culp, R. Labell, T. Matsunoga, and R. Zutshi. 2004. Therapeutic applications of lipid-coated microbubbles. Adv. Drug Deliv. Rev. 56:1291–1314. [DOI] [PubMed] [Google Scholar]
- 36.Gerber, F., M. P. Krafft, G. Waton, and T. F. Vandamme. 2006. Microbubbles with exceptionally long life—synergy between shell and internal phase components. New J. Chem. DOI: 10.1039/b600061b.
- 37.Reference deleted in proof.
- 38.Bernhard, W., J. Mottaghian, A. Bebert, G. A. Rau, H. von der Hardt, and C. Poets. 2000. Commercial versus native surfactants surface activity, molecular components, and the effect of calcium. Am. J. Respir. Crit. Care Med. 162:1524–1533. [DOI] [PubMed] [Google Scholar]
- 39.Knobler, C. M., and R. C. Desai. 1992. Phase transitions in monolayers. Annu. Rev. Phys. Chem. 43:207–236. [Google Scholar]