

Abstract
Pneumocystis carinii expresses a surface glycoprotein called MSG. Different isoforms of MSG are encoded by a gene family spread over at least 15 telomeric sites. Only one locus, called UCS, supports the production of MSG mRNA. Previous studies showed that P. carinii populations from individual rats exhibited high degrees of diversity with respect to the MSG genes attached to the UCS locus. This diversity could have been generated primarily in the rats studied. Alternatively, the rats may have been infected by P. carinii organisms that were already different at the UCS locus. To investigate this issue, we examined the UCS locus in P. carinii from rats that had been exposed to few of the microbes at a specified time, which produced a bottleneck in the microbial population. Some of the rats with bottlenecks produced P. carinii populations in which a single MSG sequence resided at the UCS locus in 80 to 90% of the organisms, showing that P. carinii can proliferate within a rat without generating the very high levels of UCS diversity previously seen. From the degree of diversity observed in the bottlenecked populations, the maximum rate of switching appeared to be 0.01 event per generation. These data also suggest that the infectious dose is as low as one organism, that rats that share a cage readily infect each other, and that the doubling time of P. carinii in vivo is ∼3 days. In addition, we found that inoculation with 107 P. carinii organisms from a population highly heterogeneous at the UCS locus reproduced this heterogeneity. By contrast, shifts in population structure occurred in rats given 104 P. carinii organisms, suggesting that a small fraction of these proliferated.
Pneumocystis pneumonia is a well-known problem in AIDS patients. Pneumocystis organisms are also found in a variety of nonhuman host species, including nonhuman primates, ferrets, rabbits, horses, shrews, pigs, mice, and rats. Until approximately 1990, a single genus and species name, Pneumocystis carinii, was used in reference to all Pneumocystis organisms regardless of the host species in which they are found. Then, DNA sequence analysis showed that the Pneumocystis organisms found in different host species are quite different from one another and appear to be host species specific. To distinguish these different organisms, a trinomial nomenclature system was adopted (2, 38; S. Keely, H. J. Pai, R. Baughman, C. Sidman, S. M. Sunkin, J. R. Stringer, and S. L. Stringer, abstract from Third International Workshops on Pneumocystis, Cryptosporidium, Microsporidia, and Toxoplasma 1994, J. Eukaryot. Microbiol. 41:94S, 1994). Under this system, Pneumocystis organisms are distinguished as special forms of P. carinii. For example, the Pneumocystis organism used in the studies described in this report was named Pneumocystis carinii f. sp. carinii. The idea that special forms are actually separate species has gained support from the large amount of DNA sequence data now available, and the trinomial names are being phased out. In 1999, two special forms were renamed. P. carinii f. sp. carinii was renamed P. carinii, and the human pathogen was renamed Pneumocystis jiroveci (16). Thus, the organism we call P. carinii here is the same organism previously called P. carinii f. sp. carinii but not the same organism present in hosts other than rats, even though all of those microbes were called P. carinii in the past.
DNA sequence comparisons place the genus Pneumocystis in the kingdom Fungi (14, 15, 39). While the sequence data firmly establish that it is a fungus and strongly suggest that it is an ascomycete, P. carinii is unlike other major fungal pathogens of mammals in several respects, including a lack of ergosterol, fragility of the cell wall, and poor proliferation in culture, the last greatly hampering research progress (37, 38). Culture systems have been described that can transiently sustain populations of P. carinii, but only when the culture is inoculated with millions of cells obtained from an infected animal (9). A culture system that supports clonal proliferation has not been described (30).
Because in vitro cultivation of P. carinii is difficult and limited, researchers have primarily studied organisms obtained from chemically immunosuppressed laboratory rats (3). Heavily infected rats can be obtained in three ways. In the natural transmission method, rats are exposed to airborne P. carinii from birth by being bred in a colony containing infected animals. Immunosuppression of animals in such a colony generally leads to severe infection, making possible the routine recovery of >100 million P. carinii per animal (8, 10, 17, 20). Rats infected by the natural-transmission method probably encounter P. carinii continuously. The second method for producing rats with heavy P. carinii infections is to allow latent P. carinii organisms to proliferate by suppressing the immune system (5). This can be done while keeping animals isolated from environmental P. carinii by housing them under a physical barrier. The provocation of latent P. carinii is sometimes problematical because the proportion of rats that develop heavy infections can be more variable than in the natural-transmission model. A third method of producing Pneumocystis pneumonia in rats is inoculation. Previous reports have established that inoculation of an immunosuppressed rat with a million or more P. carinii organisms reliably produces a heavy infection (4, 7; M. T. Cushion, M. J. Linke, M. Collins, S. P. Keely, and J. R. Stringer, abstract from Sixth International Workshops on Opportunistic Protists 1999, J. Eukaryot. Microbiol. 46:111S, 1999).
While all three methods (natural transmission, provocation of latency, and inoculation) can produce Pneumocystis pneumonia in laboratory rats and are therefore useful for studies of organism burden and the possible benefits of treatment, these methods may not be equivalent when it comes to studies of the nature of the P. carinii organisms in a given rat. This issue became particularly important as we sought to pursue studies of the genetic system that controls expression of the P. carinii major surface glycoprotein (MSG) gene family. Different isoforms of MSG are encoded by the members of a gene family containing as many as 100 genes (18, 19, 24-29, 31-33, 35, 42, 43, 46). Only one MSG appears to be expressed in an individual organism at any given time (40, 41; J. K. Schaffzin and J. R. Stringer, abstract from Sixth International Workshops on Opportunistic Protists 1999, J. Eukaryot. Microbiol. 46:127S, 1999). Previous studies of rats in an open-air colony showed that P. carinii populations taken from individual rats exhibited a high degree of variation at the locus involved in MSG gene transcription (the UCS locus) (Fig. 1).
FIG. 1.

Map of the UCS-MSG locus and locations of PCR primers. The UCS region of the locus is invariant and marks a unique telomeric site in the genome. Adjacent to the UCS, there is always a sequence encoding a protein called MSG. The MSG gene sequence at the UCS locus can be different in different organisms, presumably because recombination installs different MSG-encoding sequences at the UCS locus. The genome carries many copies of MSG-encoding sequences, which reside at the ends of all chromosomes and differ from one another. Only the MSG-encoding sequence that is linked to the UCS locus is represented by mRNA. The CRJE is a 25-bp conserved sequence located between the UCS and its adjacent MSG gene. A copy of the CRJE is also at the beginning of all known MSG-encoding sequences not attached to the UCS locus.
The presence of many different MSG genes at the UCS locus implies that MSG genes can move from a silent site to the UCS locus by recombination (41, 44). However, the frequency of such MSG switches has not been determined because such a determination would require knowing the numbers and kinds of organisms contributing to an infection and the time when each contributing organism entered the rat. Hence, while the variation observed at the UCS locus can be attributed to switching the MSG gene at that locus, it is not possible to determine when the switch occurred. A switch might have occurred in the rat from which the P. carinii organisms were harvested, in a rat that contributed to the cloud of airborne organisms that pervaded the animal room, or in a previous rat, ad infinitum.
In order to examine the stability of the UCS locus as a function of time, it is necessary to study populations of P. carinii formed within a single rat. Theoretically, it should be possible to obtain such populations in either of two ways: provocation of latent organism or inoculation. Here, we report results obtained using both of these methods.
We found that P. carinii organisms from latently infected rats varied with respect to UCS locus diversity. Some were similar to P. carinii organisms from rats kept in open-air colonies and had many different MSG genes at the UCS locus. Others were much simpler at the UCS locus, showing that pneumonia can develop without great diversity at the UCS locus. Experiments with inoculation used a population of P. carinii that had at least 27 different MSG genes at the UCS locus. Three different doses of P. carinii were given. Rats that received the highest dose, 107 organisms, produced P. carinii populations that were as heterogeneous at the UCS locus as the population that contributed the inocula. By contrast, rats inoculated with the lowest dose, 10 organisms, tended to have a single MSG gene sequence predominant at the expression locus, suggesting that the infectious dose may be as small as 1 organism. An intermediate dose, 104 organisms, produced P. carinii populations heterogeneous at the UCS locus but with signs of possible genetic drift. From the degree of diversity observed in the populations that went through the bottleneck imposed by low-dose inoculation, the maximum rate of switching appeared to be 0.01 event per generation. The data from inoculated rats also suggest that rats that share a cage readily infect each other and that the doubling time of P. carinii in vivo is ∼3 days.
We conclude that heterogeneity at the UCS locus can be slow to develop within a population founded by a low number of P. carinii. Hence, it is possible that the high levels of heterogeneity at the UCS locus previously seen in P. carinii from rats that have been exposed to a high number of airborne P. carinii organisms for a long time were due to infection with P. carinii that were already different at this locus. The results obtained with low-dose rats suggest that it may be possible to clone P. carinii via this method.
MATERIALS AND METHODS
Acquisition and housing of rats.
For latency studies, rats presumed to have been infected with P. carinii as neonates were purchased from either Charles River Laboratories, Inc. (Hollister, Calif.), Hilltop Inc. (Scottsdale, Pa.), or Harlan Inc. (Indianapolis, Ind.). The animals in the study included males and females from five strains, Lewis CD, Sprague-Dawley, Long Evans, Wistar, and Fisher. Inoculation studies used Lewis CD rats (125 to 150 g) from a colony believed to be free of P. carinii (Charles River Laboratories, Inc.). The animals were shipped in filtered transport cages. Upon receipt, the animals were immediately placed in shoebox cages fitted with microisolator tops (3-μm exclusion) on horizontal-flow filter racks. Two animals were placed in each cage, and they received irradiated food (Tekmar Irradiated Chow; Harlan Industries) and autoclaved water. To prevent the rats from contracting secondary bacterial infections, cephadrine (Velosef; ER Squibb and Sons, Inc., Princeton, N.J.) was added to the water to achieve a final concentration of 0.200 mg/ml. All animals to be used in inoculation studies were tested for evidence of latent P. carinii infection by screening sera for anti-Pneumocystis antibodies via immunoblotting. The rat sera were diluted 1:10 and 1:40 and incubated with a blot containing separated proteins from a standard preparation of P. carinii organisms, as described previously (46, 48). Three of the 31 animals received were sacrificed, and lung tissues were harvested and processed for DNA analysis, which was performed by PCR using primers targeted to the large-subunit rRNA of the mitochondrial genome (34). The PCR assay could detect as few as 104 P. carinii organisms per rat (22).
Provocation of latent P. carinii.
Rats were immunosuppressed for 12 weeks by weekly subcutaneous injections of 4 mg of methylprednisolone (Pharmacia and Upjohn Co., Kalamazoo, Mich.). Each injection produces a dose of approximately 20 mg/kg of animal weight. The animals were kept under barrier housing conditions at all times prior to and during immunosuppression.
Inoculation.
The P. carinii organisms to be used to inoculate rats were from a cryopreserved stock of P. carinii karyotype form 1 (stock M46-5) (10). To determine the number of P. carinii organisms in the stock, nuclei were counted by light microscopy after being stained with HEMA-3 (Curtis Matheson, Inc.), a rapid variant of the Giemsa stain. Cyst forms were visualized by staining them with cresyl echt violet (6). Once counted, the P. carinii preparation was serially diluted with RPMI 1640 (GIBCO) to produce suspensions with different numbers of organisms per milliliter. Prior to inoculation, the rats were immunosuppressed for 2 weeks by administration of methylprednisolone as described above. Intratracheal inoculation was performed on anesthetized animals by instilling 0.2 ml of RPMI 1640 containing P. carinii per animal into the trachea using a feeding canula introduced through the oral cavity according to the method of Boylan and Current (7). The inoculated rats were maintained in an immunosuppressed state by weekly subcutaneous injections of 4 mg of methylprednisolone.
Assessment of infection by microscopy.
After 12 weeks of immunosuppression, the rats were euthanized and lung tissues were homogenized, as previously described (10). Once homogenized, the tissue from a pair of rat lungs typically occupies a volume of 10 ml. A small aliquot was removed for staining of P. carinii with cresyl echt violet and HEMA-3. The organism burdens per animal were measured by counting the average number of P. carinii cysts in at least 30 oil immersion fields, which is approximately 1.4 × 10−5 ml (47). Enumeration of cysts rather than nuclei is preferred because of their unambiguous morphology in lung homogenates (11). The limit of detection was approximately 10,000 cysts per rat (12, 22). Another aliquot of the lung homogenate was processed for pulsed-field gel electrophoresis, and karyotype profiles were produced by contour-clamped homogeneous electric field as described previously (21). A third aliquot of the lung homogenate was processed for PCR as described below.
Assessment of infection and DNA recovery using PCR.
The primers used in PCR are listed in Table 1. A region of the gene encoding the large-subunit mitochondrial rRNA was amplified with primers PAZ102-H and PAZ102-E under the following conditions: 95°C hot start for 5 min and 30 cycles of incubation at 95°C for 60 s, 50°C for 120 s, and 72°C for 60 s. The internal transcribed spacer region 2 of the nuclear rRNA locus was amplified using primers ITS2U and ITS2L under the same conditions described above, except that the annealing temperature was 50°C and the number of cycles was 40 (23). To measure the number of UCS templates in P. carinii preparations, DNA was analyzed with a Cepheid (Sunnyvale, Calif.) Smart Cycler using Smart Cycler software version 1.01. Real-time PCR was performed under the following conditions: 95°C hot start for 120 s and 40 cycles of incubation at 95°C for 15 s, 45°C for 15 s, 72°C for 15 s, and 80°C for 10 s with optics set to detect SYBR Green fluorescence. The reaction mixture volumes were 25 μl and contained 100 μM (each) dATP, dCTP, dGTP, dTTP; 3 U of Tfl polymerase (Epicenter, Madison, Wis.); 5 mM MgCl2; 1 μl of a 1:20,000 dilution of SYBR Green I (BioWhitaker Molecular Applications); and 20 ng each of primers −145 and anti-AUG (Table 1 and Fig. 1). The amplicons produced from this primer pair were 165 bp in size. The specificity of amplification was monitored in two ways. First, SYBR Green fluorescence was measured in the Smart Cycler as a function of increasing temperature (60 to 95°C at 0.2°C/s). Such melting curves reveal when SYBR Green fluorescence is not attributable to the production of double-stranded DNA of the expected base composition and size. None of the real-time PCR results reported here failed the melting curve test. Second, the amplified DNA was subjected to electrophoresis through an agarose gel and detected by staining it with SYBR Green. The concentration of UCS DNA in samples was calculated by comparing the number of cycles required to reach a threshold level of SYBR Green fluorescence to the same parameter for various amounts of a standard plasmid containing the UCS locus. To prepare the standard curve, the amount of DNA in the standard plasmid was measured by optical absorbance at 260 nm. The plasmid was then diluted by serial 10-fold dilutions. A standard curve relating amplification to the DNA amount was produced by observing the number of cycles required to reach a threshold level of SYBR Green fluorescence in reactions containing diminishing amounts of standard plasmid.
TABLE 1.
Primers used in PCR amplification experiments
| Primer | Sequence | Target | Reference |
|---|---|---|---|
| −145 | 5′-TAGACGATATGAAGGGAGAAT-3′ | UCS | 41 |
| Anti-AUG | 5′-CTCTTAACCGGCCGTGCCAT-3′ | CRJE | 41 |
| AUG | 5′-ATGGCACGGCCGGTTAAGAG-3′ | CRJE | 41 |
| C2 | 5′-ATACATTTTTCTTCATGTTTT-3′ | MSG | 41 |
| C5 | 5′-CATGAAAGACTTGAGAAATGT-3′ | MSG | |
| C7 | 5′-GTCTTGTCCCTTTTTATAGCA-3′ | MSG | |
| ASP | 5′-GTTCTTTAACTTTTTCATCC-3′ | MSG 271.2 | |
| ITS2U | 5′-GTGGCAAAATGCGATAAGTA-3′ | 5.8S rRNA gene | |
| ITS2L | 5′-TATGCTTAAGTTCAGCGGGT-3′ | 26S rRNA gene | |
| PAZ102H | 5′-GTGTACGTTGCAAGTACTC-3′ | Mitochondrial rRNA gene | 34 |
| PAZ102E | 5′-GATGGCTGTTTCCAAGCCCA-3′ | Mitochondrial rRNA gene | 34 |
Amplification of UCS-MSG junctions.
An aliquot of lung homogenate (1 ml) was treated with proteinase K, and genomic DNA was isolated by isopropanol extraction (13). The DNA was dissolved in 0.01 M Tris-0.001 M EDTA, pH 8. One microliter of the DNA was subjected to PCR using primer −145 paired with C2 (Table 1 and Fig. 1) under the following conditions: 95°C hot start for 5 min and 40 cycles of incubation at 95°C for 60 s, 45°C for 120 s, and 72°C for 60 s. The reaction mixture volumes were 25 μl and contained dATP, dCTP, dGTP, and dTTP, each at 100 μM; 1 μl of [α-32P]dATP (10 mCi/ml; 3,000 Ci/mmol); 1.25 U of Tfl polymerase (Epicenter); 1.5 mM MgCl2; and 20 ng of each primer (40).
In some experiments, a heminested PCR assay was used, in which the first amplification was with primers −145 and C2 (Table 1 and Fig. 1), followed by reamplification with primers AUG and C2 as previously described (40). However, most of the data in this study were acquired without the heminested-PCR step because this step was rendered unnecessary by using a 4% polyacrylamide gel to separate amplicons. The radioactive amplicons were heated at 95°C for 3 min and then cooled on ice prior to being loaded on a fresh 4% sequencing gel (36). The gels were run at 80 W for 6 h, dried, and exposed to X-ray film and to PhosphorImager plates (Molecular Dynamics).
For PCR amplification targeted to the 271.2 sequence, a putative allele-specific primer (primer ASP [Table 1]) was made by synthesizing an oligonucleotide that matched a site in sequence 271.2 but did not match any of the sequences already known to be in the input population. PCRs were performed using the ASP primer paired with the −145 primer (Table 1), and the results were monitored in real time using the Cepheid Smart Cycler. Reactions were performed under the following conditions: 95°C hot start for 120 s and 40 cycles of incubation at 95°C for 15 s, 59°C for 15 s, 72°C for 15 s, and 80°C for 10 s with optics set to detect SYBR Green fluorescence. Measured amounts of DNA from P. carinii were subjected to amplification, and SYBR Green fluorescence was monitored as a function of reaction cycles. The abundance of DNA templates amplified by the −145-ASP primer pair was calculated by comparison to reactions performed with known amounts of a plasmid carrying a copy of the 271.2 sequence.
Sequence analysis.
When total PCR products were to be cloned, the PCR was performed in the absence of radioactive nucleotide triphosphate, and the amplicons were purified by agarose gel electrophoresis (1.5%; 3 parts Nusieve, 1 part SeaPlaque [FMC, Rockland, Maine]) and Geneclean (Bio 101, Vista, Calif.). The amplicons were cloned into the plasmid pCRII-TOPO (Invitrogen, Carlsbad, Calif.), which was introduced into the strain of Escherichia coli provided with the vector.
When the goal was to determine the sequence of the MSG genes in the radioactive DNA in bands separated on the 4% sequence gels, the bands were excised and then eluted in distilled sterile water and reamplified with primers AUG and C2 (Table 1) under the following conditions: 95°C hot start for 5 min and 40 cycles of incubation at 95°C for 60 s, 48°C for 120 s, and 72°C for 60 s. These PCR products were then cloned in pCRII-TOPO.
We also produced MSG gene sequences by amplifying genomic DNA with an AUG primer paired with primers C5 and C7 (Table 1). These reactions were performed under the conditions described above for amplification of the excised gel bands.
DNA sequences were determined by the Sequencing Facility at the University of Cincinnati College of Medicine. Most sequences were determined from both strands. Sequences were aligned with DNAMAN software (Lynnon BioSoft, Vaudreuil, Quebec, Canada) using the default settings. The alignments were optimized by introducing a limited number of gaps, which were not counted in relatedness calculations. The relatedness of pairs of aligned sequences was calculated using the observed divergence method, which counts the number of directly unmatched residues and divides this number by the total number of residues compared. The calculated values were used to construct a distance matrix. A homology tree was made from the distance matrix using the unweighted pair group method with arithmetic mean.
Statistical analysis.
The statistical significance of differences in diversity of UCS-MSG junctions among P. carinii from latently infected rats was tested by Fisher's exact test. The data from each rat were categorized as follows. Plasmids with a sequence seen once in a given rat were placed in one category, and plasmids with a sequence seen more than once in that rat were placed in another category. For each test, the number of plasmids in each category in the test rat was compared to the number of plasmids in each category in rat LAT 24, the rat with the most homogeneous UCS locus, in which only one sequence was seen among the eight plasmids analyzed. To illustrate, the numbers when LAT 3 was compared to LAT 24 were nine and zero (LAT 3) and zero and eight (LAT 24).
RESULTS
PCR detects a broad spectrum of UCS-MSG junctions.
PCR was the method of choice to obtain UCS-MSG junctions because it is allows rapid analysis of many P. carinii populations, even in cases where the number of microbes in the sample is too low to support other techniques, such as cloning from genomic libraries. Furthermore, PCR can amplify numerous UCS-MSG junctions because the members of the MSG gene family share certain nucleotide sequences, and these conserved sequences can be used as primer sites. In previous studies, one such primer, C2, had been used (41).
The C2 primer had previously supported amplification of numerous MSG sequences, implying that a large fraction of MSG genes have the C2 sequence. To test this idea, we inspected the GenBank database. To identify MSG genes in the database, we searched for entries that had the 25-bp conserved sequence called the CRJE (conserved recombination junction element), which is located at the 5′ end of all MSG genes and defines the border between MSG and UCS sequences (Fig. 1) (44). There were 41 different CRJE-linked MSG sequences in the database. All but 3 of these 41 sequences had a sequence that matched at least 24 of the 25 bp in the canonical C2 sequence. The other three MSG gene sequences matched 23 of the 25 bp in the C2 sequence.
We also produced an additional 22 MSG gene sequences by amplifying genomic DNA with an AUG primer paired with primers that target conserved sequences downstream of the C2 region (C5 and C7 [Fig. 1]). Ten of these MSG gene sequences had a sequence identical to the C2 sequence. Ten of the remaining 12 sequences matched 24 of the 25 bp in C2. The other two sequences matched 23 of 25 bp, but one mismatched position was at the 5′ end of the C2 primer binding site, reducing the chance that it adversely affected the amplification of such genes.
We tested the capacity of the C2 primer to amplify cloned genes that matched only 24 of the 25 bp in the primer. Two MSG genes of this type and two that matched C2 perfectly were tested, and all four genes were amplified (data not shown). These data predicted that the C2 primer will support amplification of at least 90% of the MSG genes in the genome. Previous amplification experiments had produced results consistent with this expectation. We had observed that amplification using the C2 primer paired with primer −145 (Table 1 and Fig. 1) produced an amplicon containing >26 different MSG sequences (41). Further work along the same lines, some of which is described below, has shown that these two primers can amplify >80 different MSG sequences.
P. carinii populations that emerge from latency can be relatively uniform at the UCS locus.
To obtain populations of P. carinii formed in a single rat, 10 rats that were presumed to be latently infected were kept under a physical barrier and treated with immunosuppressive drugs. Our studies included animals obtained from three commercial animal vendors (see Materials and Methods). Nine of the 10 rats developed lung burdens on the order of 109 cysts per animal. The 10th rat had >108 cysts.
To characterize the UCS locus, the region was amplified and the amplicons were cloned into a plasmid vector to produce libraries. Multiple colonies were picked from each library, and the plasmid inserts were sequenced.
The collection of latently infected rats produced P. carinii organisms that exhibited different degrees of diversity at the UCS locus (Table 2). In three cases, LAT 3, LAT 9, and LAT 16, all of the sequenced inserts were different from each other. In three others, LAT 7, LAT 1, and LAT 5, the same insert was seen in more than one plasmid, but no single sequence was present in >50% of the plasmids. In LAT 19, LAT 21, and LAT 14, one sequence was present in >50% of the plasmids. In one of the 10 latently infected rats, LAT 24, a single UCS-MSG junction was present in eight of eight plasmids sequenced.
TABLE 2.
Diversity of UCS-MSG junctions in P. carinii from latently infected rats
| Rat | No. of plasmids sequenced | No. of sequences observed | % Plasmids with a sequence that was observed once | % Plasmids with a sequence that was observed more than once | Pg |
|---|---|---|---|---|---|
| Lat 3 | 9 | 9 | 100 | 0 | <0.001 |
| Lat 9 | 5 | 5 | 100 | 0 | <0.001 |
| Lat 16 | 5 | 5 | 100 | 0 | <0.001 |
| Lat 7 | 8 | 7 | 75 | 25a | 0.003 |
| Lat 1 | 8 | 6 | 50 | 50b | 0.029 |
| Lat 5 | 10 | 7 | 50 | 50c | 0.038 |
| Lat 19 | 7 | 4 | 43 | 57d | 0.077 |
| Lat 21 | 8 | 4 | 38 | 62e | 0.099 |
| Lat 14 | 9 | 3 | 23 | 77f | 0.260 |
| Lat 24 | 8 | 1 | 0 | 100 | |
| Total | 77 | 51 |
One sequence was in two plasmids.
Two sequences were each in two plasmids.
One sequence was in three plasmids; another was in two plasmids.
One sequence was in four plasmids.
One sequence was in five plasmids.
One sequence was in seven plasmids.
P values calculated via Fisher's exact test by comparing data from each rat to those from rat Lat 24.
The data from LAT 24 indicate that the UCS locus in the P. carinii organisms dwelling in this animal was not highly heterogeneous and might have been homogeneous. Probability analysis suggests that at least 70% of the organisms in this rat had the same UCS-MSG sequence, because the probability of seeing a minority sequence in eight trials when this sequence comprises 30% of the population is 95% (1 − 0.78 = 0.95). However, the sample of eight plasmids was not large enough to rule out the possibility of a smaller minority fraction. To illustrate, there is a 43% chance of seeing the observed result (eight plasmids with the same sequence) when the population of plasmids sampled is 90% homogeneous.
When compared to LAT 24 by Fisher's exact test, P. carinii populations from six rats (LAT 1, 3, 5, 7, 9, and 16) were found to be statistically significantly less homogeneous at the UCS locus (the P values are shown in Table 2). The other three P. carinii populations (LAT 14, 19, and 21) were clearly not homogeneous, but the differences between the data from these populations and those from LAT 24 were not statistically significant.
Comparison of the UCS-MSG sequences from different rats showed that each population of P. carinii was different from the others (Fig. 2). This observation suggests that emergence from latency is not associated with expression of any particular MSG.
FIG. 2.

Relatedness of MSG genes adjacent to the UCS locus in populations of P. carinii from latently infected rats. MSG gene sequences were aligned by pairwise comparisons. The horizontal branch lengths represent divergence, and the bar above the dendrogram shows percent identity.
In summary, the data on rats with latent infections showed that emergence from latency can be accompanied by high diversity at the UCS locus. However, emergence from latency does not require such diversity, nor is it associated with the presence of a particular MSG at the UCS locus.
While not required, heterogeneity at the UCS locus was common among the P. carinii populations emergent from the rats with latent infections studied. The source of this heterogeneity is not clear. One possibility is that rats can become latently infected by multiple P. carinii organisms that differ at the UCS locus. An alternative is that infection is established by a single P. carinii organism and that changes occur at the UCS locus as the population of fungi expands. Distinguishing between these alternatives requires control over the nature of the progenitor P. carinii. The latently infected rat model does not lend itself to such control. Therefore, we explored the utility of inoculation as a means to control infection.
Rats inoculated with as few as 10 organisms developed fulminate pneumonia.
Rats were obtained from a pathogen-free commercial colony, which should be free of latent P. carinii. The rats were tested for evidence of latent P. carinii infection immediately after their arrival. All animals were tested by screening their sera for anti-Pneumocystis antibodies. In addition, 3 of the 31 animals in the shipment were sacrificed, and lung tissues were taken and analyzed by PCR. Both of these tests indicated that the animals were not carrying P. carinii.
Prior to inoculation, animals were rendered susceptible to P. carinii infection by injections of methylprednisolone acetate. The animals were then inoculated by the method of Boylan and Current (7), which entails injection of P. carinii organisms into the trachea using a feeding canula introduced through the oral cavity. The animals were inoculated with either 107, 104, 10, or 0 organisms. The number of organisms introduced was estimated from the total number of nuclei in an aliquot from the most concentrated suspension of organisms. As is usually the case for P. carinii, about half of the nuclei were in cysts, which carry up to eight nuclei each, and the other half were in the uninucleate trophic form of the organism. The inoculated animals were housed (two per cage) under barrier conditions to prevent exposure to other P. carinii organisms. The animals were sacrificed either when they appeared moribund or after 12 weeks, whichever came first. The lungs were homogenized and screened for P. carinii by light microscopy.
Inspection of the lung homogenates by microscopy showed that all of the animals given P. carinii were heavily infected. Table 3 lists the number of P. carinii cysts per animal as determined by counting an aliquot of lung homogenate. The average numbers of P. carinii cysts per lung were 1 × 109, 3 × 108, and 2 × 108 in animals inoculated with 107, 104, and 10 organisms, respectively. Hence, the average number of cysts was only weakly related to the dose, suggesting that as few as 10 organisms can initiate infections that give rise to lung burdens as great as those in rats given 107 organisms.
TABLE 3.
P. carinii organisms and DNA in inoculated rats
| Rat | Dose of P. cariniia | Results
|
||||
|---|---|---|---|---|---|---|
| No. of cysts per lung | Standard PCRb
|
Quantitative PCR (no. of P. carinii genomes per lung) | ||||
| ITS2c | mtrRNAd | UCS-MSG | ||||
| 255 | 0 | 7 × 105 | Negative | Negative | Negative | <1 × 104 |
| 256 | 0 | <1 × 104 | Negative | Negative | Negative | <1 × 104 |
| 257 | 0 | 2 × 106 | Negative | Negative | Negative | <1 × 104 |
| 260 | 0 | <1 × 104 | Negative | Negative | Negative | <1 × 104 |
| 261 | 0 | <1 × 104 | Negative | Negative | Negative | <1 × 104 |
| 262 | 0 | <1 × 104 | Negative | Negative | Negative | <1 × 104 |
| 267 | 10 | 3 × 107 | Positive | Positive | Positive | 3 × 107 |
| 268 | 10 | 8 × 108 | Positive | Positive | Positive | 4 × 109 |
| 269 | 10 | 3 × 105 | ND | ND | Positive | ND |
| 270 | 10 | 5 × 106 | ND | ND | Negative | ND |
| 271 | 10 | 2 × 107 | Positive | ND | Positive | 8 × 106 |
| 272 | 10 | 2 × 108 | Positive | ND | Positive | 3 × 108 |
| 273 | 10 | 2 × 108 | ND | ND | Positive | ND |
| 274 | 10 | 1 × 107 | ND | ND | Positive | 7 × 107 |
| 282 | 104 | 8 × 108 | Positive | Positive | Positive | ND |
| 283 | 104 | 2 × 108 | Positive | Positive | Positive | ND |
| 284 | 104 | 6 × 107 | Positive | ND | Positive | ND |
| 285 | 104 | 1 × 108 | Positive | ND | Positive | ND |
| 286 | 104 | 3 × 108 | Positive | ND | Positive | ND |
| 287 | 107 | 5 × 109 | Positive | ND | Positive | ND |
| 288 | 107 | 2 × 109 | Positive | ND | Positive | ND |
| 290 | 107 | 3 × 109 | Positive | ND | Positive | ND |
| 291 | 107 | 6 × 108 | Positive | ND | Positive | ND |
| 292 | 107 | 6 × 107 | Positive | ND | Positive | ND |
| 294 | 107 | 6 × 107 | Positive | ND | Positive | ND |
| 295 | 107 | 3 × 108 | Positive | ND | Positive | ND |
| 296 | 107 | 9 × 108 | Positive | ND | Positive | ND |
| 298 | 107 | 2 × 109 | Positive | ND | Positive | ND |
Counts are total number of P. carinii nuclei in 0.200 ml, which was the volume injected into each rat. Nuclei in both cysts and trophic forms of P. carinii were counted with a light microscope.
Standard PCR was performed for 40 rounds under conditions described in Materials and Methods. The primers used are listed in Table 1. ND, not done. The results of a standard PCR were scored as positive if a band of the expected size was visible after an agarose gel was stained with ethidium bromide.
ITS2 is a single-copy locus located in the rRNA gene of P. carinii. The primers used are listed in Table 1.
mtrRNA is the gene in the P. carinii mitochondrial genome encoding the RNA in the large ribosome subunit of mitochondria. The primers used are listed in Table 1.
While infection was easily discernible in rats given only 10 P. carinii organisms, no cysts were seen in four of the six rats that had been inoculated with medium lacking P. carinii (sham inoculated). One putative cyst was seen in the lung homogenate from one of the other two sham-inoculated rats, and three were seen in the other. The identity of these cyst-like objects is uncertain because the PCR showed no evidence of P. carinii DNA in the lung homogenates from any of the sham-inoculated rats (see below). Nevertheless, it is possible that the observed objects were P. carinii cysts, presumably derived from latent organisms. If it is assumed that latent infections were the source of the putative cysts, then these latent organisms produced very few progeny compared to the number seen in the rats given 10 or more P. carinii organisms.
To estimate the amount of P. carinii DNA recovered from lung homogenates, at least one sample from each rat was tested for P. carinii DNA by standard PCR, which entailed 40 rounds of cycling followed by analysis of the products on an agarose gel. Every animal that had received at least 104 P. carinii organisms produced a PCR product detectable by ethidium bromide staining (Table 3). All but one of the rats given 10 P. carinii organisms produced a PCR product visible by ethidium bromide staining. By contrast, lung homogenates from the six sham-inoculated rats did not produce a PCR product from any of the three loci tested, including the gene encoding the large-subunit mitochondrial rRNA, which is the most sensitive target for PCR, presumably because there are multiple copies of mitochondrial DNA per organism (45).
Similar results were obtained when the PCR was performed in real time, which allowed estimation of the amount of amplifiable DNA. In this assay, a 165-bp region of the UCS locus was amplified using primers −145 and anti-AUG (Table 1 and Fig. 1). As shown in Table 3, none of the sham-inoculated rats produced a detectable signal in the real-time PCR assay, which in our hands detected as few as 10 templates per reaction. The real-time PCR results showed that the numbers of UCS DNA molecules in the lungs of rats that were inoculated with 10 P. carinii were high, ranging from approximately 8 × 106 to 4 × 109 genomes per lung. In general, the organism burdens estimated by counting cysts were consistent with the results of real-time PCR. Deviations from perfect congruence may have been caused by factors such as inefficiency in DNA recovery and variation in the ratio of the cysts to trophic forms.
High-dose inoculation produced P. carinii populations that closely resembled the population from which the inocula were drawn.
To study UCS-MSG junctions, the C2 and −145 primers were used to amplify this region (Table 1). The amplification products were first analyzed by gel electrophoresis. As shown in Fig. 3, lanes 1 and 20, the UCS-MSG junction in the P. carinii population used to inoculate rats (input population) was quite heterogeneous. There were 10 obvious bands, as well as several fainter bands. These bands ranged in size from ∼510 to 450 bp. Analysis of DNA sequences present in these amplicons showed that there were at least 27 different MSG gene sequences at the UCS locus in the input P. carinii population (see below). All of the animals inoculated with 107 organisms produced the same band pattern as the input population (Fig. 3, lanes 11 to 19).
FIG. 3.

Sizes of UCS-MSG amplicons from rats inoculated with different doses of P. carinii. UCS-MSG junctions were amplified from genomic DNA extracted from different P. carinii populations. The radiolabeled amplicons were separated by electrophoresis through 4% acrylamide. Lanes 1 and 20, template DNA from the population from which inocula were drawn. The dots in lane 1 indicate the 10 strongest bands in the input pattern. Lanes 2 to 5, 6 to 10, and 11 to 19, template DNAs from rats inoculated with 101, 104, and 107 organisms, respectively. The DNA bands in lanes 2, 3, 4, and 5 were produced from animals 267, 268, 271, and 272. The arrowhead next to lane 6 marks band 8, which was one of several bands that varied in intensity among the rats inoculated with 104 organisms. The band in lane 21 was generated by amplification from a plasmid containing a single UCS-MSG junction. The numbers on the left are DNA sizes in base pairs.
The animals inoculated with 104 organisms also produced a UCS-MSG band pattern that had all of the bands seen in the input population (Fig. 3, lanes 6 to 10). However, the band intensities within patterns varied more than when animals were inoculated with 107 P. carinii. One band that exhibited this trait is marked in Fig. 3. Such fluctuations are expected in small populations, which are more subject to the effects of genetic drift.
Low-dose inoculation produced P. carinii with little heterogeneity at the UCS locus.
By contrast with the results obtained in rats inoculated with 104 or more organisms, seven of the eight rats inoculated with 10 P. carinii organisms produced a band pattern that was much simpler than that of the input population. (The eighth rat in the group, rat 270, did not produce a PCR product.) Amplification of lung homogenates from five of the seven rats, 271 and 272 (Fig. 3, lanes 4 and 5), and 269, 273, and 274 (data not shown) yielded a single strong band. Rats 267 and 268 produced three and two strong bands, respectively (Fig. 3, lanes 2 and 3). No bands were present after amplification of DNA prepared from lung homogenates of sham-inoculated rats (data not shown). These results are consistent with the results of quantitative PCR described above.
To confirm that the amplified UCS-MSG junctions accurately represent the UCS locus, we explored the possible influence of PCR artifacts in these experiments. One possibility that must be excluded in any PCR experiment is DNA contamination, which is usually caused by DNA from previous PCR experiments. We can rule out DNA contamination for the following reasons. (i) Real-time PCR showed that the number of P. carinii genomes in the lung homogenates corresponded to the number of cysts seen in the microscope. Therefore, the amplicons were not produced from a rare template, as would be expected if contamination were the source of that template. (ii) Contamination would not be expected to produce the many different UCS-MSG junction bands produced by amplification of lung homogenates. (iii) Control reactions that lacked lung homogenates did not produce a product. (iv) No P. carinii DNA was detected in sham-inoculated rats. (v) The sequences produced from the low-dose rats did not match anything we had amplified and sequenced before.
A second possible source of artifacts would be failure of the PCR to amplify all of the UCS-MSG junctions present in the sample. This was a particular concern in the experiments with rats that had been inoculated with <107 organisms, because these animals tended to be somewhat less infected (Table 3). Taken to the extreme, a low template concentration might cause only the most common UCS-MSG junction to amplify. We tested this possibility by amplifying diminishing amounts of DNA from the lung homogenate of rat 292, which had relatively few cysts (Table 3) but had produced a pattern with 10 major bands when undiluted DNA was amplified (Fig. 3, lane 15). These experiments showed that a reaction mixture that contained a 32-fold dilution of sample 292 produced the pattern of 10 bands. Rats 267, 268, 271, 272, 273, and 274 all contained at least five times as many organisms as the 32-fold dilution of sample 292 (Table 3). Therefore, the simple UCS-MSG band patterns produced from these rats were not artifacts imposed by low DNA template concentrations
Another concern associated with PCR is the possibility of production of artifactual bands. This possibility was examined by experiments on plasmids carrying UCS-MSG junctions of various sizes. As expected, amplifications of cloned UCS-MSG junctions each produced a single band, which migrated in the expected manner (Fig. 4). In addition, amplification of a mixture of these plasmids produced the pattern expected, and no new bands were generated (data not shown). These control experiments showed that amplification of genomic DNA from P. carinii would be expected to faithfully reproduce the UCS-MSG junctions in the genome and not to generate junctions that do not exist. Further evidence in support of the fidelity of the PCR assay was obtained by sequencing the amplicons, which showed that all of them had an open reading frame encoding an MSG (see below). Preservation of the open reading frame is inconsistent with the hypothesis that fragments of different sizes were produced by PCR mistakes that either deleted, added, or recombined DNA, because such mistakes would be expected to often disrupt the reading frame.
FIG. 4.

Sizes of UCS-MSG amplicons produced from control plasmids. UCS-MSG junctions were amplified from six plasmids, each of which carried a different sequence. The radiolabeled amplicons were separated by electrophoresis through 4% acrylamide. Lanes 1 to 6, amplicons from six different plasmids, each carrying a different UCS-MSG junction. On the right are DNA sizes in base pairs.
Each UCS-MSG junction band from low-dose-inoculated rats contained predominantly one sequence.
To quickly determine if bands from low-dose rats might contain a single sequence, the largest and smallest bands from 267, the smallest band from 268, and the band in 271 (shown in Fig. 3, lanes 2, 3, and 4) were excised from the gel, reamplified, and sequenced directly. In each case, an unambiguous sequence could be read, indicating that each band contained predominantly one UCS-MSG sequence. By contrast, the same analysis of bands from the input population indicated that these bands were comprised of multiple sequences. To confirm the sequence obtained by direct sequencing, DNA molecules from the four bands from rats 267, 268, and 271 were reamplified with primer AUG and primer C2, and the products were cloned by insertion into a plasmid vector. At least three plasmid clones from each band were sequenced. All plasmids made from a given band contained the same sequence, and the cloned DNA sequence matched that read directly from the DNA purified from the gel.
UCS-MSG sequences predominant in inoculated rats matched sequences in the input organism population.
The inoculation model is based on the premise that the procedure causes P. carinii infections. If this is the case, then the sequences of UCS-MSG junctions found in inoculated rats would be expected to match the UCS-MSG sequences in the input population. To test this hypothesis, the UCS-MSG junctions in the input organism population were subjected to sequence analysis. In the first stage of this analysis, we cloned the DNA in four of the bands (bands 2, 3, 4, and 6) in the input band pattern (Fig. 3, lanes 1 and 20). We sequenced 11 plasmid inserts, 3 each from bands 2, 3, and 4 and 2 from band 6. These 11 plasmid inserts were all different in sequence, but all 11 sequences encoded an MSG protein. The percent identities of the MSG genes from different bands were 85.9 to 86.9 (band 2), 83 to 96 (band 3), 85.2 to 98.4 (band 4), and 99% (band 6).
To further evaluate the UCS-MSG junctions in the input population, an aliquot of the organisms in this population was amplified and the amplicons were cloned directly into plasmids without first purifying bands of different sizes via electrophoretic separation. Plasmids from 25 bacterial clones were sequenced. When these sequences were pooled with the data obtained by sequencing plasmids made from gel-purified bands, 36 sequences were available for comparison. Figure 5 shows the relatedness of these 36 sequences. One sequence was found four times, two sequences were found three times, and two sequences were found twice. Twenty-two sequences were present a single time. Therefore, there were 27 different MSG genes among the 36 UCS-MSG junctions sequenced. The sequences in Fig. 5 include those previously seen in five of the low-dose rats (267, 271, 272, 273, and 274). The absence of sequences seen in the other two low-dose rats may have been due to the incompleteness of the data from the input population. Very few sequences were seen in more than one plasmid, indicating that the plasmid library made from the input population contains more than the 27 sequences observed in the 36 plasmids sequenced.
FIG. 5.

Relatedness of MSG genes adjacent to the UCS locus in the population of P. carinii used to prepare inocula. Inserts in 36 plasmids carrying UCS-MSG junctions amplified from the input population were sequenced. Twenty-seven different sequences were present. Five sequences were present in more than one plasmid. These are numbered, e.g., 1.1 and 1.2. The sequences were aligned by pairwise comparisons, and percent identities were calculated. These are displayed in the dendrogram. The horizontal branch lengths represent divergence, and the bar above the dendrogram shows percent identity. To illustrate, sequences and 17 and 18 differed by ∼17%. Sequence 13 matched that predominant in inoculated rats 271 and 272. Sequence 23 matched 13, which matched that predominant in inoculated rats 273 and 274. The sequence in plasmids 15.1, 15.2, and 15.3 was the same as that seen in inoculated rat 267.
Low-dose inoculation radically reduced but did not eliminate variation at the UCS locus.
Analysis of gel-purified bands produced by amplification of the UCS locus in homogenates from rats inoculated with 10 organisms had detected just one sequence in each band. However, these data did not rule out the possibility of minor heterogeneity at the UCS loci in these samples. For example, minor junctions might be a different size than the gel-purified bands. To address this issue, the PCR products produced from rats 271, 272, and 273 were inserted directly into plasmids without first being subjected to gel electrophoresis. Multiple plasmids were isolated and sequenced (eight from 271 and seven each from 272 and 273).
This procedure showed that the UCS locus from each of the three rats was not homogeneous (Fig. 6). Six of the eight cloned PCR products from rat 271 had the sequence expected from previous sequencing analyses, but the other two plasmids each carried a different MSG sequence. Similarly, one of the seven plasmids carrying the PCR product from rat 272 was different from the other six. In rat 273, one of the seven plasmids was different from the other six.
FIG. 6.

Relatedness of UCS-linked MSG genes from rats inoculated with 10 P. carinii organisms and housed either together or separately. Twenty-five plasmids carrying UCS-MSG junctions amplified from three rats (271 and 272, which were cagemates, and 273, which was housed in a different cage) were aligned by pairwise comparisons. Percent identities were calculated and are displayed as a dendrogram. The horizontal branch lengths represent divergence, and the bar above the dendrogram shows percent identity. To illustrate, sequences 271.1 and 271.2 differed by ∼17%.
In each of the three rats, the minority sequence(s) was quite different from the predominant sequence (13 to 22% divergent). To illustrate this divergence, Fig. 7 shows a nucleotide alignment of majority and minority sequences from rat 271. The two sequences differ at 44 positions. This degree of divergence cannot be due to PCR or sequencing error, the frequency of which, in our hands, was one mistake per 4,000 bp amplified (data not shown).
FIG. 7.

Alignment of two UCS-linked MSG genes from rat 271. The sequence labeled 271.12 was in six of eight sequenced plasmids produced by cloning the amplicons from rat 271. Sequence 271.1 was in one of these plasmids. In both sequences, the first 25 bases are the CRJE (44), which is a conserved sequence found at the beginning of all known MSG genes and at all known USC-MSG junctions. The dots represent identity, and the dashes represent gaps introduced to optimize the number of matches. The number at the end of each line of sequence indicates the number of nucleotides from the beginning of that sequence. The two sequences differ at 54 positions (17%).
Minority sequences were not found among the 36 plasmids bearing UCS-MSG junctions amplified from the input population. A sample size of 36 is large enough to suggest that none of these sequences comprised more than 10% of that population. To confirm this suggestion, a more sensitive test was applied. Allele-specific PCR (ASP) was used to test the input population for the presence of sequence 271.2, one of the two minority sequences found in rat 271. Sequence 271.2 was selected as the target of this experiment because it contained an appropriately located region in which the sequence was different from the 27 sequences observed in the input population. Hence, an oligonucleotide that matched this region of 271.2 (primer ASP in Table 1) would be expected to amplify the 271.2 sequence and not to amplify most, if not all, of the other sequences in the input population DNA.
PCRs were performed using primer ASP paired with the −145 primer (Table 1), and the results were monitored in real time. The ASP produced a product, but only after >30 cycles. By contrast, control experiments showed that the UCS locus amplified much faster. The delay in ASP product formation indicated that the template for this product constituted no more than 0.6% of the UCS-MSG junctions in the input population. Subsequent sequence analysis, however, showed that the DNA amplified in the ASP did not match the 271.2 sequence but instead matched a sequence that had been seen once in the collection of plasmids carrying UCS-MSG junctions amplified from the input population. This sequence matched the ASP primer at all but one position, which was near the 5′ end of the primer, so it is not surprising, in retrospect, that the ASP supported amplification of this gene. Because the 271.2 sequence was not detected under conditions that allowed amplification of a related gene that constituted no more than 0.6% of the UCS-MSG junctions, we conclude that if the 271.2 sequence is in the input population, it is less abundant than 0.6%.
Cagemates tended to have the same UCS-MSG junction.
Before and after inoculation, rats had been kept two to a cage. The eight rats in the low-dose group were housed as four pairs of cagemates as follows: 267 with 268, 269 with 270, 271 with 272, and 273 with 274. Rat 270 did not produce a PCR product, leaving three pairs of cagemates for analysis. In two of the three pairs of cagemates, 271-272 (Fig. 3, lanes 4 and 5) and 273-274 (data not shown), the UCS-MSG junction band was the same size in both members of the pair. In the remaining pair (267-268), the band patterns were more complex, with three bands in 267 and two in its cagemate, 268, but the two smaller bands of 267 comigrated with the bands in 268 (Fig. 3, lanes 2 and 3). Sequence analysis showed that each cage had a different MSG predominant at the UCS locus but that the two rats in a cage had the same predominant UCS-MSG junction.
These results are unlikely to occur by chance. To illustrate, in rat pair 271-272 one UCS-MSG junction predominated. The probability that this particular genotype will be the one to expand to form the predominant USC-MSG genotype in a rat is no greater than 1/27, or 0.037, because there were at least 27 different UCS-MSG junctions in the input population. The probability that both rats in a cage will have this particular genotype predominate is 0.0372. However, because there are 27 different genotypes to choose from, the probability that any two rats will have the same genotype is the sum of all probabilities of this happening for a particular genotype, 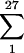 0.0372 = 0.037. Hence, the probability of seeing two rats with the same genotype is low, and the probability of seeing two sets of cagemates that share a genotype is extremely unlikely to occur by chance.
0.0372 = 0.037. Hence, the probability of seeing two rats with the same genotype is low, and the probability of seeing two sets of cagemates that share a genotype is extremely unlikely to occur by chance.
Sharing of genotypes between cagemates was also seen by comparing the minority sequences. One of the minority DNA sequences in rat 271 was 99.7% identical to the minority sequence of rat 272.
DISCUSSION
A previous study of rats from open-air colonies had shown that variation at the UCS locus was pronounced and common (41). In the previous study, all 37 rats analyzed contained P. carinii organisms that were heterogeneous at the UCS locus. In exhibiting this high degree of variation, the UCS locus is extraordinary compared to the remainder of the P. carinii genome, where sequence variation is very rarely observed, even at loci that would be expected to be prone to mutation due to their structure, such as the presence of microsatellites. The studies presented here showed that, in contrast to those in rats from the open-air colony, the UCS locus was much less complex in some of the rats that developed pneumonia following the provocation of latent P. carinii organisms and in all of the rats that received a low-dose of P. carinii via inoculation. The association between high variation at the UCS locus and the natural-transmission method suggests that high variation may be caused by infection of a given rat by numerous P. carinii organisms, each with a different UCS-MSG junction. The presence of P. carinii with different UCS-MSG junctions may be due to the accumulation of different UCS-MSG junctions in the population of P. carinii being produced by the open-air colony as a whole. A long period of accumulation is possible, because this colony has been maintained for years, during which time animals from different commercial colonies have been introduced. Given the high frequency of latency in commercial colonies, most, if not all, of the rats introduced into the open-air colony can be assumed to have been harboring latent P. carinii organisms.
The UCS diversity in P. carinii obtained by immunosuppression of latently infected rats may have come from infection with multiple P. carinii organisms prior to being placed under a barrier. Alternatively, diversity may be generated after infection. Latent infections are probably initiated in neonates, which are immunonaive but immunocompetent (1). With time, the animals might detect the presence of P. carinii and mount a response, which could select for switching at the UCS locus. This scenario differs from what would be expected in the rats given a low dose of P. carinii, because inoculated animals were immunosuppressed before and after inoculation.
To determine whether the cause of UCS diversity in latent infections is due to events that happen before or after infection, it is necessary to control the source of the P. carinii organisms that cause infections. The results of the low-dose inoculation experiments suggest that such control can be exerted via inoculation.
The data obtained from the low-dose inoculation experiments suggest a maximum frequency for switching the MSG gene at the UCS locus. This rate, r, can be estimated from the relationship f = (1 − r)d, where f is the fraction of organisms that have the predominant MSG at the UCS locus and d is the number of population doublings. f is known, and d can be estimated from the difference between the number of organisms introduced and the final population size. Inoculation with 10 P. carinii organisms typically produced a population numbering approximately 108 organisms. To increase the population size from 10 to 108 would require at least 23 doublings (223 = 0.84 × 107). However, it is probable that >23 doublings transpired, because most of the 10 P. carinii organisms introduced did not appear to measurably contribute to the population present at the end of the experiment. (That few organisms proliferated can be inferred from the relative uniformity of the UCS locus at the end of the experiment compared to the heterogeneity at the UCS locus in the input population from which inocula were drawn.) If only one organism proliferated, then d would be 27 doublings. When 23 < d < 27 and f (the fraction of UCS-MSG junctions with the predominant MSG) is 0.8, the calculated rate of switching, r, is approximately 0.01 events per P. carinii organism per doubling, or one switch for every 100 organisms. It could be argued that it is inappropriate to base such calculations on population doublings because the manner by which P. carinii reproduces is not known and may not involve simple mitotic cell division. If mitotic reproduction were not the proliferation mechanism, the presumptive alternative would be via mating followed by sporulation, in which case proliferation would proceed by using two mating partners to produce eight spores, thereby increasing the population in fourfold increments rather than twofold. If such a mating process were the sole means of reproduction, the population size (p) would be related to the number of mating generations (m) as follows: p = 22m − 1. To increase from 10 to 108 organisms via mating would require 11 mating generations. Therefore, since f = (1 − r)m, the calculated rate of change at the UCS (r), as a function of mating generations (m), is 0.02 event per mating generation, or one switch for every 50 mating events.
Admittedly, estimating the switching rate is speculative at this point, especially since the minority sequences seen at the UCS locus need not have been formed by switching in the inoculated rat. Alternative sources of the minority sequences seen at the UCS locus include proliferation of more than one of the organisms introduced by inoculation and a contribution by latent P. carinii organisms. While we cannot conclude that switching was observed, it is interesting to consider the theoretical long-term ramifications of a switching rate of 0.01 event per organism per doubling. Starting from a single P. carinii organism, switching at this rate over the course of 100 doublings would generate a population in which most (two-thirds) of the members would have an MSG at the UCS locus that is different from that in the P. carinii organism that founded the population. Hence, switching at this rate for a period of months or years would seem to be sufficient to generate the high levels of heterogeneity at the UCS locus observed in the open-air colony.
While the UCS locus can be highly heterogeneous in certain populations of P. carinii, the rate at which it changes is low enough to allow UCS diversity to be used as a tool to understand population dynamics in inoculated rats. It has been known for years that inoculation with high doses of P. carinii (on the order of 106 organisms) causes heavy infection in most rats, but the details of the infection process had not been examined. The possibilities span two extremes. At one extreme is the possibility that only one organism reproduces to eventually cause disease. At the other extreme is the possibility that all of the input organisms reproduce themselves. Tracking the UCS-MSG junctions present in input and output populations in rats that received at least 104 P. carinii organisms suggests that neither of these two extremes is the case. Rather, it seems that multiple organisms contribute to the infection when the dose is in the tens of thousands. However, the fraction of introduced organisms that contributes to proliferation may be as low as 1 in 1,000.
Multiple organisms must have contributed to population growth in rats inoculated with 107 P. carinii, because these rats produced populations with the same complex collection of UCS-MSG junctions as the input population; at least this appeared to be true based on the sizes of the UCS-MSG junctions amplified. It is not possible to say at this point if the populations produced in the nine rats given 107 P. carinii organisms were identical to the input, but it seems probable. This issue can be addressed by subjecting the output P. carinii populations to further analysis by sequencing UCS-MSG junctions. However, such an analysis would require the sequencing of hundreds of cloned UCS-MSG junctions, which was beyond the scope of this study.
While all of the rats inoculated with 107 P. carinii organisms produced the same band pattern upon amplification of the UCS-MSG junction, the patterns produced from rats inoculated with 104 organisms varied with respect to the intensities of the bands. Fig. 3, lane 8 shows an example of intensity variation. Band 8 is very faint in this lane. The near loss of this band suggests that only a small fraction of the input organisms proliferated. This fraction can be estimated from the following. In the input band pattern, band 8 is one of five bands that stand out as being most intense. Therefore, the fraction of P. carinii organisms in the input population that have a UCS-MSG junction that produces a PCR product the size of band 8 is between 10 and 20%. If we assume that this fraction is 15%, then the probability that this band would not be represented among the organisms that proliferated is given by P = 0.85x, where x is the number of organisms that proliferate. If we assume that 1,000 of the 104 input organisms contributed to the population, then P = 0.851,000 = 2 × 10−71. Hence, it is very improbable that the band in question would be missing if 1,000 organisms were to proliferate. It follows that the chance of seeing variation in the intensity of this band would also be negligibly small. By contrast, if only 10 of the 104 input organisms founded the population, then P = 0.8510 = 0.2, and it is probable that the band would be missing in ∼20% of the rats. Therefore, it is reasonable to speculate that few of the input P. carinii organisms, perhaps only 10, proliferated in rats inoculated with 104 organisms. It is interesting that if the efficiency of proliferation in rats given 107 P. carinii organisms were similarly low, i.e., 10 in 104, approximately 1,000 organisms would contribute to the infection, in which case one would expect to see no variation in band intensities, as was observed.
While multiple organisms appear to have proliferated in the rats that received 104 or more P. carinii organisms, results with rats that received only 10 P. carinii organisms suggest that only 1 may be required to cause disease. In four of these animals (rats 271, 272, 273, and 274), the P. carinii organisms exhibited characteristics that would be expected of a clonally derived population, i.e., a single predominant UCS-MSG junction, which was also present in the input population. On the other hand, the UCS-MSG junction was not completely homogeneous, which would seem to argue against clonality. However, the small fraction of organisms with a different UCS-MSG junction may have been generated by switching at the UCS locus. The possibility of switching is supported by the observation that the minority UCS-MSG junctions were not found in the input population. This observation is as would be expected if new MSG genes were to be installed at the UCS in some organisms as the population expanded. While it would be premature to conclude that switching generated the heterogeneity at the UCS locus, the consistent production of populations of P. carinii in which most (80 to 90%) of the members have the same UCS-MSG junction fits well with what would be expected from switching. By contrast, proliferation of two organisms might be expected to produce populations in which two genotypes are present in roughly equal numbers. Regardless of whether switching occurred, the heterogeneity at the UCS locus was low enough to suggest that just a few of the 10 P. carinii organisms introduced proliferated.
The rate at which the P. carinii population increases in vivo has not been well studied. Inoculation helps address this question by delivering a measured number of organisms at a fixed time. The rats that received 10 P. carinii organisms carried an average of 2 × 108 P. carinii organisms per animal at the end of the experiments (12 weeks [84 days] postinoculation). The minimum number of doublings required to increase from 1 to 108 organisms is approximately 27. Therefore, if only one input organism contributed to the infection, then the apparent doubling time was approximately 3.1 days (84 days/27 doublings). If all 10 of the input organisms proliferated, then the calculated doubling time increases to 3.5 days. These doubling time estimates are subject to caveats. The doubling time could have been <3.1 days if the proliferation period were shorter than 12 weeks. This parameter can be analyzed in the future. The only way that the true doubling time could have been much more than 3.5 days would be if many more than 10 P. carinii contributed to population growth. For example, if doubling the population actually required 5 days, then the production of 108 P. carinii organisms in 12 weeks would require a founding population of 1,000 P. carinii organisms. Since only 10 P. carinii organisms were introduced, the only way the founding population could have been larger would be via the contribution of latent organisms. The presence of such a large number of latent P. carinii organisms seems very unlikely. The sham-inoculated rats produced no detectable P. carinii DNA, indicating that if latent organisms were present, they were at a very low level. Therefore, all indications are that the doubling time of P. carinii in low-dose rats was <3.5 days.
Another interesting observation that emerged from the inoculation studies was the tendency of low-dose rats kept in the same cage to have the same MSG gene at the UCS locus. This observation suggests that one animal may have infected the other in the cage. Even though both animals in each pair of cagemates were inoculated, it is possible that only one was infected by the inoculation because the dose was low. The transmission scenario fits with the large difference in organism burden between rats 271 and 272, for instance. The alternative to the transmission scenario is that the sharing of genotypes in cagemates was due to chance. Indeed, it is nearly certain that the two aliquots of P. carinii used to infect the pair of rats in a given cage shared at least one genotype. Nevertheless, the probability that both rats in a cage would have the same predominant genotype by chance is only 0.037. Hence, the presence of the same predominant UCS-MSG junction in multiple pairs of cagemates seems unlikely to have been due to chance.
A more surprising aspect of the cagemate data was the presence of the same minority UCS-MSG junctions in one pair of cagemates. This situation may have been caused by transmission of the majority UCS-MSG junction, followed by a programmed switch to the minority MSG gene. Alternatively, it is possible that one cagemate was infected by one UCS-MSG genotype and the other by the other genotype, followed by transmission between the cagemates in both directions. Another possibility is that both UCS-MSG junctions were transmitted from one rat to the other, perhaps independently, perhaps by a transmissible form of the organism that carries both genotypes.
In summary, the P. carinii populations in rats that received 10 organisms were much less complex at the UCS locus than the input population. The populations of P. carinii observed in the low-dose rats were at least 80% homogeneous with respect to the UCS locus. One explanation of these results is that only one organism founded the population and that switching occurred as the population grew. Whether or not this explanation proves to be correct, the presence of populations that are at least quasiclonal strongly suggests that inoculation with one organism can be used to clone P. carinii, provided that one can obtain rats that are free of P. carinii prior to being inoculated. In any event, our data suggest that when the objective is to obtain P. carinii that are relatively homogeneous with respect to MSG, it would be advantageous to use low-dose inoculation rather than rats infected via constant exposure to airborne P. carinii. Alternatively, latently infected rats can be screened for populations of P. carinii that are relatively simple at the UCS locus.
Acknowledgments
We thank Mario Medvedovic and Gary Weiss for advice on statistics and probability.
This work was supported by grants R01AI36701 (J.R.S.) and R01AI29839 (M.T.C.), both from the National Institute of Allergy and Infectious Diseases.
Editor: J. M. Mansfield
REFERENCES
- 1.Aliouat, E. M., R. Escamilla, C. Cariven, C. Vieu, C. Mullet, E. Dei-Cas, and M. C. Prevost. 1998. Surfactant changes during experimental pneumocystosis are related to Pneumocystis development. Eur. Respir. J. 11:542-547. [PubMed] [Google Scholar]
- 2.Anonymous. 1994. Revised nomenclature for Pneumocystis carinii. The Pneumocystis Workshop. J. Eukaryot. Microbiol. 41:121S-122S. [PubMed] [Google Scholar]
- 3.Armstrong, M. Y., and M. T. Cushion. 1994. Animal models, p. 181-222. In P. D. Walzer (ed.), Pneumocystis carinii pneumonia. Marcel Dekker, New York, N.Y.
- 4.Bartlett, M. S., J. A. Fishman, S. F. Queener, M. M. Durkin, M. A. Jay, and J. W. Smith. 1988. New rat model of Pneumocystis carinii infection. J. Clin. Microbiol. 26:1100-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartlett, M. S., S. F. Queener, M. A. Jay, M. M. Durkin, and J. W. Smith. 1987. Improved rat model for studying Pneumocystis carinii pneumonia. J. Clin. Microbiol. 25:480-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowling, M. C., I. M. Smith, and S. L. Wescott. 1973. A rapid staining procedure for Pneumocystis carinii. Am. J. Med. Technol. 39:267-268. [PubMed] [Google Scholar]
- 7.Boylan, C. J., and W. L. Current. 1992. Improved rat model of Pneumocystis carinii pneumonia: induced laboratory infections in Pneumocystis-free animals. Infect. Immun. 60:1589-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandler, F. W., Jr., J. K. Frenkel, and W. G. Campbell, Jr. 1979. Pneumocystis pneumonia. Animal model: Pneumocystis carinii pneumonia in the immunosuppressed rat. Am. J. Pathol. 95:571-574. [PMC free article] [PubMed] [Google Scholar]
- 9.Cushion, M. T., and D. Ebbets. 1990. Growth and metabolism of Pneumocystis carinii in axenic culture. J. Clin. Microbiol. 28:1385-1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cushion, M. T., M. Kaselis, S. L. Stringer, and J. R. Stringer. 1993. Genetic stability and diversity of Pneumocystis carinii infecting rat colonies. Infect. Immun. 61:4801-4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cushion, M. T., J. J. Ruffolo, M. J. Linke, and P. D. Walzer. 1985. Pneumocystis carinii: growth variables and estimates in the A549 and WI-38 VA13 human cell lines. Exp. Parasitol. 60:43-54. [DOI] [PubMed] [Google Scholar]
- 12.Cushion, M. T., D. Stanforth, M. J. Linke, and P. D. Walzer. 1985. Method of testing the susceptibility of Pneumocystis carinii to antimicrobial agents in vitro. Antimicrob. Agents Chemother. 28:796-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DePrimo, S. E., P. J. Stambrook, and J. R. Stringer. 1996. Human placental alkaline phosphatase as a histochemical marker of gene expression in transgenic mice. Transgenic Res. 5:459-466. [DOI] [PubMed] [Google Scholar]
- 14.Edman, J. C., J. A. Kovacs, H. Masur, D. V. Santi, H. J. Elwood, and M. L. Sogin. 1988. Ribosomal RNA sequence shows Pneumocystis carinii to be a member of the fungi. Nature 334:519-522. [DOI] [PubMed] [Google Scholar]
- 15.Eriksson, O. E. 1994. Pneumocystis carinii, a parasite in lungs of mammals, referred to a new family and order (Pneumocystidaceae, Pneumocystidales, Ascomycota). Systema Ascomycetum 13:165-180. [Google Scholar]
- 16.Frenkel, J. K. 1999. Pneumocystis pneumonia, an immunodeficiency-dependent disease (IDD): a critical historical overview. J. Eukaryot. Microbiol. 46:89S-92S. [PubMed] [Google Scholar]
- 17.Frenkel, J. K., J. T. Good, and J. A. Shultz. 1966. Latent Pneumocystis infection of rats, relapse, and chemotherapy. Lab. Investig. 15:1559-1577. [PubMed] [Google Scholar]
- 18.Gigliotti, F. 1992. Host species-specific antigenic variation of a mannosylated surface glycoprotein of Pneumocystis carinii. J. Infect. Dis. 165:329-336. [DOI] [PubMed] [Google Scholar]
- 19.Gigliotti, F., L. R. Ballou, W. T. Hughes, and B. D. Mosley. 1988. Purification and initial characterization of a ferret Pneumocystis carinii surface antigen. J. Infect. Dis. 158:848-854. [DOI] [PubMed] [Google Scholar]
- 20.Hendley, J. O., and T. H. Weller. 1971. Activation and transmission in rats of infection with Pneumocystis. Proc. Soc. Exp. Biol. Med. 137:1401-1404. [DOI] [PubMed] [Google Scholar]
- 21.Hong, S. T., P. E. Steele, M. T. Cushion, P. D. Walzer, S. L. Stringer, and J. R. Stringer. 1990. Pneumocystis carinii karyotypes. J. Clin. Microbiol. 28:1785-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Icenhour, C. R., S. L. Rebholz, M. S. Collins, and M. T. Cushion. 2001. Widespread occurrence of Pneumocystis carinii in commercial rat colonies detected using targeted PCR and oral swabs. J. Clin. Microbiol. 39:3437-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keely, S. P., and J. R. Stringer. 1997. Sequences of Pneumocystis carinii f. sp. hominis strains associated with recurrent pneumonia vary at multiple loci. J. Clin. Microbiol. 35:2745-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovacs, J. A., J. L. Halpern, J. C. Swan, J. Moss, J. E. Parrillo, and H. Masur. 1988. Identification of antigens and antibodies specific for Pneumocystis carinii. J. Immunol. 140:2023-2031. [PubMed] [Google Scholar]
- 25.Linke, M. J., M. T. Cushion, and P. D. Walzer. 1989. Properties of the major antigens of rat and human Pneumocystis carinii. Infect. Immun. 57:1547-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linke, M. J., and P. D. Walzer. 1989. Analysis of a surface antigen of Pneumocystis carinii. J. Protozool. 36:60S-61S. [DOI] [PubMed] [Google Scholar]
- 27.Linke, M. J., and P. D. Walzer. 1991. Identification and purification of a soluble species of gp120 released by zymolyase treatment of Pneumocystis carinii. J. Protozool. 38:176S-178S. [PubMed] [Google Scholar]
- 28.Lundgren, B., C. Koch, L. Mathiesen, J. O. Nielsen, and J. E. Hansen. 1993. Glycosylation of the major human Pneumocystis carinii surface antigen. APMIS 101:194-200. [PubMed] [Google Scholar]
- 29.Lundgren, B., G. Y. Lipschik, and J. A. Kovacs. 1991. Purification and characterization of a major human Pneumocystis carinii surface antigen. J. Clin. Investig. 87:163-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merali, S., U. Frevert, J. H. Williams, K. Chin, R. Bryan, and A. B. Clarkson, Jr. 1999. Continuous axenic cultivation of Pneumocystis carinii. Proc. Natl. Acad. Sci. USA 96:2402-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura, Y., K. Tanabe, and K. Egawa. 1989. Structure of major surface determinants and DNA diagnosis of Pneumocystis carinii. J. Protozool. 36:58S-60S. [DOI] [PubMed] [Google Scholar]
- 32.Paulsrud, J. R., S. F. Queener, M. S. Bartlett, and J. W. Smith. 1991. Isolation and characterization of rat lung Pneumocystis carinii gp120. J. Protozool. 38:10S-11S. [PubMed] [Google Scholar]
- 33.Pesanti, E. L., and J. D. Shanley. 1988. Glycoproteins of Pneumocystis carinii: characterization by electrophoresis and microscopy. J. Infect. Dis. 158:1353-1359. [DOI] [PubMed] [Google Scholar]
- 34.Pixley, F. J., A. E. Wakefield, S. Banerji, and J. M. Hopkin. 1991. Mitochondrial gene sequences show fungal homology for Pneumocystis carinii. Mol. Microbiol. 5:1347-1351. [DOI] [PubMed] [Google Scholar]
- 35.Radding, J. A., M. Y. Armstrong, E. Ullu, and F. F. Richards. 1989. Identification and isolation of a major cell surface glycoprotein of Pneumocystis carinii. Infect. Immun. 57:2149-2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook, J. F., E. F. Fristch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 37.Sloand, E., B. Laughon, M. Armstrong, M. S. Bartlett, W. Blumenfeld, M. Cushion, A. Kalica, J. A. Kovacs, W. Martin, E. Pitt, et al. 1993. The challenge of Pneumocystis carinii culture. J. Eukaryot. Microbiol. 40:188-195. [DOI] [PubMed] [Google Scholar]
- 38.Stringer, J. R. 1996. Pneumocystis carinii: what is it, exactly? Clin. Microbiol. Rev. 9:489-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stringer, S. L., J. R. Stringer, M. A. Blase, P. D. Walzer, and M. T. Cushion. 1989. Pneumocystis carinii: sequence from ribosomal RNA implies a close relationship with fungi. Exp. Parasitol. 68:450-461. [DOI] [PubMed] [Google Scholar]
- 40.Sunkin, S. M., and J. R. Stringer. 1996. Translocation of surface antigen genes to a unique telomeric expression site in Pneumocystis carinii. Mol. Microbiol. 19:283-295. [DOI] [PubMed] [Google Scholar]
- 41.Sunkin, S. M., and J. R. Stringer. 1997. Residence at the expression site is necessary and sufficient for the transcription of surface antigen genes of Pneumocystis carinii. Mol. Microbiol. 25:147-160. [DOI] [PubMed] [Google Scholar]
- 42.Sunkin, S. M., S. L. Stringer, and J. R. Stringer. 1994. A tandem repeat of rat-derived Pneumocystis carinii genes encoding the major surface glycoprotein. J. Eukaryot. Microbiol. 41:292-300. [DOI] [PubMed] [Google Scholar]
- 43.Tanabe, K., S. Takasaki, J. Watanabe, A. Kobata, K. Egawa, and Y. Nakamura. 1989. Glycoproteins composed of major surface immunodeterminants of Pneumocystis carinii. Infect. Immun. 57:1363-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wada, M., S. M. Sunkin, J. R. Stringer, and Y. Nakamura. 1995. Antigenic variation by positional control of major surface glycoprotein gene expression in Pneumocystis carinii. J. Infect. Dis. 171:1563-1568. [DOI] [PubMed] [Google Scholar]
- 45.Wakefield, A. E., F. J. Pixley, S. Banerji, K. Sinclair, R. F. Miller, E. R. Moxon, and J. M. Hopkin. 1990. Amplification of mitochondrial ribosomal RNA sequences from Pneumocystis carinii DNA of rat and human origin. Mol. Biochem. Parasitol. 43:69-76. [DOI] [PubMed] [Google Scholar]
- 46.Walzer, P. D., and M. J. Linke. 1987. A comparison of the antigenic characteristics of rat and human Pneumocystis carinii by immunoblotting. J. Immunol. 138:2257-2265. [PubMed] [Google Scholar]
- 47.Walzer, P. D., R. D. Powell, Jr., K. Yoneda, M. E. Rutledge, and J. E. Milder. 1980. Growth characteristics and pathogenesis of experimental Pneumocystis carinii pneumonia. Infect. Immun. 27:928-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walzer, P. D., D. Stanforth, M. J. Linke, and M. T. Cushion. 1987. Pneumocystis carinii: immunoblotting and immunofluorescent analyses of serum antibodies during experimental rat infection and recovery. Exp. Parasitol. 63:319-328. [DOI] [PubMed] [Google Scholar]