

Abstract
We report on the development of the F64L/S65T/T203Y/L231H GFP mutant (E2GFP) as an effective ratiometric pH indicator for intracellular studies. E2GFP shows two distinct spectral forms that are convertible upon pH changes both in excitation and in emission with pK close to 7.0. The excitation of the protein at 488 and 458 nm represents the best choice in terms of signal dynamic range and ratiometric deviation from the thermodynamic pK. This makes E2GFP ideally suited for imaging setups equipped with the most widespread light sources and filter settings. We used E2GFP to determine the average intracellular pH (pHi) and spatial pHi maps in two different cell lines, CHO and U-2 OS, under physiological conditions. In CHO, we monitored the evolution of the pHi during mitosis. We also showed the possibility to target specific subcellular compartments such as nucleoli (by fusing E2GFP with the transactivator protein of HIV, (Tat) and nuclear promyelocytic leukemia bodies (by coexpression of promyelocytic leukemia protein).
INTRODUCTION
Intracellular pH is an important modulator of cell function. The activity of most proteins is affected by even small changes of proton concentration, and a number of cellular mechanisms exist that finely regulate the intracellular pH (pHi) value (1). Indeed, pHi is much higher than expected if H+ were passively distributed between the extracellular and intracellular space. All cells have mechanisms for H+ extrusion that maintain pHi above equilibrium value, as required by cytoplasmic reactions (2). Furthermore, the intracellular distribution of H+ is not uniform and depends on the nature of subcellular domains (3). Consequently monitoring pHi with high spatial resolution can help elucidate many physiological or pathogenic processes taking place within cells.
Fluorescent pH indicators represent a valuable option for the determination of pHi. Nowadays, many organic dyes with pH-dependent optical properties are available for pHi monitoring by means of fluorescence microscopy or other techniques relying on cell fluorescence analysis (4–7). Conventional dyes, however, must be loaded from the external medium and cannot be directed to specific subcellular locations except through endocytic pathways (8). In addition, the actual cell penetration varies significantly with different dyes and cell types (9). In the search for more effective pHi probes, much attention was recently given to protein-based fluorescent pH indicators. These are particularly promising for the selective targeting of subcellular compartments by genetic engineering methodologies (10).
The green fluorescent protein (GFP) represents a naturally evolved nanosized optical device whose use as fluorescent probe in molecular and cell biology is well established (11,12). GFP has the remarkable property that the formation of its chromophore is genetically encoded and is a spontaneous process in physiological conditions with no need for specific cofactors (13). It can be expressed fused to a protein of interest within living cells without losing its optical activity. Furthermore, molecular engineering of GFP allows for the modification of fluorophore characteristics in terms of absorption efficiency, quantum yield, and emission quenching by a number of ligands (14).
The presence of one protonation site in the chromophore of most GFP mutants makes these proteins interesting candidates for the implementation of fluorescence-based pHi indicators (8). Measuring the fluorescence intensity, however, does not lead to reliable pHi determination owing to the difficulty in the control of the actual number of fluorescent proteins expressed within a cell. Coexpression of two GFP mutants, one pH-dependent and one not, was shown to be a possible way to overcome this problem (9). However, a simpler and more attractive approach is represented by ratiometric GFP mutants. Fluorescent ratiometric indicators are characterized by multiple excitation or emission maxima with distinct pH dependence. The pioneering work by Miesenbock led to the development of some ratiometric GFP mutants (pHluorins) and showed the successful pHi determination in some biological conditions (15–17). The first pH indicators were ratiometric by excitation, i.e., the measurement relies on the changes in the excitation spectrum upon pH. Remington and co-workers introduced a number of interesting GFP-based pH indicators that are ratiometric by emission (deGFP) (18–20). The latter are very promising for bioimaging applications, particularly for the case of two-photon excitation (18). Unfortunately, most of the GFP-based ratiometric pH indicators developed so far suffer from significant drawbacks such as: pK values unsuitable for physiological conditions (an interesting exception is reported in (21)); noisy fluorescence signals in one of the emission intervals; or excitation wavelengths in the far blue/UV region of the electromagnetic spectrum, where much cellular autofluorescence and photodamaging occur. Often the role of excitation and emission wavelengths selected in determining the main indicator characteristics is not clearly outlined. This is particularly significant for the apparent pK of the fluorescence-ratio calibration curve.
In an effort to address these issues, in this article we describe for the first time the application of the GFP mutant F64L/S65T/T203Y/L231H GFP (E2GFP) as an effective targetable excitation and emission ratiometric pH indicator for in vivo studies on mammalian cells. We have a long-lasting interest in E2GFP properties and demonstrated its optical switching between a bright and a dark state down to the single-molecule level (24). A molecular mechanism to explain this photoinduced reversal of photobleaching was also proposed (22–24).
Here, we show that E2GFP displays marked changes in both excitation and emission fluorescence spectra upon pH variations. Its pK is well-within physiological range, and we shall demonstrate ratiometric independence from the presence of cellular binding quenchers of fluorescence. Importantly, the spectral characteristics of E2GFP allow the use of widespread light-excitation sources without affecting the linear response range of pH and make it an intracellular pH probe perfectly matched for the most widespread cell-microimaging setups.
MATERIALS AND METHODS
Cell culture
Chinese Hamster Ovary (CHO) and U 2-OS (human bone osteosarcoma) cells were cultured on 3.5-cm glass bottom petri dishes (WillCo-Dish; WillCo Wells, Amsterdam, The Netherlands) in Dulbecco's Modified Eagle Medium (Invitrogen, Carlsbad, CA) supplemented with 10% Fetal Bovine Serum, 1.0 mg/ml glucose, 2 mM glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin (all from Invitrogen). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere.
Plasmid construction
For prokaryotic expression, the full-length protein coding region of a pGEX-E2GFP (F64L/S65T/T203Y/L231H GFP) (23) was inserted by PCR amplification into the pPR-IBA2 (IBA GmbH, Göttingen, Germany) vector by using the following primers: 5′-ATGGTAGGTCTCAGGCCATGGTGAGCAAGGGCGAGGAG-3′ and 5′-ATGGTAGGTCTCAGCGCTCTTGTACAGCTCGTCCATGCCG-3′.
The plasmid for eukaryotic expression of E2GFP was obtained by subcloning the protein sequence from pPR-IBA2-E2GFP into pcDNA3.1+ (Invitrogen) by using the following primers: 5′-CCCAAGCTTGGGATGGCTAGCTGGAGCCACCCG-3′ containing a HindIII site (underlined), and 5′-CCGGAATTCCGGTTACGCGGATCCGCGCTTGTA-CAGCTCGTCCAT-3′, containing an EcoRI site (underlined).
The plasmid for eukaryotic expression of Tat-E2GFP was a generous gift by Prof. Mauro Giacca (ICGB, Trieste, Italy), and was obtained by site-directed mutagenesis on the previously fabricated pcDNA3-Tat-EGFP plasmid (25).
Cell transfection
Cells were transfected 1 day after plating (at 60–80% confluence) by the Effectene Transfection Kit (Qiagen, Hilden, Germany) as described by the manufacturer for adherent cell lines. Transfected cells were observed 24–48 h after transfection to obtain maximal levels of gene expression.
E2GFP expression and purification
Expression of Strep-tagged E2GFP was induced by IPTG into Escherichia coli strain BL21(DE3) (Invitrogen). Starter cultures were grown overnight in Luria-Bertani Medium (10 of Bacto tryptone, 5 g of yeast extracts, 5 g NaCl, and 1 ml of 1 M NaOH in 1 liter of water) containing 100 μg/ml ampicillin at 37°C. The maximum yield was obtained at 30°C 30 h after induction, as determined by SDS-PAGE gel of the total lysate. Proteins were purified to homogeneity at 4°C by an affinity chromatography step with a streptactin column. A further anion exchange purification step was carried out using a ResourceQ column (Pharmacia, Bridgewater, NJ) fitted in an FPLC (AKTA) system (Amersham, Little Chalfont, Buckinghamshire, England). Approximately 25–30 mg of recombinant E2GFP were purified from 1 liter bacterial culture. The purity was >95%, as judged by silver-stained SDS-PAGE gels and mass spectrometry analysis.
Spectroscopic measurements in vitro
Absorption spectra were recorded at 23°C on a Jasco V550 spectrophotometer (JASCO, Easton, MD) by setting the monochromator slits to 2 nm, the scanning speed to 100 nm/min, the data resolution to 0.5 nm, and the time collection average on each wavelength interval to 0.25 s. The absorption spectrum of each sample was corrected for background and normalized to the measured optical density at 278 nm.
Fluorescence spectra were recorded at 23°C on a Cary Eclipse spectrofluorometer (Varian, Palo Alto, CA) by setting the excitation and emission monochromator slits to 2.5 nm, the scanning speed to 150 nm/min, the data resolution to 0.5 nm, and the time collection average on each wavelength interval to 0.2 s. Emission spectra were taken by exciting at 278, 350, 395, 408, 421, 434, 447, 460, 473, 486, and 499 nm. Excitation spectra were taken by collecting the fluorescence at 508 and 523 nm. The fluorescence spectrum of each sample was corrected for background and normalized to the previously-measured optical density at 278 nm.
Titrations of E2GFP absorbance and fluorescence versus pH were performed on 2.1 μM protein samples dissolved in a citrate (2 mM)/phosphate (10 mM) buffer whose pH was adjusted to the desired value by addition of 1 M NaOH. The extrapolated absorption and fluorescence spectra at [H+]→∞ (acid form) and [H+]→0 (alkaline form) were calculated by means of the PCA-based chemometric method developed by Kubista et al. (26). The same method afforded also the equilibrium pK.
Comparison of the absorption spectrum of E2GFP (pH = 6.5, 23°C) with that of the protein after exposure to 6M guanidine-HCl/80°C for 10 min (final pH = 6.5, 23°C) afforded the optical density ratio at 278 nm between the folded and the unfolded state. Assuming 26,730 M−1cm−1 as the extinction coefficient at 278 nm for the unfolded state (from ProtParama, available at www.expasy.org), we obtained 27,180 M−1cm−1 as the extinction coefficient for the folded state at the same wavelength. This value yielded, in turn, the extinction coefficients of the spectral bands of the protonated and deprotonated forms.
Fluorescence quantum yields of the protonated and deprotonated forms of E2GFP were calculated by using fluorescein as standard, assuming 0.92 as fluorescein quantum yield (18).
Fluorescence microscopy
Cell fluorescence was measured using a Leica TCS SP2 inverted confocal microscope (Leica Microsystems, Wetzlar, Germany). Glass-bottom petri dishes containing transfected cells were mounted in a thermostated chamber (Leica Microsystems) and viewed with a 40 × 1.25 NA oil immersion objective (Leica Microsystems). The 488 and 458 nm excitation sources were provided by the inline Ar laser of the microscope; the 406 nm excitation source was provided by a frequency-doubled Mira 900 76 MHz pulsed NIR laser (Coherent, Santa Clara, CA). All images were collected simultaneously during excitation at 488 and 458 nm or 406 nm while monitoring the emission in the selected wavelength range (see Results) by means of AOBS-based built-in detectors of the confocal microscope. Each image was the average of four frames. Scan speed was set to 400 Hz. For each ratiometric couple of images (excitation or emission), care was taken to operate at the same photomultiplier voltage.
Data were analyzed by Leica Imaging package version 2.61 and ImageJ software (NIH Image, http://rsb.info.nih.gov/nih-image/index.html). For the determination of intracellular pH maps, background-subtracted images were processed by the median filter option of the ImageJ software, assuming a pixel radius comparable to the highest achievable optical resolution of the image (500 nm, as experimentally measured on 100 nm fluorescent microspheres, λx = 488 nm). Then, a threshold level was determined interactively for each cell. The threshold was determined only for the images recorded at λx = 458 or 406 nm (ratiometric excitation setups) or λe = 475–515 nm (ratiometric emission setups), because they had the lower signal/noise ratio.
Spectroscopic measurements in vivo
In vivo pH calibrations were carried out by perfusing the cells initially for 10 min with an enriched K+ buffer: 120 mM potassium gluconate, 40 mM sodium gluconate, 20 mM HEPES, 0.5 mM calcium chloride, 0.5 mM magnesium sulfate, pH = 7.2. Then, the cells were perfused with the same buffer, adjusted to the desired pH by 0.1 M NaOH, and supplemented with the following compounds: 5 μM nigericin, 5 μM valinomycin, 5 μM carbonyl cyanide p-chlorophenyl hydrazone, 10 μM tributyltin chloride, and 20 μM forskolin (27). To monitor the effect of carbonate or chloride on the calibration curve, a fraction of gluconate anion was replaced by bicarbonate or chloride. Images were taken 10 min after the perfusion with ionophores.
Physiological alkalinization/acidification experiments were carried out by initially perfusing the cells with PBS: 136 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 1.47 mM KH2PO4, and 8.1 mM Na2HPO4, pH = 7.4. Then, the cells were perfused by the same buffer where 30 mM NaCl had been replaced by 30 mM NH4Cl, pH = 7.4. Images were taken every 1 min.
RESULTS
Theoretical analysis of the fluorescence ratiometric signal
We consider a fluorescent molecule whose fluorophore has more than one protonation state and focus on the characteristics that can make this molecule a pH indicator within the cell. Assuming a linear relationship between collected emission and fluorophore concentration, the fluorescence depends on pH according to
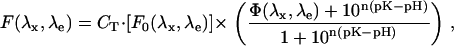 |
(1) |
where
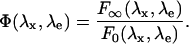 |
(2) |
Here, λx (λe) is the excitation (collection) wavelength, F0(λx,λe) (F∞(λx,λe)) is the extrapolated fluorescence emission at [H+]→0 ([H+]→∞), pK is the negative Log of the global protonation constant, and CT is the concentration of the fluorophore. The value n is the Hill number, and contains information on the number of protonation states: when only two protonation states are present, n = 1. The value Φ(λx,λe) represents the fluorescence ratio of the two forms at (λx,λe).
Equation 1 shows that the fluorescence of the molecule is affected by changes of H+ concentration in its environment only when Φ(λx,λe) ≠ 1. In this case, the excitation spectrum of the completely protonated form does not fully overlap with that of the deprotonated form. Nonetheless, the photon emission intensity is dependent also on the total number of fluorophores through their concentration (CT). Such dependence is lost when the ratio of fluorescence obtained at two excitation and/or emission wavelengths is considered,
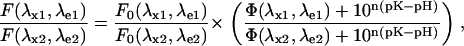 |
(3) |
where (λx1,λe1) and (λx2,λe2) are the two sets of excitation-emission wavelengths and the other terms have the usual meanings. Equation 3 can be recast in a more compact form as
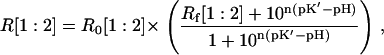 |
(4) |
where
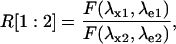 |
(5) |
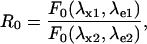 |
(6) |
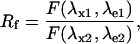 |
(7) |
and
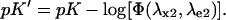 |
(8) |
In Eq. 4, R0 is the ratiometric offset, whereas Rf reflects the difference in behavior between the emission and/or the excitation spectra of the molecule upon pH changes. It represents the dynamic range of the ratiometric signal. If Rf ≠ 1 for the two couples (λx1,λe1) and (λx2,λe2), the fluorescent ratio R[1:2] will be dependent on H+ concentration. In this case, we can speak of a ratiometric pH indicator.
Due to its relevance for the ratiometric measurements, we stress that the fluorescent ratio R[1:2] is not a function of the actual pK of the protein, but of the apparent pK′ given in Eq. 8. Indeed, the maximum sensitivity of the ratiometric probe is displayed in the pH range ((pK′-1/n); (pK′+1/n)), where its response to pH is nearly linear. It is worth mentioning that pK′ and Rf depend only on the nature of the fluorescent molecule and on the selection of the excitation-emission couples (λx1,λe1) and (λx2,λe2). Conversely, R0 is also a function of instrumental characteristics such as the excitation light intensity(ies) and the detector efficiency(ies), as it represents a fluorescence ratio taken with two distinct excitation/emission setups.
In vitro experiments
Spectral and thermodynamic characteristics of E2GFP
Similarly to most mutants of the green fluorescent protein class (11), the absorption spectrum of E2GFP in the visible region of the electromagnetic spectrum is characterized by two absorption bands (Fig. 1 a). At pH 4.6–5.0, E2GFP shows only one peak in the visible region, centered at 425 nm. As the pH is raised, a second absorption band appears near 510–514 nm and grows to a maximum, whereas the high-energy peak decreases its intensity and is blue-shifted to 400 nm. An isosbestic point is detectable around 475 nm and indicates the presence of two absorbing forms in equilibrium. Below pH = 4.6 or above pH = 9.8 the protein unfolds: the absorption spectrum resembles that of the free chromophore at low or high pH, respectively (data not shown).
FIGURE 1.

(a) Absorption spectra of E2GFP at different pH. Fluorescence spectra of E2GFP at different pH: (b) excitation at 408 nm, (c) excitation at 473 nm, (d) emission at 508 nm, and (e) emission at 523 nm. (f) Dependence of the protonated (solid circles) and deprotonated (open squares) form of E2GFP upon pH.
Also, the fluorescence spectra are consistent with the presence of two pH-interconverting forms. Excitation at 408 and 473 nm yields pH-dependent emission spectra which are red-shifted as the pH is raised (Fig. 1, b and c). Indeed, the fluorescence emission peaks at 508–510 nm at low pH and at 523 nm at high pH (Fig. 1, b and c). The fluorescence excitation spectra at 508 nm emission wavelength displayed similar pH-dependent behavior to the blue region (400–470 nm) of the absorption spectrum (Fig. 1 d), whereas the excitation spectra collected at 523 nm paralleled the changes of the cyan/green region (470–515 nm) of absorption (Fig. 1 e). Remarkably, an isosbestic point is clearly defined in both the emission and the excitation spectra. The comparison of the absorption and excitation spectra shows a higher quantum yield for the green-absorbing species. In agreement with literature results (28), we attribute the band at high (low) energy to the protonated (deprotonated) chromophore on Tyr66.
Although the residual presence of the protonated band at high pH can be explained only by a multisite protonation model, the absorption and fluorescence spectral changes can be described by a simple two-state apparent protonation equilibrium that follows Eq. 1 with n = 1.1 and pK = 6.78 ± 0.05 (Fig. 1 f). No significant deviation from this behavior was found for several excitation wavelengths in the range 395–512 nm. Table 1 is a summary of the photophysical characteristics of the low-pH and high-pH forms of E2GFP.
TABLE 1.
Photophysical characteristics of the low pH and high-pH forms of E2GFP
| PH | λ1 (abs) (cm−1) | ɛ1 (abs) (M−1 · cm−1) | λ2 (abs) (cm−1) | ɛ2 (abs) (M−1 · cm−1) | λ (Flu) (cm−1) | QY* |
|---|---|---|---|---|---|---|
| <5.0 | 424 | 31,560 | — | — | 510 | 0.22 |
| >8.0 | 401 | 29,280 | 515 | 22,400 | 523 | 0.91 |
Quantum yield.
Evaluation of E2GFP as an excitation and emission ratiometric pH indicator
Following a classical chemometric method of spectral analysis that allows the determination of the fluorescent emission spectra at [H+]→0 and [H+]→+∞ (26), we determined the Φ(λx,λe) values for a number of excitation wavelengths (395–486 nm) while keeping constant the emission range. Five different emission ranges were analyzed: 475–515 nm, 475–530 nm, 515–600 nm, 530–600 nm, and 500–600 nm (Table 2). As expected, the calculated Φ(λx,λe) values showed a predominant fluorescent signal from the blue-absorbing protonated form in the first two emission ranges (Φ(λx,λe) <1). The green-absorbing deprotonated form was the dominant emitting fluorophore in the last three emission intervals for high excitation wavelengths (Φ(λx,λe) >1). In view of the subsequent ratiometric characterization, we calculated the correspondent pK′ for each fluorescent ratio Φ(λx,λe) by means of Eq. 8.
TABLE 2.
Fluorescence ratios Φ(λx,λe) calculated at different excitation and emission wavelengths
| λe* | 475–515
|
475–530
|
515–600
|
530–600
|
500–600
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| λx* | Φ | pK′ | Φ | pK′ | Φ | pK′ | Φ | pK′ | Φ | pK′ |
| 395 | 0.20 | 7.53 | 0.35 | 7.29 | 0.63 | 7.03 | 0.59 | 7.06 | 0.49 | 7.14 |
| 408 | 0.13 | 7.72 | 0.25 | 7.43 | 0.46 | 7.17 | 0.45 | 7.18 | 0.36 | 7.28 |
| 421 | 0.08 | 7.93 | 0.17 | 7.60 | 0.34 | 7.30 | 0.33 | 7.31 | 0.25 | 7.42 |
| 434 | 0.05 | 8.13 | 0.13 | 7.72 | 0.29 | 7.37 | 0.28 | 7.38 | 0.21 | 7.51 |
| 447 | 0.06 | 8.05 | 0.19 | 7.55 | 0.45 | 7.18 | 0.45 | 7.18 | 0.31 | 7.34 |
| 460 | 0.20 | 7.53 | 0.57 | 7.07 | 1.28 | 6.72 | 1.30 | 6.72 | 0.91 | 6.87 |
| 473 | — | — | — | — | 1.43 | 6.20 | 4.31 | 6.10 | 3.03 | 6.35 |
| 486 | — | — | — | — | 10.5 | 5.81 | 10.8 | 5.80 | 7.57 | 5.95 |
Expressed in nanometers.
Both excitation and emission ratiometric operations were investigated. In an excitation ratiometric setup, only the excitation wavelength of the probe is switched, whereas the emission range is kept constant. Since most of the E2GFP fluorescence is in the 500–600 nanometer range, we considered the Φ(λx,λe) values relevant to this emission interval. Remarkably, Φ(λx,λe) is close to 1 at λx = 460 nm, and pK′ is only slightly higher than pK (6.9 vs. 6.78). This wavelength is close to 458 nm, which is one of the emitting lines of the Argon laser—a common light source for most modern microscopes. Other emission lines of the Ar laser are located at 476 and 488 nm. Inspection of Φ(λx,λe) values suggests that ratiometric excitation measurements with λx1 = 488 nm and λx2 = 458 nm by means of an Ar laser source would provide a good dynamic range Rf = 8.3 (from Eq. 7). A much higher dynamic range (above 20) may be obtained by adopting a deep-blue laser source (406 nm) as λx2. This choice, however, would lead to a significant pK′ increase.
In an emission ratiometric setup, the excitation wavelength of the probe is kept constant, whereas the emission range is changed. We chose the two couples of complementary emission intervals 475–515/515–600 nm and 475–530/530–600 nm. Inspection of the Φ(λx,λe) values showed that the best compromise of dynamic range (∼6.4) and pK′ (7.50) is obtained when λx= 460 nm, λe1 = 515–600 nm, and λe2 = 475–515 nm. Not surprisingly, the excitation wavelength nearly corresponds to that of the isosbestic point detectable in the excitation spectra of E2GFP. More importantly, the 458 nm Ar line can still be considered a primary light source to collect the fluorescence signal.
Independence of the ratiometric signal from fluorescence quenchers of E2GFP
The T203Y mutation makes E2GFP a member of the class of Yellow Fluorescent Proteins (YFPs) (12). Similarly to other YFPs (27,29), we found that the fluorescence emission of E2GFP is dependent on the concentration of small ions, such as chloride, iodide, and bromide. Comprehensive studies by Remington and co-workers clearly showed the existence of at least one binding site for such ions, whose affinity is modulated by the protonation state of the chromophore (30). When the ion is bound, the fluorescence emission of the YFP mutant is substantially quenched. Considering only one binding site, the pattern of fluorescence emission can be expressed by a simple hyperbolic function,
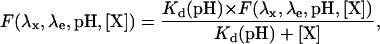 |
(9) |
where [X] is the concentration of the quenching ligand and Kd (pH) is the pH-dependent dissociation constant. Typical values of Kd (pH) are in the low mM range (30). Owing to the mathematical form of Eq. 9, it is easy to show that the fluorescence ratiometric signal R[1:2] is independent of ligand concentration and equal to the value obtained when [X] = 0, provided [X] is the same for the two fluorescence measurements. Thus, when the in vivo fluorescence measurements of the excitation (or emission) ratio are taken in a time shorter than the physiological temporal changes of the ligand, the measured values depend only on pH.
To provide experimental evidence to this theoretical result, we examined whether chloride ions affect the ratiometric response of E2GFP. We plotted the F(488,523):F(458,523) ratio, obtained as an average of several in vitro measurements at different chloride concentrations, versus pH (Fig. 2 a). At each pH, the F(488,523):F(458,523) ratio is statistically indistinguishable for the different concentrations of chloride and is fitted by Eq. 4 to give pK′ = 6.97 ± 0.02, Rf = 9.4 ± 0.01, and n ≈ 1.03 ± 0.02, in good agreement with the values inferred from spectroscopic measurements at [Cl−] = 0 (see Table 2).
FIGURE 2.

Dependence of fluorescence ratio on pH. (a) In vitro titration: each point is an average of fluorescence ratios taken at different chloride concentrations (0–200 mM). (b) Excitation calibration of E2GFP in CHO cells at 23°C. (Solid circles, no chloride added; open circle, 70 mM chloride added; open square, 5 mM chloride added.) (c) Excitation calibration of Tat-E2GFP in U-2 OS cells at 37°C. (Solid squares, no bicarbonate/CO2; open squares, 20 mM bicarbonate/5% pCO2.) (d) Emission calibration of Tat-E2GFP in U-2 OS cells at 37°C. (Solid circles, no bicarbonate/CO2; open circles, 20 mM bicarbonate/5% pCO2.)
In vivo experiments
To evaluate E2GFP as a ratiometric pH indicator in vivo, we produced a cDNA vector encoding for the E2GFP sequence in eukaryotic cells. The protein was transiently expressed in the CHO and the U-2 OS cell lines. Similarly to other GFP mutants in other cell lines (31,32), we found that E2GFP has a diffused intracellular expression pattern with lower accumulation in the nucleolar regions (Fig. 3, a, b, e, and f). In addition, we realized a second expression DNA vector, encoding for E2GFP linked to the human immunodeficiency virus type 1 (HIV-1) transactivator protein Tat (Tat-E2GFP). Tat is a small arginine- and lysine-rich polypeptide (86 amino acids) whose sequence is known to contain a localization domain for the nuclear region of the cell, with accumulation being most evident in the nucleolus (33). For our purposes, Tat-E2GFP represents an example of an E2GFP-based ratiometric pH indicator targeting a specific intracellular body. As the nucleolar/nuclear localization pattern is particularly evident in the U-2 OS cells (Fig. 4, a, b, d, and e), the in vivo experiments with Tat-E2GFP were carried out only with this cell line. Furthermore, some of us showed that the coexpression of the promyelocytic leukemia protein (PML) was found to relocate Tat-EGFP into the PML accumulation bodies, a class of subnuclear compartments presently of high interest in molecular biology (25,34,35). Accordingly, when we coexpressed Tat-E2GFP with PML in U-2 OS cells, Tat-E2GFP was effectively recruited into the PML bodies with an expression pattern identical to that reported for Tat-EGFP (Fig. 4, g and h). Thus, Tat-E2GFP was used also to probe pHi in PML bodies.
FIGURE 3.

(a) Transmission image, (b) intensity image, (c) ratiometric pH map by excitation, and (d) ratiometric pH map by emission of four resting CHO cells transfected with E2GFP and maintained under physiological medium. (e) Transmission image, (f) intensity image, (g) ratiometric pH map by excitation, and (h) ratiometric pH map by emission of a CHO cell transfected with E2GFP and kept in HEPES buffer supplemented with ionophores to clamp extracellular and intracellular pH to 7.25. (i) Normalized frequency histograms of spatial pHi maps by excitation of the lowest cell displayed in c (red bars) and the cell displayed in g (black bars).
FIGURE 4.

(a) Transmission image, (b) intensity image, (c) ratiometric pH map by excitation of one resting U-2 OS cell transfected with Tat-E2GFP and maintained under physiological medium. (d) Transmission image, (e) intensity image, (f) ratiometric pH map by excitation of a U-2 OS cell transfected with Tat-E2GFP and kept in HEPES buffer supplemented with ionophores to clamp extracellular and intracellular pH to 7.25. (g) Transmission image, (h) intensity image, (i) ratiometric pH map by excitation of a U-2 OS cell cotransfected with Tat-E2GFP/PML and maintained under physiological medium. (j) Normalized frequency histograms of spatial pHi maps by excitation of the cells displayed in c (red bars), Fig. 3 f (black bars), and Fig. 3 i (blue bars).
pH calibration curves in cultured cells expressing E2GFP or Tat-E2GFP
In vivo pH calibration of E2GFP or Tat-E2GFP fluorescence ratios was performed by intracellular acid-base titrations, according to a procedure that involves the use of molecular ionophores to clamp the intracellular and extracellular pH at the same value (27). For all titrations, E2GFP or Tat-E2GFP excitation ratiometric curves were well fitted by Eq. 4 (Table 3, Fig. 2, b–d). We set the Hill coefficient to 1 under the assumption that two protein states contribute to the acid-base equilibrium, as demonstrated for E2GFP in steady-state measurements. Nonetheless, we also fitted the ratiometric curves by letting n free to vary. We obtained n-values in the 1.0–1.2 range, though slightly wider confidence limits were found for the other parameters (results not shown). Reversible dynamics of E2GFP fluorescence upon pH was always observed.
TABLE 3.
Experimental conditions of the intracellular titrations and relevant parameters obtained by fitting the ratiometric curves to Eq. 4 with n = 1
| Protein | Setup | Cell line | T (°C) | Confocal aperture* | Rf | pK′ |
|---|---|---|---|---|---|---|
| E2GFP | Ex488:458 | CHO | 23 | 3 | 12.6 ± 1.7 | 6.99 ± 0.04 |
| E2GFP | Ex488:458 | CHO | 23 | 1 | 12.5 ± 1.5 | 7.01 ± 0.04 |
| E2GFP | Ex488:458 | CHO | 37 | 3 | 10.2 ± 1.2 | 6.89 ± 0.04 |
| E2GFP | Ex488:458 | CHO | 37 | 1 | 10.4 ± 0.9 | 6.91 ± 0.04 |
| Tat-E2GFP | Ex488:458 | U-2 OS | 37 | 3 | 14.2 ± 3.4 | 6.83 ± 0.06 |
| Tat-E2GFP | Ex488:458 | U-2 OS | 37 | 1 | 14.5 ± 4.5 | 6.91 ± 0.09 |
| E2GFP | Ex488:406 | CHO | 37 | 3 | 11.0 ± 1.2 | 7.37 ± 0.07 |
| E2GFP | Em515:475 | CHO | 37 | 3 | 5.4 ± 0.2 | 7.48 ± 0.05 |
| Tat-E2GFP | Em515:475 | U-2 OS | 37 | 3 | 6.3 ± 0.3 | 7.21 ± 0.05 |
Airy units (1 airy = 81.44 μm for our setup).
For the excitation ratiometric characteristics, two excitation-wavelength setups were analyzed: λx1 = 488 nm, λx2 = 458 nm (Ex488:458 setup), and λx1 = 488 nm, λx2 = 406 nm (Ex488:406 setup). The emission interval was set to 500–600 nm and measurements were taken at two confocal apertures of the microscope, to identify the best compromise for axial resolution and signal intensity.
In CHO cells at T = 23°C we found for E2GFP a pK′ value comparable to what expected from the in vitro steady-state experiments (pK′ = 6.87, see Table 2). On the contrary Rf in vivo is higher than in vitro (Table 2). Raising the temperature to 37°C led to a slight decrease of both pK′ and Rf (Table 3). At 37°C, Tat-E2GFP showed pK′ values comparable to E2GFP, whereas the dynamic range was somewhat higher. Noticeably, the calibration of Tat-E2GFP was characterized by an increased variability of the single ratiometric measurements (and fitting parameters) with respect to E2GFP, most likely owing to its spatial confinement into smaller cellular compartments that must be imaged by the microscope.
As expected from steady-state measurements, when the second excitation wavelength was switched to 406 nm, higher pK′ values were found (Table 3). These calibration experiments were carried out adopting the larger confocal aperture, to compensate for the lower quantum yield of the blue-absorbing form of the E2GFP chromophore. We did not detect, however, any influence of the confocal aperture on the parameter values in all ratiometric calibrations performed.
We enriched the extracellular medium in K+ and Cl− to increase the intracellular chloride concentration over the physiological value while still equalizing the intracellular and extracellular pH at the same value. Remarkably, no significant changes of the ratiometric curve of E2GFP were observed (Fig. 2 b). Furthermore, the calibration curve of Tat-E2GFP was not affected by the presence in the external buffer of 20 mM sodium bicarbonate in equilibrium with 5% pCO2 (Fig. 2 c). The effect of bicarbonate on the ratiometric characteristics of E2GFP and Tat-E2GFP is worth investigating, since bicarbonate is a membrane permeant anion that plays a significant role in the pH regulation of most eukaryotic cells.
We investigated the ratiometric emission characteristics of E2GFP or Tat-E2GFP, by setting λx = 458 nm, λe1 = 515–600 nm, and λe2 = 475–515 nm (Em515:475 setup). Like for the Ex488:406 setup, we carried out the emission calibration experiments at larger confocal aperture to provide a better signal/noise ratio. As expected from steady-state measurements, the emission pK′ value resulted higher than that determined by excitation on the same protein (+0.6 pH unit for E2GFP, +0.4 pH unit for Tat-E2GFP), while the opposite held for the dynamic range (Table 3). Surprisingly, the addition of sodium bicarbonate to the calibration buffer in equilibrium with 5% pCO2 at pH around the physiological value led to a slight but significant decrease of the fluorescence ratio for Tat-E2GFP (Fig. 2 d).
Determination of intracellular pH in CHO and U-2 OS cells expressing E2GFP
Based on the calibration parameters determined, we measured the pHi in resting CHO and U-2 OS cells under physiological conditions (Table 4). For CHO, we found pHi (Ex488:458) = 7.21 ± 0.13 (at confocal aperture = 3 airy units) and pHi (Em515:475) = 7.24 ± 0.08. These values are in excellent agreement with the average pHi = 7.2 ± 0.1 indicated in the literature for resting CHO (5). Under the same conditions, we found pHi (Ex488:458) = 7.33 ± 0.13 and pHi (Em515:475) = 7.31 ± 0.04 for U-2 OS cells. We never detected any significant difference between the pHi values of whole cells, of the nucleus, and of the cytoplasm (Table 4).
TABLE 4.
Average intracellular pH values of resting CHO and U-2 OS cells determined in different subcellular regions
| Protein | Cell line* | Subcellular region | pK′ (Ex488:458) (CA† = 3) | pK′ (Ex488:458) (CA† = 1)† | pK′ (Em515:475) (CA† = 3) |
|---|---|---|---|---|---|
| E2GFP | CHO (25) | Whole cell | 7.21 ± 0.13 | 7.26 ± 0.14 | 7.24 ± 0.08 |
| E2GFP | CHO (18) | Nucleus | 7.16 ± 0.11 | 7.18 ± 0.11 | 7.23 ± 0.09 |
| E2GFP | CHO (18) | Cytoplasm | 7.19 ± 0.11 | 7.26 ± 0.12 | 7.24 ± 0.09 |
| E2GFP | U-2 OS (22) | Whole cell | 7.33 ± 0.13 | 7.38 ± 0.17 | 7.31 ± 0.04 |
| E2GFP | U-2 OS (19) | Nucleus | 7.33 ± 0.10 | 7.31 ± 0.11 | 7.32 ± 0.04 |
| E2GFP | U-2 OS (19) | Cytoplasm | 7.36 ± 0.13 | 7.42 ± 0.20 | 7.31 ± 0.04 |
| Tat-E2GFP | U-2 OS (12) | Nucleolus | 7.57 ± 0.19 | 7.59 ± 0.19 | — |
| Tat-E2GFP | U-2 OS (7) | Nucleus | 7.71 ± 0.15 | — | — |
| Tat-E2GFP | U-2 OS (12) | PML body | 7.39 ± 0.13 | 7.36 ± 0.11 | — |
Number of averaged cells are in parentheses.
CA = confocal aperture (in Airy units, 1 airy = 81.44 μm for our setup).
We also determined the spatial distribution of pHi in resting CHO cells (Fig. 3). The typical pH frequency histogram of a resting CHO cell obtained by the excitation setup was found to show a rather broad pH distribution (full width at half-maximum, FWHM ≈ 0.5 pH unit, Fig. 3 i). By comparison, the pH frequency histogram of a cell clamped to pHi = 7.25 by means of ionophores displayed a much sharper distribution (FWHM ≈ 0.2 pH unit, Fig. 3 i). When the emission setup was adopted, similar results were obtained (data not shown). This difference reflects the natural spatial nonuniformity of pHi in CHO cells under physiological conditions. The images reported in a recent article concerning the determination of pHi by means of lifetime imaging indicated a similar spatial nonuniformity of pHi in resting CHO cells (5). It is worth noting that pHi was not calculated along the far periphery of the cells, as this region is typically characterized by a much lower signal/noise ratio and higher mobility compared to the remaining part of the cellular body (7).
pHi maps were also monitored at different time instants of the CHO mitotic cycle. We observed a progressive alkalinization as the cell goes on in the division process (Fig. 5, a and b, from time 0′ to 18′). The exit from the mitotic M state and the reentry in the G1 state parallels a significant decrease (∼0.2 pH units) of intracellular pH (Fig. 5, a and b, from time 0′ to 110′). However, no organized pH spatial pattern could be observed during the division process (Fig. 5 a). The intracellular alkalinization during the mitotic cycle is related to the changes in the Na+/H+ antiport activity during DNA synthesis and it has been described in the past for several cell lines by using organic dyes as pH probes (36). However, to the best of our knowledge this is the first time that this process is monitored by means of a GFP-based indicator.
FIGURE 5.

(a) Time evolution of the transmission image (left column), intensity image (center column), and ratiometric pH map by excitation (right column) of one dividing CHO cell transfected with E2GFP and maintained in physiological medium (see Materials and Methods). (b) Normalized frequency histograms of spatial pHi maps by excitation of the dividing cell, at time 0′ (black bars), +18′ (blue bars), and +110′ (red bars).
Finally, the response dynamics of E2GFP was tested by monitoring the changes of average pHi induced by addition and subsequent removal of  ion from extracellular medium (37). Fig. 6 shows that NH4Cl addition to the external buffer of the cells (under isomolar conditions) produced a rapid rise in the fluorescence ratio of expressed E2GFP due to intracellular alkalinization, followed by a slower decrease resulting from
ion from extracellular medium (37). Fig. 6 shows that NH4Cl addition to the external buffer of the cells (under isomolar conditions) produced a rapid rise in the fluorescence ratio of expressed E2GFP due to intracellular alkalinization, followed by a slower decrease resulting from 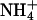 transport and pH regulation. Replacement of NH4Cl by NaCl in the external medium produced a rapid intracellular acidification again followed by a slower regulatory phase.
transport and pH regulation. Replacement of NH4Cl by NaCl in the external medium produced a rapid intracellular acidification again followed by a slower regulatory phase.
FIGURE 6.

Time course of intracellular pH in CHO (average of five cells) upon exposure to NH4Cl (solid circles) and after its removal (open circles).
Determination of intracellular pH in U-2 OS cells expressing Tat-E2GFP
We performed imaging experiments on resting U-2 OS cells in physiological conditions adopting the Ex488:458 ratiometric setup with or without PML coexpression (Fig. 4). The average pHi was 7.59 ± 0.19 for the nucleolar region, 7.71 ± 0.15 for the nucleus, and 7.36 ± 0.11 for the PML bodies (Table 4). The lower expression level of Tat-E2GFP in the nucleoplasm allowed the determination of nuclear pHi only for a subset of the transfected U-2 OS cells and required the adoption of the wider confocal aperture only. The nucleoplasm pHi resulted in 0.4 pH units higher than the value observed in the same cellular region by means of E2GFP (Table 4). On the contrary, the PML-body value approached the nuclear pHi value measured by E2GFP.
By the same procedure followed for E2GFP in CHO, we determined the spatial pHi distribution map in the nucleolar and PML regions of resting or pH-clamped U-2 OS cells (Fig. 4, c, f, and j). Again, the frequency distribution histograms of resting U-2 OS cell displayed much broader pH distribution compared with that of a cell clamped to physiological pH by means of ionophores, indicating a natural heterogeneity of pH in the imaged organelles (Fig. 4 k). In addition, a larger FWHM value was found for PML bodies (≈0.6 pH unit) than nucleoli (≈0.5 pH unit).
DISCUSSION
General remarks on the pH-dependent E2GFP photophysics
F64L/S65T/T203Y/L231H GFP belongs to the yellow fluorescent protein (YFP) class. YFPs are characterized by the replacement of Thr203 with aromatic amino-acid residues, typically Tyr (12). This substitution leads to the π–π stacking between the mutated residue and the chromophore. This in turn produces a marked red-shift of excitation and emission wavelengths with respect to wtGFP (38). Furthermore, Thr203 replacement usually changes the hydrogen-bonding network, and steric packing around the chromophore leading to a modified chromophore acid-base equilibrium (29,30). In particular, T203Y strongly favors the protonated state of the chromophore. Conversely, S65T substitution is believed to stabilize the anionic state of the GFP chromophore, as a result of the destabilization of a hydrogen-bonding network involving Glu222 (39,40). The concomitant introduction of the T203Y and S65T mutations leads to a YFP variant with peculiar thermodynamic and spectral characteristics. Although a full photophysical description of the fundamental states of E2GFP is beyond the scope of this work and will be described elsewhere (R. Bizzarri, unpublished data), we notice that the stabilization/destabilization effects of these mutations on the chromophore protonation somewhat balance each other. In fact, the absorption peaks of the two chromophore states have comparable magnitude, while they exhibit opposite pH dependence (Fig. 1, a, d, and e). pH-driven changes are observed also in the emission spectra, since the deprotonated form of the protein has a red-shifted emission maximum (∼15 nm) compared to the protonated form (Fig. 1, b and c). pK calculation from absorption or fluorescence measurements yields always the some value 6.78, which is significantly higher than the average pK of GFP mutants and close to the physiological value 7 (Fig. 1 f). These characteristics prompted us to exploit E2GFP to implement a ratiometric excitation or emission pH indicator tailored for the physiological pH range typical of in vivo studies (pH = 6.5–8.5).
Factors affecting the properties of E2GFP as a ratiometric pH indicator
Our theoretical analysis of fluorescent ratiometric pH indicators highlighted the influence of the selected excitation and emission wavelengths on the probe properties. In particular, the modulation of the dynamic range may be accompanied by large deviations of the ratiometric apparent pK′ from the actual protein pK value. The linear-response range of the indicator, however, is determined by pK′, not by the actual pK. Ideally, the excitation and emission wavelengths should be chosen to yield a good compromise between dynamic range and a pK′ well within the pH range of interest.
From a series of in vitro steady-state measurements of E2GFP fluorescence spectra at several excitation and emission wavelengths, we determined the fluorescence ratios Φ(λx,λe) of the fully protonated and fully deprotonated spectral forms of the protein, in different conditions of excitation and emission wavelengths (Table 2). Experimental Φ(λx,λe) values showed that E2GFP can be used as an effective ratiometric excitation pH indicator by adopting λx1 = 486 nm, λx2 = 460 nm, and collecting the emission in the 500–600 nm range. Such a selection of λx2 is extremely favorable, since it leads to minor differences between pK′ and pK, thanks to the proximity of λx2 and the isosbestic point in the excitation spectra of the protein (Table 2). Furthermore, the selected excitation wavelengths nearly correspond to two of the emission lines of the Ar laser, one of the most widespread light sources in microimaging setups, allowing for a straightforward exploitation of the present pH indicator. Decreasing the second excitation wavelength was shown to yield wider dynamic ranges at the expense of a pK′-shift toward higher pH values. Although this effect does not really hinder the capability of E2GFP to respond linearly to pH in the physiological range, the use of far blue or UV light certainly raises some problems in terms of higher autofluorescence and cell damage when tested in vivo.
The in vitro steady-state measurements showed also that E2GFP works effectively as a ratiometric emission pH indicator. We found a good compromise between the dynamic range (6.4) and the pK′ (7.5) by choosing the emitted photon collection intervals 475–515 and 515–600 nm, and the excitation wavelength at 460 nm. Again, the use of Ar-ion laser proves a favorable option for in vivo studies, although we are currently trying to extend the application of E2GFP as ratiometric emission pH indicator by means of two-photon excitation.
Analogously to other YFP variants (27,30), the fluorescence of E2GFP is quenched by the protein binding of certain anions. Among these, chloride is certainly the most relevant from a biological viewpoint, since resting cells usually display significant intracellular chloride concentrations (∼5 mM). We showed both theoretically and experimentally that the single-binding curve typical of chloride quenching leads to chloride having no effect on the fluorescence ratio. The latter is equal to the value determined at [Cl−] = 0 (Eq. 9 and Fig. 2 a). The symmetric issue of pH influence on the ratiometric measurement of chloride was the subject of some interesting observations in a recent article (41).
In vivo experiments
To assess the capability of our mutant to act as an in-vivo diffused or targeted pH indicator, we produced DNA vectors codifying for E2GFP alone or covalently bound to the HIV-1 transactivator protein Tat (Tat-E2GFP). Due to its diffused expression pattern, E2GFP was meant to operate as a cytoplasmic as well as nuclear pH probe. Conversely, the presence on Tat of a functional domain specific for nucleolar and, with less affinity, the cell nucleoplasm (33), makes Tat-E2GFP an interesting example of nucleolus-targetable E2GFP-based pH indicator (Fig. 4 a, b, d, and e). More importantly, the intracellular localization of Tat is tunable upon overexpression of proteins that are known to influence the transcriptional activity of HIV mRNA (25,34). Indeed, it was demonstrated that Tat is the key factor of a complex assembly of cellular proteins at the HIV long-terminal repeat promoter that is regulated by subnucleolar compartments such as nuclear speckles or PML accumulation bodies (35). When PML was coexpressed with Tat-E2GFP, we obtained the typical expression pattern of the PML bodies and observed the relocalization of the fluorescent fusion protein (Fig. 4, g and h).
Intracellular titrations yielded the calibration of excitation and emission ratiometric response of both proteins. We demonstrated that the fluorescence ratio signal could be adequately fitted to the theoretical equation developed for a general ratiometric indicator (Eq. 5). The ratiometric dynamics was fully reversible and the dependence of pK′ upon the excitation/emission setup characteristics followed what expected from the in vitro steady-state experiments. Significantly, raising the temperature to 37°C led to the decrease of both pK′ and Rf. This indicates a higher thermodynamic stability and a reduced quantum yield of the deprotonated form of the chromophore. A similar behavior was recently displayed for other GFP-based pH indicators (20). Minor deviations in the found Rf values between the in vitro and in vivo calibrations seem explainable taking into account the difference between the excitation setup of the two experiments (lamp and spectrophotometer monochromator versus laser and microscope optics) as well as the effect of the heterogeneous cellular environment on the thermodynamics of the present GFP mutant.
Both in excitation and in emission, Tat-E2GFP was characterized by a noticeably higher Rf value than E2GFP. Emission data in vivo also showed a significant variation of pK′ values between the two proteins, whereas similar pK′ values were found after the excitation calibrations. Such behavior suggests some influence of the positively charged amino-acid structure of Tat on the emission spectrum of E2GFP rather than on the thermodynamics of chromophore equilibrium. One should keep in mind that this phenomenon could also be caused by some specific interactions of Tat with cell proteins and nucleic acids. However, we never observed any difference of fluorescence ratio in distinct cellular compartments (e.g., nucleoplasm and nucleoli) during the calibration experiments, thus suggesting a predominant role of the Tat structure on the spectral characteristics of the fusion protein.
Importantly, the in vivo studies confirmed that the presence of chloride ions does not affect the ratiometric signal. Apparently, the excitation setup is also insensitive to the presence of the bicarbonate anion, a membrane-permeant metabolite that plays a considerable role in the homeostasis of intracellular pH. Bicarbonate, however, was found to reduce slightly the fluorescence emission ratio of Tat-E2GFP, at least near the physiological pH. We speculate that the bicarbonate anion may shield the positively charged Tat chain, thus reducing its influence on the linked-chromophore photophysical characteristics.
E2GFP was used to obtain pHi maps of resting (CHO and U-2 OS) and dividing (CHO) cells under physiological conditions (Figs. 3 and 5). Significantly, the measured average pHi values displayed no dependence on the ratiometric setup (Ex488:458 vs. Em515:475) or on the degree of confocality of the imaging process (Table 4). Whereas, for the CHO line at rest, the determined on-average pHi was in close agreement with previously reported data (5), the detected pHi for the U-2 OS line was slightly higher (+0.25 pH unit) than the value recently published for resting U-2 OS cells in suspension (42). This discrepancy can be linked to the different cell culture conditions during the pH measurement (adherent versus suspended cells).
At variance with some authors that reported a significant pH gradient between the cytoplasm and nucleus (43–46), we did not detect any pH differences between the two regions. Our results are consistent with other studies on the CHO and similar cell lines by fluorescence pH intracellular probes, which did not show any evidence of cytoplasmic/nuclear pH gradient (7) or indicated minor deviations close to the limit of the technique (36). The significant pH gradient detected by Seksek and Bollard (43) between cytoplasm and nucleus in CHO and other mammalian cells (0.3 pH unit) seems hardly explainable, as it would imply a large free energy difference (0.43 Kcal/mol) of the proton in the two cellular compartments whose origin is unclear. Indeed, as it was correctly pointed out by the same authors, the presence of such a pH gradient can be attributed neither to a Donnan effect nor to macroscopic differences of the dielectric constant, since, in the presence of nigericin (which promotes the K+/H+ exchange), the pH gradient is abolished. Thus, the observed gradient was attributed to a diffusion resting potential of the nuclear membrane toward protons, a phenomenon whose existence is quite controversial (43). Even more striking was the observation in the same experiment that replacement of culture medium with phosphate buffer destroyed the pH gradient (43). While we do not rule out the possibility of minor nuclear-cytoplasm pH gradients in experimental condition or in cell lines different from those adopted in this work, we believe that our results are truly representative of the proton thermodynamic state in the two compartments.
The localization pattern of Tat-E2GFP yielded the average pHi in nucleoli and, upon coexpression of PML, in the PML bodies of resting U-2 OS cells (Table 4). When the expression level in the nucleoplasm was high enough, we were able to determine also the nuclear pHi. For such measurements, we always adopted the Ex488:458 setup since the emission calibration curve of Tat-E2GFP is affected by the presence of bicarbonate anion.
Remarkably, the pHi of U-2 OS nucleus determined by Tat-E2GFP is much higher than the value obtained for the same intracellular compartment by means of E2GFP, while it does not appear statistically different from the nucleolar pHi (Table 4). Conversely, the pHi of PML bodies is close to the value found for the nucleoplasm and cytoplasm. The similarity between the pK′ values of E2GFP and Tat-E2GFP and the observed independence of the calibration parameters of both indicators upon the subcellular localization indicates Tat as the primary factor responsible for the nuclear alkalinization when PML is not coexpressed. This Tat-induced change of pHi would be intriguingly related to the different protein environment around Tat in the absence or in presence of large nuclear bodies. Indeed, a recently-proposed model of Tat activity highlights the inhibitory role of nuclear body formation on HIV transcription by sequestering a variety of factors (including Cyclin T1, Cdk9, p300, and PML) that are required for transcriptional activation (35). Under this hypothesis, the higher degree of pH heterogeneity displayed by PML bodies rather than nucleoli (Fig. 4) may reflect the larger variability of Tat-protein aggregates in the former organelles.
Finally, the ability of E2GFP to respond in vivo to rapid physiological pHi changes was demonstrated by inducing intracellular alkalinization and acidification by means of the 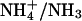 couple, according to a classical procedure (10,37). We observed a quite reproducible nonmonotonic pH-decay in the first alkalinization phase, which results from the complex interplay of molecular diffusion under the adopted experimental conditions and active cellular mechanisms. Analysis of this behavior is of interest, but is not within the scope of this work.
couple, according to a classical procedure (10,37). We observed a quite reproducible nonmonotonic pH-decay in the first alkalinization phase, which results from the complex interplay of molecular diffusion under the adopted experimental conditions and active cellular mechanisms. Analysis of this behavior is of interest, but is not within the scope of this work.
CONCLUSIONS
The F64L/S65T/T203Y/L231H GFP mutant E2GFP was demonstrated to implement an effective ratiometric pH indicator for intracellular studies. Indeed, E2GFP displays very attractive characteristics for fluorescent ratiometric analysis: two distinct different forms that are convertible upon pH changes both in excitation and in emission, high quantum yields, a pK close to the physiological value 7.0, and insensitivity to molecules that reversibly quench its fluorescence upon binding.
E2GFP can work as a ratiometric pH probe either by excitation or by emission, although with a smaller dynamic range in the latter case. Furthermore, the spectral properties of E2GFP make it an optimized pH indicator for imaging setups supplied with Ar-laser excitation sources. Indeed, excitation of the protein at 488 and 458 nm represents the best compromise of signal dynamic range and ratiometric deviation from the thermodynamic pK. The excitation or photon collection at different wavelengths was demonstrated not to degrade the linear response of the probe in the physiological range.
When tested on cells, E2GFP allowed the determination of average pHi and spatial pHi maps in two different cell lines. For the first time with a GFP-based pH indicator, we observed the cellular alkalinization upon mitosis and the subsequent acidification upon entry in the G1 phase. The possibility of targeting our pH probe to specific subcellular regions was demonstrated by the realization of the fusion protein of E2GFP with the transactivator protein of HIV-1. Tat-E2GFP shows only minor changes in the ratiometric properties compared to E2GFP, likely attributable to some intramolecular electrostatic effects of the Tat chain on the fluorescent protein. Tat-E2GFP is prevalently localized in nucleoli and, to a less extent, in the nucleoplasm, but can be relocalized to PML bodies upon PML coexpression. Accordingly, we were able to determine the average pHi and spatial pHi maps for these subcellular compartments. We observed a significant alkalinization of the nucleoplasm and nucleoli after Tat-E2GFP expression that was reversed upon recruitment of the indicator in the PML bodies. This effect may be linked to the different aggregation status of Tat with other proteins involved in the transcriptional activity of HIV in absence or in presence of expression pattern modulators such as PML.
The extension of the emission ratiometric properties of E2GFP to two-photon excitation cell imaging, and the determination of possible relations between the nuclear pH and the transcriptional modulation of the HIV long-terminal repeat promoter by Tat-E2GFP, will be the subjects of forthcoming studies.
Acknowledgments
The authors thank Dr. Riccardo Nifosì for useful discussions. Skillful technical assistance with protein characterization and cell imaging by Abimanyu Rana and Sara Guidi is gratefully acknowledged.
The study was partially supported by MIUR under FIRB project No. RBNE01YSR8-003.
References
- 1.Roos, A., and W. F. Boron. 1981. Intracellular pH. Physiol. Rev. 61:296–434. [DOI] [PubMed] [Google Scholar]
- 2.Frelin, C., P. Vigne, A. Ladoux, and M. Lazdunski. 1988. The regulation of the intracellular pH in cells from vertebrates. Eur. J. Biochem. 174:3–14. [DOI] [PubMed] [Google Scholar]
- 3.Ro, H. A., and J. H. Carson. 2004. pH microdomains in oligodendrocytes. J. Biol. Chem. 279:37115–37123. [DOI] [PubMed] [Google Scholar]
- 4.Sanders, R., A. Draaijer, H. C. Gerritsen, P. M. Houpt, and Y. K. Levine. 1995. Quantitative pH imaging in cells using confocal fluorescence lifetime imaging microscopy. Anal. Biochem. 227:302–308. [DOI] [PubMed] [Google Scholar]
- 5.Lin, H. J., P. Herman, and J. R. Lakowicz. 2003. Fluorescence lifetime-resolved pH imaging of living cells. Cytom. A. 52:77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinlich, M., C. Theiss, C. T. Lin, and R. K. H. Kinne. 1998. BCECF in single cultured cells: inhomogeneous distribution but homogeneous response. J. Exp. Biol. 201:57–62. [DOI] [PubMed] [Google Scholar]
- 7.Bright, G. R., G. W. Fisher, J. Rogowska, and D. L. Taylor. 1987. Fluorescence ratio imaging microscopy—temporal and spatial measurements of cytoplasmic pH. J. Cell Biol. 104:1019–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kneen, M., J. Farinas, Y. Li, and A. S. Verkman. 1998. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 74:1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Awaji, T., A. Hirasawa, H. Shirakawa, G. Tsujimoto, and S. Miyazaki. 2001. Novel green fluorescent protein-based ratiometric indicators for monitoring pH in defined intracellular microdomains. Biochem. Biophys. Res. Commun. 289:457–462. [DOI] [PubMed] [Google Scholar]
- 10.Llopis, J., J. M. McCaffery, A. Miyawaki, M. G. Farquhar, and R. Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 95:6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- 12.Tozzini, V., V. Pellegrini, and F. Beltram. 2004. Green fluorescent proteins and their applications to cell biology and bioelectronics. In CRC Handbook of Organic Photochemistry and Photobiology. W. Horspool and F. Lenci, editors. CRC Press, Boca Raton, FL. 1–15.
- 13.Reid, B. G., and G. C. Flynn. 1997. Chromophore formation in green fluorescent protein. Biochemistry. 36:6786–6791. [DOI] [PubMed] [Google Scholar]
- 14.Lippincott-Schwartz, J., and G. H. Patterson. 2003. Development and use of fluorescent protein markers in living cells. Science. 300:87–91. [DOI] [PubMed] [Google Scholar]
- 15.Hess, S. T., A. A. Heikal, and W. W. Webb. 2004. Fluorescence photoconversion kinetics in novel green fluorescent protein pH sensors (pHluorins). J. Phys. Chem. B. 108:10138–10148. [Google Scholar]
- 16.Sankaranarayanan, S., D. De Angelis, J. E. Rothman, and T. A. Ryan. 2000. The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 79:2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miesenbock, G., D. A. De Angelis, and J. E. Rothman. 1998. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394:192–195. [DOI] [PubMed] [Google Scholar]
- 18.Hanson, G. T., T. B. McAnaney, E. S. Park, M. E. Rendell, D. K. Yarbrough, S. Chu, L. Xi, S. G. Boxer, M. H. Montrose, and S. J. Remington. 2002. Green fluorescent protein variants as ratiometric dual emission pH sensors. 1. Structural characterization and preliminary application. Biochemistry. 41:15477–15488. [DOI] [PubMed] [Google Scholar]
- 19.McAnaney, T. B., E. S. Park, G. T. Hanson, S. J. Remington, and S. G. Boxer. 2002. Green fluorescent protein variants as ratiometric dual emission pH sensors. 2. Excited-state dynamics. Biochemistry. 41:15489–15494. [DOI] [PubMed] [Google Scholar]
- 20.McAnaney, T. B., X. H. Shi, P. Abbyad, H. Jung, S. J. Remington, and S. G. Boxer. 2005. Green fluorescent protein variants as ratiometric dual emission pH sensors. 3. Temperature dependence of proton transfer. Biochemistry. 44:8701–8711. [DOI] [PubMed] [Google Scholar]
- 21.Abad, M. F. C., G. Di Benedetto, P. J. Magalhaes, L. Filippin, and T. Pozzan. 2004. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J. Biol. Chem. 279:11521–11529. [DOI] [PubMed] [Google Scholar]
- 22.Cinelli, R. A. G., A. Ferrari, V. Pellegrini, A. Signorelli, M. Tyagi, M. Giacca, and F. Beltram. 2001. Engineering single-molecule fluorescence dynamics for advanced biomolecular applications. Aust. J. Chem. 54:107–111. [Google Scholar]
- 23.Cinelli, R. A. G., V. Pellegrini, A. Ferrari, P. Faraci, R. Nifosi, M. Tyagi, M. Giacca, and F. Beltram. 2001. Green fluorescent proteins as optically controllable elements in bioelectronics. Appl. Phys. Lett. 79:3353–3355. [Google Scholar]
- 24.Nifosi, R., A. Ferrari, C. Arcangeli, V. Tozzini, V. Pellegrini, and F. Beltram. 2003. Photoreversible dark state in a tri-stable green fluorescent protein variant. J. Phys. Chem. B. 107:1679–1684. [Google Scholar]
- 25.Marcello, A., R. A. Cinelli, A. Ferrari, A. Signorelli, M. Tyagi, V. Pellegrini, F. Beltram, and M. Giacca. 2001. Visualization of in vivo direct interaction between HIV-1 TAT and human cyclin T1 in specific subcellular compartments by fluorescence resonance energy transfer. J. Biol. Chem. 276:39220–39225. [DOI] [PubMed] [Google Scholar]
- 26.Kubista, M., R. Sjoback, and B. Albinsson. 1993. Determination of equilibrium-constants by chemometric analysis of spectroscopic data. Anal. Chem. 65:994–998. [Google Scholar]
- 27.Jayaraman, S., P. Haggie, R. M. Wachter, S. J. Remington, and A. S. Verkman. 2000. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J. Biol. Chem. 275:6047–6050. [DOI] [PubMed] [Google Scholar]
- 28.Ward, W. W., and S. H. Bokman. 1982. Reversible denaturation of Aequorea green-fluorescent protein: physical separation and characterization of the renatured protein. Biochemistry. 21:4535–4540. [DOI] [PubMed] [Google Scholar]
- 29.Wachter, R. M., and S. J. Remington. 1999. Sensitivity of the yellow variant of green fluorescent protein to halides and nitrate. Curr. Biol. 9:R628–R629. [DOI] [PubMed] [Google Scholar]
- 30.Wachter, R. M., D. Yarbrough, K. Kallio, and S. J. Remington. 2000. Crystallographic and energetic analysis of binding of selected anions to the yellow variants of green fluorescent protein. J. Mol. Biol. 301:157–171. [DOI] [PubMed] [Google Scholar]
- 31.Wei, X. B., V. G. Henke, C. Strubing, E. B. Brown, and D. E. Clapham. 2003. Real-time imaging of nuclear permeation by EGFP in single intact cells. Biophys. J. 84:1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wachsmuth, M., T. Weidemann, G. Muller, U. W. Hoffmann-Rohrer, T. A. Knoch, W. Waldeck, and J. Langowski. 2003. Analyzing intracellular binding and diffusion with continuous fluorescence photobleaching. Biophys. J. 84:3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruben, S., A. Perkins, R. Purcell, K. Joung, R. Sia, R. Burghoff, W. A. Haseltine, and C. A. Rosen. 1989. Structural and functional characterization of Human Immunodeficiency Virus Tat protein. J. Virol. 63:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcello, A., A. Ferrari, V. Pellegrini, G. Pegoraro, M. Lusic, F. Beltram, and M. Giacca. 2003. Recruitment of human cyclin T1 to nuclear bodies through direct interaction with the PML protein. EMBO J. 22:2156–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcello, A., M. Lusic, G. Pegoraro, V. Pellegrini, F. Beltram, and M. Giacca. 2004. Nuclear organization and the control of HIV-1 transcription. Gene. 326:1–11. [DOI] [PubMed] [Google Scholar]
- 36.Amirand, C., P. Mentre, S. van de Geijn, M. Waksmundzka, and P. Debey. 2000. Intracellular pH in one-cell mouse embryo differs between subcellular compartments and between interphase and mitosis. Biol. Cell. 92:409–419. [DOI] [PubMed] [Google Scholar]
- 37.Boron, W. F., and P. Deweer. 1976. Intracellular pH transients in squid giant-axons caused by CO2, NH3, and metabolic-inhibitors. J. Gen. Physiol. 67:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wachter, R. M., M. A. Elsliger, K. Kallio, G. T. Hanson, and S. J. Remington. 1998. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure. 6:1267–1277. [DOI] [PubMed] [Google Scholar]
- 39.Jung, G., J. Wiehler, and A. Zumbusch. 2005. The photophysics of green fluorescent protein: influence of the key amino acids at positions 65, 203, and 222. Biophys. J. 88:1932–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elsliger, M. A., R. M. Wachter, G. T. Hanson, K. Kallio, and S. J. Remington. 1999. Structural and spectral response of green fluorescent protein variants to changes in pH. Biochemistry. 38:5296–5301. [DOI] [PubMed] [Google Scholar]
- 41.Kuner, T., and G. J. Augustine. 2000. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 27:447–459. [DOI] [PubMed] [Google Scholar]
- 42.Porcelli, A. M., K. Scotlandi, R. Strammiello, G. Gislimberti, N. Baldini, and M. Rugolo. 2002. Intracellular pH regulation in U-2OS human osteosarcoma cells transfected with P-glycoprotein. Biochim. Biophys. Acta Mol. Cell Res. 1542:125–138. [DOI] [PubMed] [Google Scholar]
- 43.Seksek, O., and J. Bolard. 1996. Nuclear pH gradient in mammalian cells revealed by laser microspectrofluorimetry. J. Cell Sci. 109:257–262. [DOI] [PubMed] [Google Scholar]
- 44.Masuda, A., M. Oyamada, T. Nagaoka, N. Tateishi, and T. Takamatsu. 1998. Regulation of cytosol nucleus pH gradients by K+/H+ exchange mechanism in the nuclear envelope of neonatal rat astrocytes. Brain Res. 807:70–77. [DOI] [PubMed] [Google Scholar]
- 45.Cody, S. H., P. N. Dubbin, A. D. Beischer, N. D. Duncan, J. S. Hill, A. H. Kaye, and D. A. Williams. 1993. Intracellular pH mapping with Snarf-1 and confocal microscopy. 1. A quantitative technique for living tissues and isolated cells. Micron. 24:573–580. [Google Scholar]
- 46.Dubbin, P. N., S. H. Cody, and D. A. Williams. 1993. Intracellular pH mapping with Snarf-1 and confocal microscopy. 2. pH gradients within single cultured-cells. Micron. 24:581–586. [Google Scholar]