

Abstract
Unimolecular rate constants for blackbody infrared radiative dissociation (BIRD) were calculated for the model protonated peptide (AlaGly)n (n = 2–32) using a variety of dissociation parameters. Combinations of dissociation threshold energies ranging from 0.8 to 1.7 eV and transition entropies corresponding to Arrhenius preexponential factors ranging from very “tight” (A∞ = 109.9 s−1) to “loose” (A∞ = 1016.8 s−1) were selected to represent dissociation parameters within the experimental temperature range (300–520 K) and kinetic window (kuni = 0.001–0.20 s−1) typically used in the BIRD experiment. Arrhenius parameters were determined from the temperature dependence of these values and compared to those in the rapid energy exchange (REX) limit. In this limit, the internal energy of a population of ions is given by a Boltzmann distribution, and kinetics are the same as those in the traditional high-pressure limit. For a dissociation process to be in this limit, the rate of photon exchange between an ion and the vacuum chamber walls must be significantly greater than the dissociation rate. Kinetics rapidly approach the REX limit either as the molecular size or threshold dissociation energy increases or as the transition-state entropy or experimental temperature decreases. Under typical experimental conditions, peptide ions larger than 1.6 kDa should be in the REX limit. Smaller ions may also be in the REX limit depending on the value of the threshold dissociation energy and transition-state entropy. Either modeling or information about the dissociation mechanism must be known in order to confirm REX limit kinetics for these smaller ions. Three principal factors that lead to the size dependence of REX limit kinetics are identified. With increasing molecular size, rates of radiative absorption and emission increase, internal energy distributions become relatively narrower, and the microcanonical dissociation rate constants increase more slowly over the energy distribution of ions. Guidelines established here should make BIRD an even more reliable method to obtain information about dissociation energetics and mechanisms for intermediate size molecules.
Introduction
The development of “soft” ionization methods, such as electrospray ionization (ESI)1,2 and matrix-assisted laser desorption ionization (MALDI),3 has led to an explosive growth in the application of mass spectrometry to the investigation of the intrinsic properties of large gas-phase biomolecule ions and ionic complexes over the past decade. Of particular interest is the development of methods to obtain information about dissociation energetics and mechanisms of sequence-specific bond cleavages in peptides,4–11 proteins,11–17 and DNA.18–22 A thorough understanding of dissociation energetics and dynamics for these large ions can aid in the development of predictive fragmentation models which would make mass spectrometry an even more powerful analytical technique for obtaining sequence information. Specific noncovalent interactions, such as enzyme–substrate and antibody–antigen complexes and DNA double-helix formation, play a critical role in the function of many biological molecules. The question of whether solution-phase specificity is retained in the gas phase may have important experimental and theoretical implications for enhanced drug discovery strategies and is an active area of research.23–28 One approach would be to compare gas-phase binding energies with physiological solution binding constants for a number of different receptor–ligand complexes. Correlation between these values would indicate the degree of specificity, if any, that is retained in the absence of solvent.
Measuring accurate dissociation and binding energies for large biomolecule ions provides a difficult challenge. Methods in which a controlled quantity of energy is imparted into the precursor ion and the resulting extent and type of fragmentation measured have been used to determine the effect of sequence on relative dissociation energies. Photodissociation using different wavelengths can be used to deposit precise quantities of energy into an ion provided the number of photons absorbed can be accurately determined.29–33 Variable amounts of internal energy can be deposited into an ion via collisions with surfaces10 or neutral gas-phase atoms or molecules34 by changing center-of-mass collision velocities. Ion internal energy can also be varied using gas-phase proton-transfer (chemical ionization) reactions with bases of varying gas-phase basicity.35 Although these activation methods can produce a population of ions with a known maximum or average energy, the internal energy distribution may be poorly characterized due to uncertainties in the energy of the initial population and in the energy transferred by the activating process.36 A further complication is that large ions have many internal modes into which energy can be partitioned. To observe fragmentation during a short time frame, an amount of energy larger than the threshold dissociation energy (E0) must be deposited into the ion to ensure that the bond or bonds along the reaction coordinate have a significant probability of accumulating enough energy to become activated. This can result in a “kinetic shift” in threshold energy measurements for large ions that may be many times larger than E0 itself.37 In principle, statistical methods, such as RRKM theory, can be used to extract the true energetics from the experimental kinetic data if both the internal energy of the ion population is known and the transition state for the dissociation can be accurately modeled.
The activation energy (Ea) for a dissociation process can be obtained directly from variable temperature thermal dissociation methods via Arrhenius analysis for any size ion.38 A simple relation between E0 and Ea is given by the Tolman theorem if the ion population has a canonical internal energy distribution.39 Information about the dynamics and mechanism of the dissociation is contained in the Arrhenius preexponential factor (A).39 In a thermal method recently implemented by both Meot-Ner et al.7 and Busman et al.,40 ESI generated peptide ions were dissociated in a heated, variable temperature reaction chamber located at the electrospray interface. Activation energies were determined from the temperature dependence of the extent of reaction.
We have recently demonstrated a new thermal technique for the activation of biomolecule ions as large as myoglobin (17.0 kDa): blackbody infrared radiative dissociation (BIRD).9,16,28,41,42 This method has the advantage that precursor ions can be isolated and that potential interference from ion desolvation is eliminated. As first demonstrated for small, weakly bound clusters by McMahon and co-workers,43 ions trapped in the heated chamber of a Fourier transform mass spectrometer at pressures less than 10−8 Torr dissociate by absorbing infrared photons from the blackbody field. From the temperature dependence of the unimolecular dissociation constant, Arrhenius activation parameters in the zero-pressure limit can be obtained. A simple expression for the collisionless unimolecular rate constant (kuni,zpl) is given by
| (1) |
where k1,rad and k−1,rad are the energy-dependent rate constants of blackbody photon absorption and emission, respectively, and kd is the energy-dependent microcanonical dissociation rate constant. Three regimes given by the relative values of k−1,rad and kd can be defined.
For small ions with entropically favored dissociation channels, the rate of dissociation can greatly exceed the rate of photon emission (k−1,rad, ≪ kd). A dissociating population does not reach thermal equilibrium under these conditions. In this regime, an ion that is activated above E0 promptly dissociates. The measured zero-pressure limit Ea and A factors for these small ions are significantly lower than the values obtained from a Boltzmann distribution of ions. Dunbar has shown that the ion population can be modeled as a Boltzmann distribution which is truncated at E0 and that E0 can be obtained by adding a correction factor to the measured Ea.44 The measured A factor, however, contains no information about the dynamics of dissociation but depends only on the rate of photon absorption.44
For slightly larger ions, such as amino acid proton-bound dimers, the truncated Boltzmann analysis is no longer valid.42 For these intermediate size ions, the dissociation rates become competitive with those of photon emission (k−1,rad ≈ kd). Some energy states above E0 are populated. However, thermal equilibrium is not yet established. The population is Boltzmann-like but still depleted at higher energies. The measured Ea and A factors are smaller than those measured in the high-pressure limit. For these intermediate size ions, accurate values of E0 can be obtained from master equation analysis of the measured activation parameters.42
Under readily achievable conditions, large ions can absorb and emit photons at a rate significantly faster than they dissociate (k−1,rad ≫ kd). Ions can reach thermal equilibrium with the vacuum chamber walls, and the measured dissociation kinetics are the same as those that would be obtained in the high-pressure limit.16 We have termed this the rapid energy exchange (REX) limit and define the Arrhenius parameters in this limit as and A∞. Evidence for REX limit kinetics for large ions include both results from master equation modeling of ions as a function of size9,16 and experimental measurements of large A factors.16,45 For example, A factors of 1017 s−1 have been measured for the dissociation of the proton and alkali-metal cation-bound dimers of the pentapeptide leucine enkaphalen.45 This value is comparable to the highest values that have been reliably measured for small molecules.38 A factors of 1015–18 s−1 correspond to entropically favored direct bond cleavages and are consistent with values expected for dissociation of ion–dipole interactions.46 If dissociation of the leucine enkaphalen dimer were not in the REX limit, A∞ would have to be even higher than the measured value. While even larger values cannot be ruled out for biomolecule ion dissociations, results from master equation modeling indicate that the energy distribution of the ion population does not significantly differ from that of a Boltzmann. Both these pieces of evidence suggest that this dissociation process is in the REX limit.
Factors that influence the relative rates of photon absorption and emission to the rate of dissociation include ion type and size, E0, A∞, the experimental temperature range, and kinetic window. Here, we model dissociation kinetics of protonated oligopeptides, (AlaGly)n (n = 2–32), under typical conditions used in BIRD experiments. Effects of the above factors on the BIRD kinetics are determined, and guidelines for dissociation processes in the REX limit are established.
Calculations
Radiative and Dissociation Parameters.
Lowest energy structures of protonated (AlaGly)n (n = 2, 4, and 5) were found using the consistent valence force field (CVFF) provided in the InsightII/Discover suite of programs (Biosym Technologies, San Diego, CA) on an IBM RS/6000 computer. The proton was placed on the N-terminus. Starting geometries were randomly selected. Using a 1 fs step size, dynamics were run at 800 K for 10 ps followed by simulated annealing over a period of 4 ps to 200 K. An additional 6 kcal/mol torsional barrier was imposed on each peptide bond to prevent facile rotations about this partial double bond. The structure was then energy minimized to 0 K and used as the starting geometry for the next dynamics cycle. This process was repeated 120 times to produce an ensemble of energy-minimized structures for each peptide.
The lowest energy structure of each (AlaGly)n peptide ion obtained from the molecular dynamics was used as the starting geometry for AM1 semiempirical calculations using MOPAC 6.0 on an IBM RS/6000 computer. Minimum-energy structures were found using the eigenvector following routine. Vibrational frequencies and transition dipole moments were calculated from these minimum-energy structures.
Transition-state frequency sets were formed for (AlaGly)n (n= 2, 4) by removing the C–N stretch (1475 cm−1) of the first (AlaGly) subunit (considered to be the reaction coordinate). Five additional frequencies (23,125,159, 267, and 945 cm−1) were systematically varied to reproduce REX limit Arrhenius pre-exponential factors of 109.9, 1012.4, 1014.5, and 1016.8 s−1.
Reactant and transition-state frequency sets for (AlaGly)n (n>4) were compiled by explicitly calculating the vibrational frequencies and intensities for both (AlaGly)4 and (AlaGly)5. Vibrational modes found in the (AlaGly)5 frequency set but not in the (AlaGly)4 set are compiled into a new frequency set (including the associated transition dipole moments) that is used to expand the (AlaGly)4 model peptide by one AlaGly unit. In this fashion, a peptide of arbitrary length, (AlaGly)n, can be constructed by successively appending this new AlaGly subunit to (AlaGly)n−1. Thus, low-frequency cooperative modes can be included in the larger systems without having to calculate them explicitly.
Master Equation.
A full description of our implementation of the master equation has been previously given.42 Briefly, the master equation model numerically simulates the experiment as the solution to a set of coupled differential equations explicitly accounting for the detailed rates of all internal state-to-state transitions (ki,j) and dissociation processes (kd). To model the dissociation of these peptides, a discrete value (100 cm−1 energy grain size), finite-difference approximation to the master equation is used. The coupled equations are given by eq 2
| (2) |
where Nj(t), Ni(t), and dNi(t) are the population fraction in energy level j, i and the time-dependent change in population fraction in energy level i, respectively. The microcanonical dissociation rates (kd) are determined from RRKM theory using the reactant and transition-state frequency sets described above. Effects of angular momentum are not included. The sum and density of states were calculated using the direct count Beyer–Swinehart algorithm.46
In the zero-pressure limit, the rate of state-to-state transitions are exclusively given by the rates of radiative absorption (k1,rad) and emission (k−1,rad).
| (3) |
A weakly coupled harmonic oscillator model was used to calculate the detailed rate constants for blackbody absorption, k1,rad, and for spontaneous and stimulated emission, k−1,rad.
| (4) |
| (5) |
Where is the product of the probability of the νth oscillator having m quanta of energy in the jth internal energy state and of the increased transition probability of an excited harmonic oscillator, ρ(hν) is the radiation density at ν given by the Planck distribution, and A(hν) and B(hν) are the Einstein coefficients for stimulated and spontaneous radiative transition processes, respectively, given by
| (6) |
| (7) |
Transition frequencies (ν) calculated at the AM1 semiempirical level were used without any further scaling. AM1 transition dipole moments (μ) were multiplied by a factor of 3.
The relative rates of spontaneous vs stimulated emission are given by the ratio of the two terms in brackets in eq 5. This ratio, at a given temperature, is expressed as an exponentially increasing function of the transition frequency. For example, at 400 K the spontaneous to stimulated emission ratio is 36 and 1340 for transitions of 1000 and 2000 cm−1, respectively. At higher temperatures, stimulated emission becomes relatively more favorable due to the increased photon flux of the blackbody field. At 500 K, the ratio becomes 17 and 317 for the 1000 and 2000 cm−1 transitions, respectively. Although spontaneous processes dominate the emission kinetics under the conditions modeled here, stimulated emission rates are included for completeness.
Master equation modeling was performed with software developed in this laboratory using double-precision Fortran 77 code. The stiffly coupled set of equations given by eq 2 was solved by numerical integration using the backward differentiation formula (BDF) routine (DO2NCF) provided within the NAG Mark 17 library. A Boltzmann distribution at the temperature of each simulation was used for the initial population. Following the establishment of a steady state in the population distribution, data points at a minimum of five “reaction” times were collected to evaluate the unimolecular rate constant. This process was repeated for each system investigated at two or three temperatures spanning a minimum 50 °C range. Arrhenius parameters are obtained from the temperature dependence of these rate constants.
Results and Discussion
Radiative Rates.
The interpretation of the Arrhenius parameters measured in a BIRD experiment depends on several factors that influence the relative rates of photon absorption and emission to the rate of dissociation. To calculate rate constants for radiative absorption and emission, both the vibrational frequencies and the transition dipole moments (μ) for all optically active modes are required. These values have not been measured for gas-phase peptide ions. For protonated (AlaGly)n, these values are obtained from calculations at the AM1 semiempirical level. Values of μ obtained from these calculations are multiplied by 3 (vide infra). A calculated infrared spectrum for (AlaGly)2 is shown in Figure 1. The strongest absorption bands are at ~2000 cm−1 and correspond to the amide carbonyl stretches. These are somewhat higher in frequencies than expected (~1700 cm−1). Transition frequencies calculated at the Hartree–Fock self-consistent-field level using the harmonic approximation are typically about 10% larger than measured values.47 We are not aware of a similar comparison for semiempirically derived frequencies. The frequency of the C═O stretch appears to be consistent with a 10% overestimation. However, other vibrations with significant intensity, such as N–H stretches and bends (3000–3400 and 1500–1700 cm−1, respectively) and C–N stretches (1200–1500 cm−1) are in a reasonable range. Vibrational frequencies that overlap with the most intense portion of the Planck distribution dominate the rates of photon transfer between the ion and the blackbody field. Small shifts in frequency do not significantly change the rate at which a particular frequency absorbs photons. For example, shifting a carbonyl stretch from the calculated value of 2000 cm−1 to 1700 cm−1 increases the energy density of the field by 24%. This change is insignificant compared to error in other factors, such as the transition dipole moments. For these reasons, the calculated vibrational frequencies are not scaled.
Figure 1.
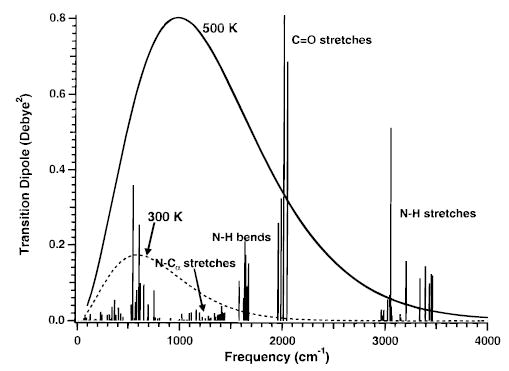
Infrared absorption spectrum of protonated (AlaGly)2 calculated at the AM1 semiempirical level. Transition dipole moments have been multiplied by 3 (see text). Also shown are the Planck blackbody energy distributions at 300 (dashed line) and 500 K (solid line).
Accurate transition dipole moments are critical for modeling BIRD kinetics of processes that are not in the REX limit. Accurate absolute values are difficult to calculate, even at the highest levels of theory, although the relative values in Figure 1 appear reasonable. For small neutral molecules, RHF/3-21g* ab initio transition intensities are larger than measured values by a factor of ~2.48,49 In contrast, transition dipole moments obtained by direct laser absorption spectroscopy of ions in fast beams compare favorably with ab initio values.50 From limited data, protonated ions have larger transition dipole moments than the analogous neutral. For example, the ν3 vibration of NH4+ is ~5 times larger than that of NH3.50
The BIRD kinetics of small ions that dissociate readily are sensitive to the integrated rate of radiative absorption. Using ab initio evaluated transition intensities, Dunbar found a high degree of accuracy between master equation modeled dissociation rates and experimental values for (H2O)nCl−(n = 2 and 3) cluster ions.51 A similar result was obtained for proton-bound dimers of the amino acid glycine and analogue N,N-dimethyl-acetamide (N,N-DMA).42 The sums of intensities over all vibrations were compared using values of μ calculated at both ab initio and semiempirical levels. On average, the values of μ at the RHF/3-21g* level for the N,N-DMA dimer are 2.5 times greater than the AM1 values.42 For protonated glycine dimers, the ab initio values (MP2/3-21g*) are 2.8 times larger. To fit the experimental data, it was necessary to multiply the AM1-derived transition dipole moments by a scaling factor of 3 using the reference value of E0 in the master equation analysis. These results suggest that multiplying the semiempirical derived values of μ by 3 for protonated (AlaGly)n is reasonable. In addition, overtone and combination frequencies which may have significant intensity are not included in these calculations, but may be partially compensated for by multiplying the other transition dipoles by 3.
The extent to which the AM1 transition dipole multiplication factor is valid for larger ions is not known. If these values are overestimated, then the ion size required for the REX limit will be shifted to larger n. To obtain an estimate of the effects of lower radiative rates on the REX limit analysis, the radiative rates used in these calculations were reduced by a factor of 2 (μ divided by 1.4), and a process with A∞ = 1016.8 s−1 and E0 = 1.2 eV was modeled for (AlaGly)n. The minimum size required for an ion to be in the REX limit is increased from (AlaGly)12 to (AlaGly)19. More accurate radiative rates could be obtained by investigating smaller ions for which both the E0 and transition-state entropy are known. Several such systems are currently under investigation.
Additional factors that effect the energy transfer into and out of peptide ions include amino acid composition as well as collisions with gas-phase molecules over the long time frame of the experiment (10–1000 s). Any collisions that occur increase the rate of energy transfer into and out of the ion which may reduce the size necessary for REX limit kinetics.
Kinetic Window.
Another factor that influences the relative rates of photon absorption and emission to the rate of dissociation is the observed dissociation rate constant. This can be controlled by varying the vacuum chamber temperature. At lower temperatures, the experimental observation window is increased to longer times. Despite the T4 power dependence of the blackbody radiation field, lowering the temperature increases the relative radiative to dissociation rates. This shifts the kinetics closer to the REX limit. This effect is illustrated in Figure 2 which shows modeled Arrhenius plots for (AlaGly)n, n = 2, 8, 16, and 24 dissociating via a pathway with a fixed E0 of 1.5 eV and A∞ = 1016.8 s−1 over a temperature range of 380–500 K. Also plotted is the Arrhenius behavior of an ion always in the REX limit, i.e., the dissociation rate constants for a Boltzmann population (solid line). At lower temperatures, little deviation is observed from the expected REX limit Arrhenius behavior even for the smallest ion. The ion population at lower temperatures has less internal energy. The microcanonical dissociation rates decrease faster with decreasing energy than does the rates of photon exchange with the blackbody field. This results in a Boltzmann distribution of internal energies. Thus, the most accurate experimental data from a BIRD experiment will be obtained when the kinetic window is shifted to longer time frames.
Figure 2.
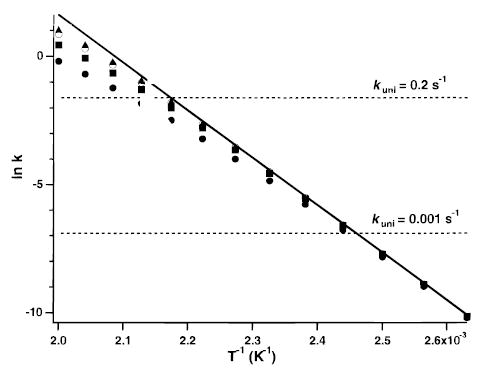
Calculated blackbody infrared radiative dissociation Arrhenius plots for protonated (AlaGly)n, where n = 4 (•), 8 (▪), 16 (○), and 32 (▴), and for an ion in the rapid energy exchange limit (–). Dissociation parameters of E0 = 1.5 eV and A∞ = 1016.8 s−1 were used. Dashed lines indicate the typical Fourier transform mass spectrometry experimental kinetic window at a magnetic field strength of 2.7 T and for electrospray-generated ions.
At higher temperatures (faster rate constants), significant deviation from the REX limit values is observed. This effect is most pronounced for the smaller ions. In this fast kinetic regime, the ion ensemble attempts to populate the higher energy states. However, the rates of microcanonical dissociation at these higher internal energies become increasingly competitive with those of photon exchange. This results in a significant depletion of the highest energy ions. Arrhenius plots evaluated in this fast kinetic window will yield activation energies and preexponential factors that are systematically lower than their true values. Less deviation occurs with increasing ion size. Thus, BIRD Arrhenius parameters will be either lower than or approximately equal to true REX values depending on the temperature range surveyed.
The kinetic window selected for calculating the Arrhenius parameters in this study was chosen to correspond to the typical range of experimental BIRD rates that can be readily measured for larger ions.9,41 These values range between 0.001 and 0.2 s−1 (dashed lines in Figure 2) and correspond to ion storage times between 10 and 1000 s. Measurements made within this range have a high degree of reproducibility and accuracy. Although slow dissociation kinetics are desirable to maintain the REX limit, practical considerations limit the long storage times necessary to measure rate constants <0.001 s−1. At long storage times, loss of ion signal due to sample diffusion out of the cell can occur. Ion loss due to magnetron excitation may be especially significant for multiply charged ions dissociating into two or more charged fragments. These dissociation processes have a significant reverse activation barrier which can couple into the translational modes of the separating products, further exciting their magnetron motion. An additional complication is that subsequent fragmentation of product ions can occur at long reaction times. This can obscure the assignment of the primary fragmentation pathway and lead to diminished signal-to-noise. Measurement of rate constants faster than 0.2 s−1 has a complication for electrosprayed ions; the ion lifetime becomes competitive with the ion accumulation times currently used in this experiment. This makes it difficult to collect a sufficient number of ions prior to their isolation and measurement of the dissociation kinetics.
For each pair of dissociation parameters (E0 and A∞), BIRD rate constants were calculated over a minimum 50 °C temperature range within our experimental temperature boundary (300–520 K). The limited accessible temperature and kinetic window available in our current experimental setup restricts the use of this method to probing dissociation processes over a relatively narrow range of E0. For example, three threshold dissociation energies were used to calculate BIRD rate constants for the model peptides with an A∞ of 1012.4 s−1. E0’s of 0.9, 1.2, and 1.4 eV give rate constants within the kinetic window over a temperature range of 300–350, 400–450, and 470–520 K, respectively.
Ion storage times increase with the square of the magnetic field strength.52 Thus, a (7.0/2.7)2 = 6.7-fold gain in the kinetic window would be obtained at a field strength of 7.0 T. An additional advantage of higher magnetic field strength is the increased ion storage capacity of the cell. This improves the S/N ratio which further increases the kinetic window of the experiment. A wider temperature range could be achieved by adding a shroud inside the vacuum chamber that surrounds the cell and can be either heated or cooled.
Dissociation Mechanisms.
The process by which an ion dissociates influences the relative radiative to dissociation rates. For a given set of parameters, increasing E0 or decreasing A∞ results in kinetics that are closer to the REX limit. The effects of these parameters are illustrated in Figure 3a–d for (AlaGly)n (n = 2–32) for four different transition-state frequencies. These frequencies were constructed to yield A∞ values between 109.9 and 1016.8 s−1. These preexponential factors correspond to dissociation mechanisms ranging from entropically unfavorable rearrangements to entropically favorable direct bond cleavages. The temperature range was constrained to match that currently accessible in our instrument (300–520 K). Three threshold energies were selected for each of the four preexponential factors such that the entire temperature range could be sampled within the kinetic window experimentally accessible.
Figure 3.
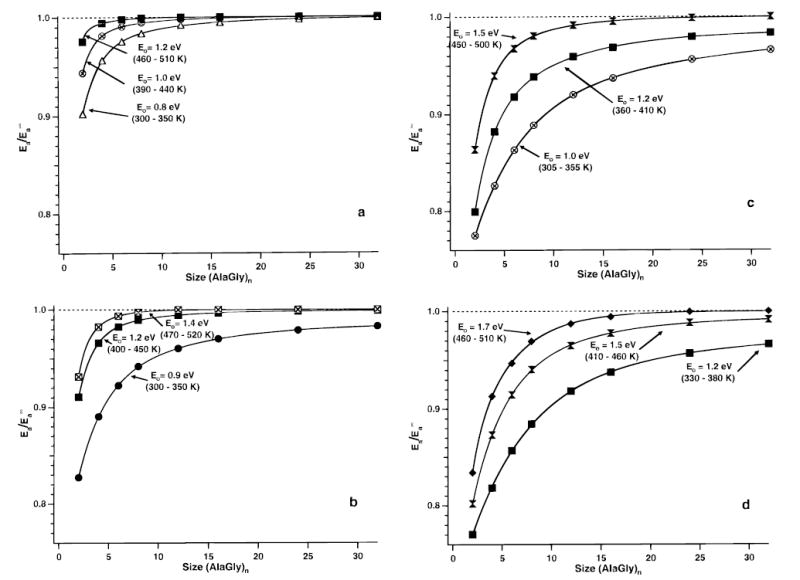
Plots of the ratio of calculated blackbody infrared radiative Arrhenius activation energy (Ea) to the calculated rapid energy exchange limit activation energy as a function of (AlaGly)n ion size. The Ea is calculated from the master equation model using a rapid energy exchange limit Arrhenius preexponential of (a) 109.9, (b) 1012.4, (c) 1014.5, and (d) 1016.8 s−1. Threshold dissociation energies (E0) and the temperature range necessary to keep the dissociation rate constants within the experimental window are labeled on the graphs.
The dependence of the measured BIRD Arrhenius activation parameters (Ea and A) on ion size for a variety of dissociation parameters (E0 and A∞) is shown relative to the REX limit activation energy . The REX limit is considered to be reached when Ea and differ by less than 0.1 eV. This criterion is based on typical experimental error in activation energies measured for large ions. This error is from a linear least-squares fit to the Arrhenius data.9 The ratios of Ea to for (AlaGly)n are plotted versus ion size (n) in Figure 3. The lines between the calculated rate constants are double-exponential fits to these values and are meant to guide the eye. An empirical relationship between these parameters is currently under investigation.
Very Tight Transition State.
The ratios for dissociation processes characterized by an A∞ = 109.9 s−1 with threshold dissociation energies of 0.8, 1.0, and 1.2 eV are shown in Figure 3a. This Arrhenius preexponential factor corresponds to a transition-state configuration that is very entropically unfavorable relative to that of the reactant ion. This would correspond to a complex reaction mechanism that requires functional groups or charge sites remote in connectivity to participate in the reaction center. For example, doubly protonated lysbradykinin ([LysArgProProGlyPheSerProPheArg + 2H]2+) has an A∞ = 1010 s−1.9 This ion dissociates by eliminating an [ArgPro]+ residue from the interior sequence. Thus, two bonds must be broken, but another bond is presumably formed since the activation energy for this process is only 1.2 eV. For n = 2 and an E0 of 0.8 eV, the ratio is 0.90 (Figure 3a). Even for this relatively small ion (275 Da), the difference between Ea and is only 0.076 eV. This difference is sufficiently small that the dissociation of this ion satisfies the stated criteria for being in the REX limit. This ratio is even closer to 1 with increasing E0 and n. Thus, the BIRD method will yield measured activation values that accurately reflect the true values for these entropically hindered dissociation mechanisms at temperatures below 520 K for peptide ions as large or larger than (AlaGly)2.
Tight Transition State.
Data calculated using an A∞ = 1012.4 s−1 are shown in Figure 3b for E0 = 0.9, 1.2, and 1.4 eV. This Arrhenius preexponential factor is characteristic of an important and commonly observed tight transition state corresponding to a simple but entropically unfavorable rearrangement. For example, dissociation of n-butylbenzene molecular ions (C10H14•+) to form C7H8•+ occurs by a six-membered ring McLafferty rearrangement. This process has a ΔS‡ = −10.9 cal/(K mol) at 1000 K,53 which corresponds to an A∞ = 1011.4 s−1. The loss of small neutral molecules from peptides and proteins has also been shown to proceed by a tight mechanism.9,16 For E0 = 0.9 eV, the ratio is greater than 0.9 for (AlaGly)6 (787 Da) so that ions with n ≥ 6 are in the REX limit. For an E0 ≥ 1.2 eV, even (AlaGly)2 would be in the REX limit. Thus, if an Ea greater than 1.2 eV is measured for a dissociation process that is known to occur by a tight transition state, this process is in the REX limit for ions as small as the (AlaGly)2.
Loose Transition State.
An A∞ of 1014.5 s−1 is characteristic of direct bond cleavages with limited opening of internal rotations in the transition state. The data calculated using this value and E0 = 1.0, 1.2, and 1.4 eV are shown in Figure 3c. The minimum ion size required to reach the REX limit is n = 12, 6, and 4 for these respective E0’s. This corresponds to molecular weights of 1555, 787, and 531 Da, respectively.
Very Loose Transition State.
The largest preexponential factors reliably measured for small neutral molecules are 1017–18 s−1.38 Entropy of activation becomes very large when internal rotors associated with the reaction center become active in the transition state. Calculations with an A∞ of 1016.8 s−1 for E0’s of 1.2, 1.5, and 1.7 eV are shown in Figure 3d. For these respective E0’s, a minimum ion size of n = 12 (1555 Da), 8 (1043 Da), and 6 (787 Da) is required in order to be in the REX limit.
REX Limit.
A key finding from these calculations is that, for ions larger than (AlaGly)12 (1.6 kDa), the measured BIRD kinetics will be in the REX limit at temperatures below 520 K irrespective of the dissociation mechanism provided that the transition-state entropy is no greater than that for a reaction with A∞ of 1017 s−1. Thus, the REX limit applies to even smaller ions dissociating by an entropically favored process than was reported previously.16 The origin of this difference is primarily due to an underestimation of transition dipoles used in the earlier calculations.16
For ions smaller than 1.6 kDa, an uncertainty in the interpretation of the measured Arrhenius parameters may occur unless an A > 1016 s−1 is measured or information about the dissociation mechanism is known. Dissociation processes that are not in the REX limit will result in measured Arrhenius parameters that are less than their true values. To determine whether the measured values are in the REX limit requires explicitly modeling the dissociation process. For example, a measured Ea and A of 1.1 eV and 1013.9 s−1 for (AlaGly)6 results from the dissociation of this ion by a process with an and A∞ of 1.3 eV and 1016.8 s−1. The accuracy with which E0 can be obtained from the experimental data by modeling depends on the accuracy of both the experimental data and the calculated radiative rate constants.42 Thus, caution must be taken in the analysis of BIRD data for ions smaller than 1.6 kDa.
Molecule Size.
The number of photons an ion exchanges with the vacuum chamber walls depends on the vibrational transitions and the overlap of the transitions with, and the intensity of, the blackbody radiation field (the Planck distribution). For polymers, such as proteins and DNA, the number of oscillators, and hence the number of photons absorbed and emitted, should increase roughly linearly with increasing molecular size. That is, an ion twice the size will exchange approximately twice the number of photons with the blackbody field. In contrast, the unimolecular dissociation constant (kuni) weighted by the Boltzmann distribution is independent of ion size for a given dissociation process. The approach to REX kinetics with ion size is even more favorable than suggested simply by the relative increase in the rates of photon exchange compared to those of dissociation. For example, the threshold of REX limit kinetics for a dissociation process with E0 = 1.2 eV and A∞ = 1016.8 s−1 ( ) occurs at (AlaGly)12. If the infrared transition rates used in these calculations are reduced to half their normal values, then this same would occur for (AlaGly)19. Thus, decreasing the photon exchange rates by a factor of 2 requires only an ~60% increase in ion size for this dissociation process to remain in the REX limit.
Two additional factors contribute to the onset of the REX limit with increasing ion size. The effect of these factors is illustrated in Figure 4 which shows the canonical energy distributions and RRKM rates (calculated for E0 = 1.5 eV and A∞ = 16.8 s−1) for both (AlaGly)4 and (AlaGly)32. First, the relative width of the population distribution becomes narrower as the ion size increases. The calculated Boltzmann populations of (AlaGly)4 at 350 and 450 K overlap extensively, whereas the (AlaGly)32 populations at these two temperatures do not. At 450 K, the average energy of the (AlaGly)32 is 16.5 eV, which is ~8 times greater than that of the (AlaGly)4 (2.05 eV). This is consistent with the linear scaling of polymer heat capacities. In contrast, the width of a Gaussian fit of the (AlaGly)32 population is 2.53 eV (fwhm), only 80.5 times that of (AlaGly)4 (0.89 eV) at this same temperature. That is, an (AlaGly)32 ion of average energy absorbs 8 times as many photons as the average energy (AlaGly)4 ion, but only requires 2.8 times as many photons to be excited into the most reactive high-energy tail of the distribution. Thus, repopulation of the critical highest energy states is more efficient and rapid for larger ions.
Figure 4.
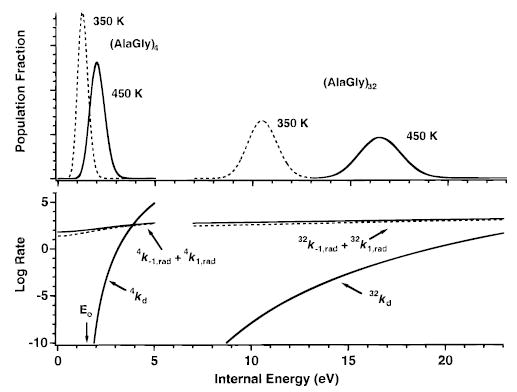
Boltzmann distributions for (AlaGly)4 and (AlaGly)32 ions, each determined at 350 (dashed line) and 450 K (solid line) (top). The sum of the absorption (k1,rad) and emission (k−1,rad) rate constants for these ions is calculated from the master equation model at 350 (dashed line) and 450 K (solid line); microcanonical dissociation rate constants are calculated from RRKM theory with E0 = 1.5 eV and transition-state frequencies modeled to fit an A∞ of 1016.8 s−1 (bottom).
An additional factor contributing to the size dependence of the REX limit is the energy dependence of the microcanonical rates relative to the population distribution. For (AlaGly)4, kd increases rapidly over the range of energies at which the ion population is significant at 450 K. Only ions in the higher energy states (the reactive region) have a finite probability of dissociating in an experimentally reasonable time frame. If, as is the case for (AlaGly)4, the rate of reactive region ion replenishment by absorption of photons is slower than the rate at which ions dissociate, then the population will be depleted at higher energies, and the observed dissociation rate constant will be less than the REX limit value. In contrast, kd for (AlaGly)32 rises relatively slowly with energy over which the ion population is significant at 450 K. Nearly the full population envelope is reactive on the time frame of the experiment. Thus, the higher energy portion of the population does not get significantly depleted relative to the lower energy portion, and a Boltzmann distribution is maintained.
This effect is more evident when the kd for these two ions are compared as a function of population fraction (Figure 5). This normalizes the energy scale of these two populations. For (AlaGly)32, kd increases by only a factor of 62 when the population fraction changes from 0.1 to 0.9. For (AlaGly)4, kd increases by a factor of ~1016 over this same range. The values of kd remains more than 2 orders of magnitude smaller than the photon exchange rates up to the 99.9999% for (AlaGly)32 but only to the 99.6941% for (AlaGly)4.
Figure 5.
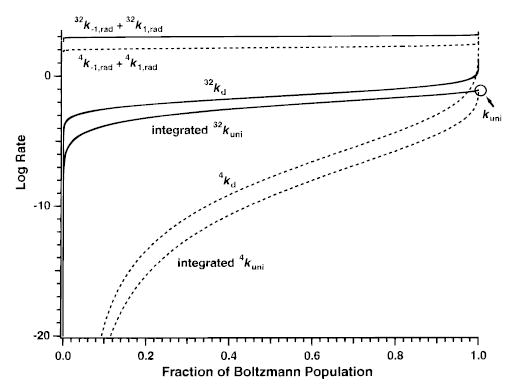
RRKM dissociation (kd), sum of the absorption (k1,rad) and emission (k−1,rad), and integrated unimolecular dissociation (kuni) rate constants at 450 K for (AlaGly)4 (dashed line) and (AlaGly)32 (solid line) ions plotted on an energy scale normalized to a Boltzmann distribution. RRKM dissociation rate constants are calculated for a dissociation process with E0 = 1.5 eV and A∞ of 1016.8 s−1. Integrated kuni is obtained by integrating the Boltzmann-weighted RRKM dissociation rates over the range from E = 0 to the specified energy fraction of a Boltzmann distribution. The integrated kuni is equal to the rapid energy exchange limit unimolecular dissociation constant (labeled on graph) when this product is integrated over all energies.
Even more revealing is the comparison of the unimolecular rate constants integrated from zero to m% of the population (kuni,m):
| (8) |
where N(E) is the normalized population fraction with internal energy E. A fixed Boltzmann distribution was used in this calculation. The log of these rate constants for (AlaGly)4 and (AlaGly)32 is shown in Figure 5. The rate constants for (AlaGly)32 do not change much over the population. This indicates that the contribution to kuni from each of these energy states is similar; i.e., the entire population is depleted roughly uniformly over occupied energy levels so that a Boltzmann distribution is maintained. For (AlaGly)4, the integrated rate constant changes dramatically over the population and approaches the REX limit value only when the contributions of the highest energy 0.05% of the population are included. Thus, this population will be Boltzmann-like but significantly depleted at the critical high-energy tail.
Conclusions
Dissociation kinetics of a protonated peptide polymer, (AlaGly)n (n = 2–32), activated by blackbody radiation were systematically modeled as a function of ion size and dissociation parameters to determine the role of these parameters on establishing kinetics in the rapid energy exchange limit. The REX limit is reached when the rates of ion photon absorption and emission are much greater than the microscopic dissociation rates for all energy levels that have a significant thermal population. In this limit, the observed dissociation rates do not depend on photon absorption and emission rates but accurately reflect the kinetics of the dissociation process of a Boltzmann distribution of ions. A simple correspondence exists between the measured Arrhenius activation energy and the threshold dissociation energy for reaction processes in the REX limit. The Arrhenius preexponential in this limit directly reflects the dynamics of the dissociation process and can be used to obtain information about reaction mechanisms.
Kinetics approach the REX limit with increasing ion size and threshold dissociation energy and decreasing reaction entropy and temperature. Three key factors responsible for the size dependence of REX limit kinetics are identified. First, the number of photons absorbed and emitted increases roughly linearly with polymer size. In contrast, the observed dissociation rate for a Boltzmann distribution of ions is independent of size for a given dissociation process. A second factor is that, although the average energy of the population energy distribution increases linearly with size, the width of this distribution increases only with the square root of size. The average energy of an (AlaGly)8 is 4 times that of (AlaGly)2, but the distribution is only 2 times as wide. Thus, a smaller fraction of the photons absorbed are required to couple the energy levels. That is, repopulation of the most reactive high-energy tail occurs more efficiently for larger ions. Finally, the microscopic dissociation rate constants increase with energy more slowly for large ions than for small ones over the energies populated by a Boltzmann distribution. Thus, a larger fraction of the population contributes to the observed dissociation rate with increasing ion size. For larger ions in the REX limit, nearly the entire population contributes to the observed dissociation rate. For small ions, only the high-energy tail contributes to this rate.
The minimum ion size required for REX limit kinetics depends on a number of factors. A simple empirical relationship between these factors has not yet been established. However, useful empirical guidelines are presented for the temperature range (300–520 K) and kinetic window (kuni = 0.001–0.2 s−1) readily accessible in these BIRD experiments. Increasing the kinetic window by lowering the temperature and increasing ion storage times would result in REX limit kinetics for even smaller ions. For an entropically favorable direct bond cleavage with a low E0, a faster rate of photon exchange is required to reach the REX limit than for an entropically less favored process with the same E0. For an A∞ and E0 of 1016.8 s−1 and 1.2 eV, respectively, ions greater than 1.6 kDa will be in the REX limit, and the measured Arrhenius parameters will be those of a true Boltzmann distribution within the typical experimental error. The largest value for A∞ that has been reliably measured is on the order of 1017–18 s−1. To the extent that this represents the most entropically favored dissociation process possible, then dissociation of peptide ions greater than this mass should be in the REX limit regardless of the dissociation mechanism.
For processes with a higher E0 and/or a tighter transition state, the REX limit can be attained with even smaller ions. However, modeling is necessary to unambiguously interpret the experimental data, unless information about the dissociation mechanism is known. The accuracy with which an E0 can be obtained depends on the accuracy of both the experimental data as well as the radiative rate constants used in the calculations. For ions not in the REX limit, kuni and activation parameters are lower than their true values but may still be useful to establish lower limits to the REX values.
BIRD appears to be a highly promising method for obtaining information about dissociation energetics and mechanisms, particularly for large ions where modeling is not required. The results of the modeling presented here should increase the reliable application of BIRD for intermediate size ions. For ions not in the REX limit, uncertainties in the absolute radiative absorption and emission rates decrease the ultimate accuracy of this method for obtaining E0. These uncertainties result in additional error that must be added to that of the actual experimental measurement. Even with this error included, the overall uncertainty from this method compares favorably to other methods even for small ions. From the investigation of smaller ions for which threshold dissociation energies and information about the dissociation mechanisms are known, more accurate information about the absolute radiative rates can be obtained. This information should further improve the general applicability of the BIRD method to biomolecules of any size.
Acknowledgments
The authors are grateful to generous financial support provided by the National Science Foundation (CHE-9258178) and the National Institutes of Health (1R29GM50336-01A2).
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 2.Smith RD, Loo JA, Ogorzalek Loo RR, Busman M, Udseth HR. Mass Spectrom Rev. 1991;10:359. [Google Scholar]
- 3.Hillenkamp F, Karas M, Beavis RC, Chait BT. Anal Chem. 1991;63:1193A. doi: 10.1021/ac00024a002. [DOI] [PubMed] [Google Scholar]
- 4.Alexander AJ, Boyd AK. Int J Mass Spectrom Ion Processes. 1989;90:211. [Google Scholar]
- 5.Marzluff EM, Campbell S, Rodgers MT, Beauchamp JL. J Am Chem Soc. 1994;116:7787–7796. [Google Scholar]
- 6.Fabris D, Kelly M, Wu Z, Fenselau C. Rapid Commun Mass Spectrom. 1994;8:791. doi: 10.1002/rcm.1290080926. [DOI] [PubMed] [Google Scholar]
- 7.Meot-Ner (Mautner) M, Dongré AR, Somogyi Á, Wysocki VH. Rapid Commun Mass Spectrom. 1995;9:829. doi: 10.1002/rcm.1290090922. [DOI] [PubMed] [Google Scholar]
- 8.Vachet RW, Winders AD, Glish GL. Anal Chem. 1996;68:522. doi: 10.1021/ac950893r. [DOI] [PubMed] [Google Scholar]
- 9.Schnier PD, Price WD, Jockusch RA, Williams ER. J Am Chem Soc. 1996;118:7178. doi: 10.1021/ja9609157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vekey K, Somogyi Á, Wysocki VH. Rapid Commun Mass Spectrom. 1996;10:911. doi: 10.1002/(SICI)1097-0231(19960610)10:8<911::AID-RCM593>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 11.Biemann K, Martin SA. Mass Spectrom Rev. 1987;6:1. [Google Scholar]
- 12.Hunt DF, Yates JR, Shabanowitz J, Winston S, Hauer CR. Proc Natl Acad Sci USA. 1986;83:6233. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loo JA, Edmonds CG, Smith RD. Anal Chem. 1991;63:2488. doi: 10.1021/ac00021a018. [DOI] [PubMed] [Google Scholar]
- 14.Loo JA, Edmonds CG, Smith RD. Anal Chem. 1993;65:425. doi: 10.1021/ac00052a020. [DOI] [PubMed] [Google Scholar]
- 15.Senko MW, Speir JP, McLafferty FW. Anal Chem. 1994;66:2801. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- 16.Price WD, Schnier PD, Jockusch RA, Strittmatter EF, Williams ER. J Am Chem Soc. 1996;118:10640. doi: 10.1021/ja961812r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jockusch RA, Schnier PD, Price WD, Strittmatter EF, Demirev PA, Williams ER. Anal Chem. 1997;69:1119. doi: 10.1021/ac960804q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLuckey SA, Van Berkel GJ, Glish GL. J Am Soc Mass Spectrom. 1992;3:60. doi: 10.1016/1044-0305(92)85019-G. [DOI] [PubMed] [Google Scholar]
- 19.McLuckey SA, Habibigoudarzi S. J Am Chem Soc. 1993;115:12085. [Google Scholar]
- 20.Little DP, Chorush RA, Speir JP, Senko MW, Kelleher NL, McLafferty FW. J Am Chem Soc. 1994;116:4893. [Google Scholar]
- 21.Little DP, McLafferty FW. J Am Chem Soc. 1995;117:6783. [Google Scholar]
- 22.Rodgers MT, Campbell S, Marzluff EM, Beauchamp JL. Int J Mass Spectrom Ion Processes. 1995;148:1. [Google Scholar]
- 23.Loo JA, Giordani AB, Muenster H. Rapid Commun Mass Spectrom. 1993;7:186. doi: 10.1002/(SICI)1097-0231(19990115)13:1<54::AID-RCM450>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 24.Loo RRO, Goodlett DR, Smith RD, Loo JA. J Am Chem Soc. 1993;115:4391. [Google Scholar]
- 25.Smith RD, Light-Wahl KJ, Winger BE, Loo JA. Org Mass Spectrom. 1992;27:811. [Google Scholar]
- 26.Smith DL, Zhang ZQ. Mass Spectrom Rev. 1994;13:411. [Google Scholar]
- 27.Tang X, Callahan JH, Zhou P, Vertes A. Anal Chem. 1995;67:4542. doi: 10.1021/ac00120a018. [DOI] [PubMed] [Google Scholar]
- 28.Gross DS, Zhao Y, Williams ER. J Am Soc Mass Spectrom. 1997;8:519. doi: 10.1016/S1044-0305(97)00010-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams ER, Furlong JJP, McLafferty FW. J Am Soc Mass Spectrom. 1990;1:288. doi: 10.1016/1044-0305(90)85003-5. [DOI] [PubMed] [Google Scholar]
- 30.Tecklenburg RE, Jr, Miller MN, Russell DH. J Am Chem Soc. 1989;111:1161. [Google Scholar]
- 31.Dunbar RC. J Chem Phys. 1991;95:2537. [Google Scholar]
- 32.van der Hart WJ. Int J Mass Spectrom Ion Processes. 1992;118/119:617. [Google Scholar]
- 33.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Anal Chem. 1994;66:2809. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 34.Klassen JS, Anderson SG, Blades AT, Kebarle P. J Phys Chem. 1996;100:4218. [Google Scholar]
- 35.Speir JP, Amster IJ. J Am Soc Mass Spectrom. 1995;6:1069. doi: 10.1016/1044-0305(95)00547-1. [DOI] [PubMed] [Google Scholar]
- 36.Vekey K. J Mass Spectrom. 1996;31:445. [Google Scholar]
- 37.Armentrout PB, Baer T. J Phys Chem. 1996;100:12866. [Google Scholar]
- 38.Benson, S. W. Thermochemical Kinetics. Methods for the Estimation of Thermochemical Data and Rate Parameters; John Wiley & Sons: New York, 1968.
- 39.Steinfeld, J. I.; Francisco, J. S.; Hase, W. L. Chemical Kinetics and Dynamics; Prentice-Hall: Englewood Cliffs, NJ, 1989.
- 40.Busman M, Rockwood AL, Smith RD. J Phys Chem. 1992;96:2397. [Google Scholar]
- 41.Price WD, Schnier PD, Williams ER. Anal Chem. 1996;68:859. doi: 10.1021/ac951038a. [DOI] [PubMed] [Google Scholar]
- 42.Price WD, Schnier PD, Williams ER. J Phys Chem B. 1997;101:664. doi: 10.1021/jp9628702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thölmann D, Tonner DS, McMahon TB. J Phys Chem. 1994;98:2002. [Google Scholar]
- 44.Dunbar RC. J Phys Chem. 1994;98:8705. [Google Scholar]
- 45.Schnier PD, Price WD, Strittmatter EF, Williams ER. J Am Soc Mass Spectrom. 1997;8:771. doi: 10.1016/S1044-0305(97)84129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilbert, R. C.; Smith, S. C. Theory of Unimolecular and Recombination Reactions; Blackwell Scientific Publications: London, 1990.
- 47.Hehre, W. J.; Radom, L.; Schleyer, P. R.; Pople, J. A. Ab initio Molecular Orbital Theory; Wiley: New York, 1986.
- 48.Yamaguchi Y, Frisch M, Gaw J, Schaefer HF, Binkley JS. J Chem Phys. 1986;84:2262. [Google Scholar]
- 49.Green WH, Willetts A, Jayatilaka D, Handy NC. Chem Phys Lett. 1990;169:127. [Google Scholar]
- 50.Keim ER, Polak ML, Owrutsky JC, Coe JV, Saykally RJ. J Chem Phys. 1990;93:3111. [Google Scholar]
- 51.Dunbar RC, McMahon TB, Thölmann D, Tonner DS, Salahub DR, Wei D. J Am Chem Soc. 1996;117:12819. [Google Scholar]
- 52.Marshall AG, Guan SH. Rapid Commun Mass Spectrom. 1996;10:1819. doi: 10.1002/(SICI)1097-0231(199611)10:14<1855::AID-RCM764>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 53.Baer T, Dutuit O, Mestdagh H, Rolando C. J Phys Chem. 1988;92:5674. [Google Scholar]