

Abstract
A role for the N-acetyltransferase 2 (NAT2) genetic polymorphism in cancer risk has been the subject of numerous studies. Although comprehensive reviews of the NAT2 acetylation polymorphism have been published elsewhere, the objective of this paper is to briefly highlight some important features of the NAT2 acetylation polymorphism that are not universally accepted to better understand the role of NAT2 polymorphism in carcinogenic risk assessment. NAT2 slow acetylator phenotype(s) infer a consistent and robust increase in urinary bladder cancer risk following exposures to aromatic amine carcinogens. However, identification of specific carcinogens is important as the effect of NAT2 polymorphism on urinary bladder cancer differs dramatically between monoarylamines and aryldiamines. Misclassifications of carcinogen exposure and NAT2 genotype/phenotype confound evidence for a real biological effect. Functional understanding of the effects of NAT2 genetic polymorphisms on metabolism and genotoxicity, tissue-specific expression and the elucidation of the molecular mechanisms responsible are critical for interpretation of previous and future human molecular epidemiology investigations into the role of NAT2 polymorphism on cancer risk. Although associations have been reported for various cancers, this paper focuses on urinary bladder cancer, a cancer in which a role for NAT2 polymorphism was first proposed and for which evidence is accumulating that the effect is biologically significant with important public health implications.
Keywords: N-acetyltransferase 2 (NAT2), urinary bladder cancer, aromatic amines, NAT2 acetylator genotype, single nucleotide polymorphisms, NAT2 haplotypes
Introduction
The N-acetyltransferase 2 (NAT2) acetylation polymorphism was discovered over fifty years ago when individual variability in isoniazid neurotoxicity was attributed to genetic variability in N-acetylation (Hughes et al., 1954). The importance increased when it was discovered that many aromatic amine and hydrazine drugs are subject to the acetylation polymorphism thus affecting therapeutic efficacy and toxicity (Weber and Hein, 1985). It was soon apparent that many environmental and occupational aromatic amine carcinogens undergo catalysis by N-acetyltransferases (Hein, 1988). Thus, a role for NAT2 acetylation polymorphism in individual risk to various cancers in which aromatic amines play an etiologic role is biologically plausible and has been the subject of numerous studies.
N-acetyltransferase isozymes NAT1 and NAT2
Whereas the N-acetylation of isoniazid and sulfamethazine divided human populations by NAT2 acetylator phenotypes, the N-acetylation of drugs such as p-aminosalicylic acid yielded apparently unimodal distribution of individuals (Jenne, 1965). The biochemical basis relates to substrate specificity and molecular genetics of two distinct N-acetyltransferase isozymes, subsequently identified as N-acetyltransferase 1 (NAT1) and NAT2 (Vatsis et al., 1995). The crystallographic structures of several prokaryotic N-acetyltransferases have been published (Sinclair et al., 2000; Sandy et al., 2002; Dupret and Rodrigues-Lima 2005). Although crystal structures of mammalian NAT1 and NAT2 have yet to be reported, molecular modeling of both human NAT1 (Rodrigues-Lima et al., 2001) and NAT2 (Rodrigues-Lima et al., 2002) have revealed a cysteine protease-like catalytic triad (Cys68-His107-Asp122). The Cys68 residue is critical for transferring the acetyl moiety from acetyl coenzyme A cofactor to acceptor substrates (Dupret and Grant, 1992). Aromatic amines and hydrazines (N-acetylation), N-hydroxyaromatic and -heterocyclic amines (O-acetylation) and N-hydroxy-N-acetylaromatic amines (N,O-acetylation) are examples of acceptor substrates for both NAT1 and NAT2 (Hein, 1988). Although both human NAT1 and NAT2 catalyze these reactions, human NAT2 has a three to four-fold higher affinity than NAT1 for urinary bladder carcinogens such as 4-aminobiphenyl (ABP) and ß-naphthylamine (BNA) (Hein et al., 1993b). This finding is consistent with the hypothesis that the effect of NAT2 polymorphism on urinary bladder cancer is more prevalent at low dose aromatic amine exposures (Vineis et al, 1994; 2004).
Animal models
NAT1 and NAT2 in animal models such as rabbit, mouse, Syrian hamster, and rat are highly homologous to both human NAT1 and NAT2 (Hein et al., 1997; Hein, 2002). Substrate specificities for Syrian hamster, mouse, and rat NAT2 may resemble human NAT1 more than they do human NAT2 (Weber and Hein, 1985). Several different mechanisms are responsible for NAT2 polymorphisms in non-human species. The molecular basis for slow acetylator phenotype is NAT2 gene deletion in the rabbit (Blum et al., 1989), a nonsense single nucleotide polymorphism (SNP) yielding a truncated NAT2 enzyme in the Syrian hamster (Ferguson et al., 1994; 1996; Nagata et al., 1994), and missense SNP(s) in the mouse (Martell et al., 1991) and rat (Doll and Hein, 1995). Both NAT1 and NAT2 have been identified and partially purified from Syrian hamster liver (Hein et al., 1985; Smith et al., 1986; Trinidad et al., 1989; Ozawa et al., 1990), intestine (Smith et al., 1986), colon (Hein et al., 1993a), prostate (Hein et al., 2003), and urinary bladder (Yerokun et al., 1989) cytosols. Expression of NAT1 and NAT2 isozymes also has been reported in rapid and slow acetylator mouse (Hein et al., 1988) and rat (Hein et al., 1991a) liver cytosols.
Phenotypic expression of the NAT2 polymorphism
In a congenic Syrian hamster model in which all slow acetylators are homozygous for a single slow NAT2 allele or haplotype and obligate heterozygotes all possess the same combination of rapid and slow NAT2 allele or haplotype, the NAT2 acetylation polymorphism clearly segregates the N-acetylation of aromatic amine urinary bladder carcinogens such as ABP and BNA into three phenotypes in hepatic and extrahepatic tissues (Figure 1). This trimodal distribution of rapid, intermediate and slow acetylator phenotypes in Syrian hamsters congenic at NAT2 also is clearly evident in vivo (Figure 2). Although many human studies often exhibit bimodal distributions of rapid and slow acetylator NAT2 phenotypes, studies with hydrazine drugs such as isoniazid (Parkin et al., 1997; Smith et al., 1997), aromatic amine drugs such as sulfamethazine (Chapron et al., 1980; Lee and Lee, 1982), and caffeine, a compound with a metabolite that is N-acetylated (Gross et al., 1999; Cascorbi et al., 1999; Grant et al., 2004), yield rapid, intermediate, and slow acetylator phenotypes. The unequivocal detection of three phenotypes can be confounded by various factors including catalysis by NAT1. Since isoniazid has high selectivity for catalysis via NAT2, rapid, intermediate, and slow acetylator phenotypes can be readily and unequivocally discerned as illustrated in Figure 3.
Figure 1.
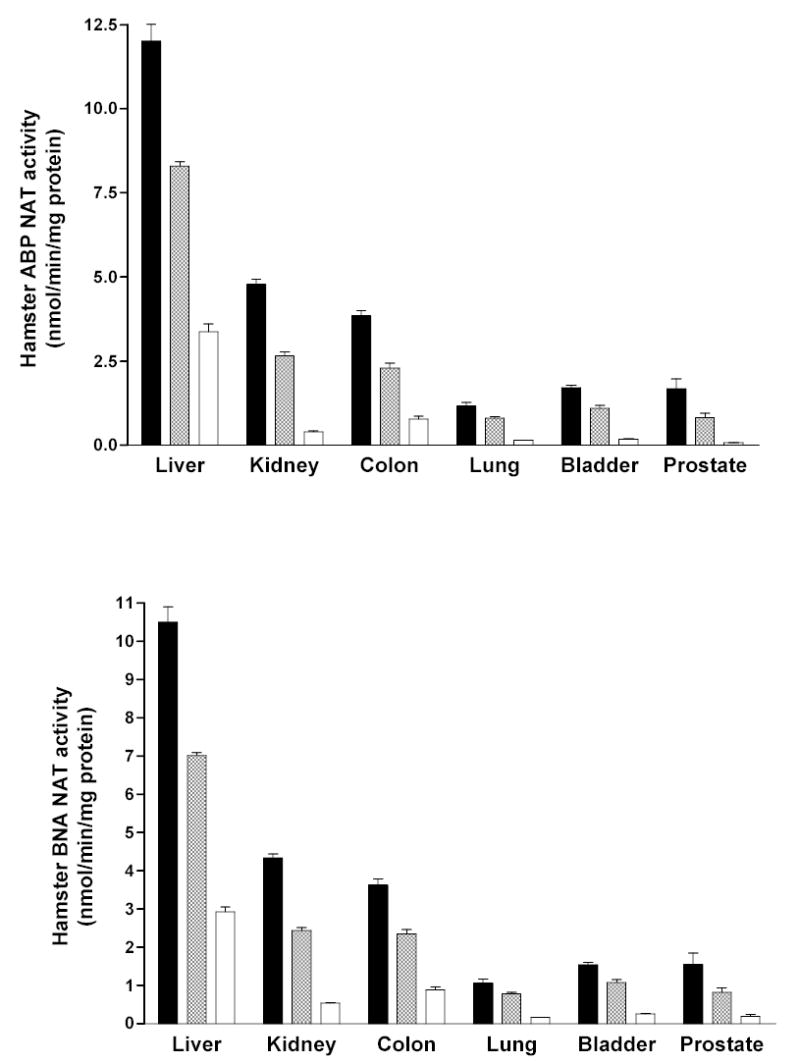
Each bar represents Mean ± SE for cytosolic N-acetyltransferase activities towards the aromatic amine urinary bladder carcinogens 4-aminobiphenyl (ABP) and ß -naphthylamine (BNA) in congenic Syrian hamsters with homozygous rapid acetylator genotype (black), heterozygous acetylator genotype (gray) or homozygous slow acetylator genotype (white). Differences among the genotypes were highly significant (p<0.0001) following one way analysis of variance. Adapted from Hein et al., 1994a; 2003.
Figure 2.
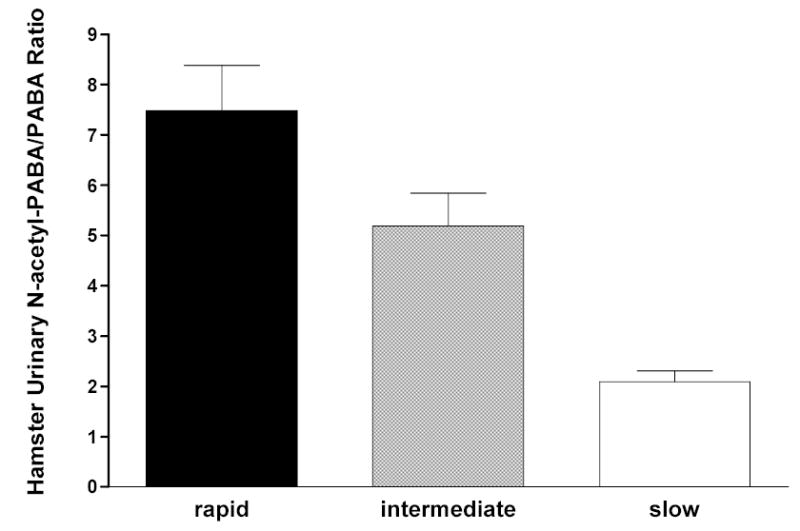
Each bar represents Mean ± SE for urinary excretion ratio of N-acetyl-p-aminobenzoic acid to p-aminobenzoic acid in Syrian hamsters with homozygous rapid acetylator genotype (black), heterozygous acetylator genotype (gray) or homozygous slow acetylator genotype (white). Differences among the genotypes were highly significant (p<0.0001) following one way analysis of variance. Adapted from Hein et al., 1994a.
Figure 3.
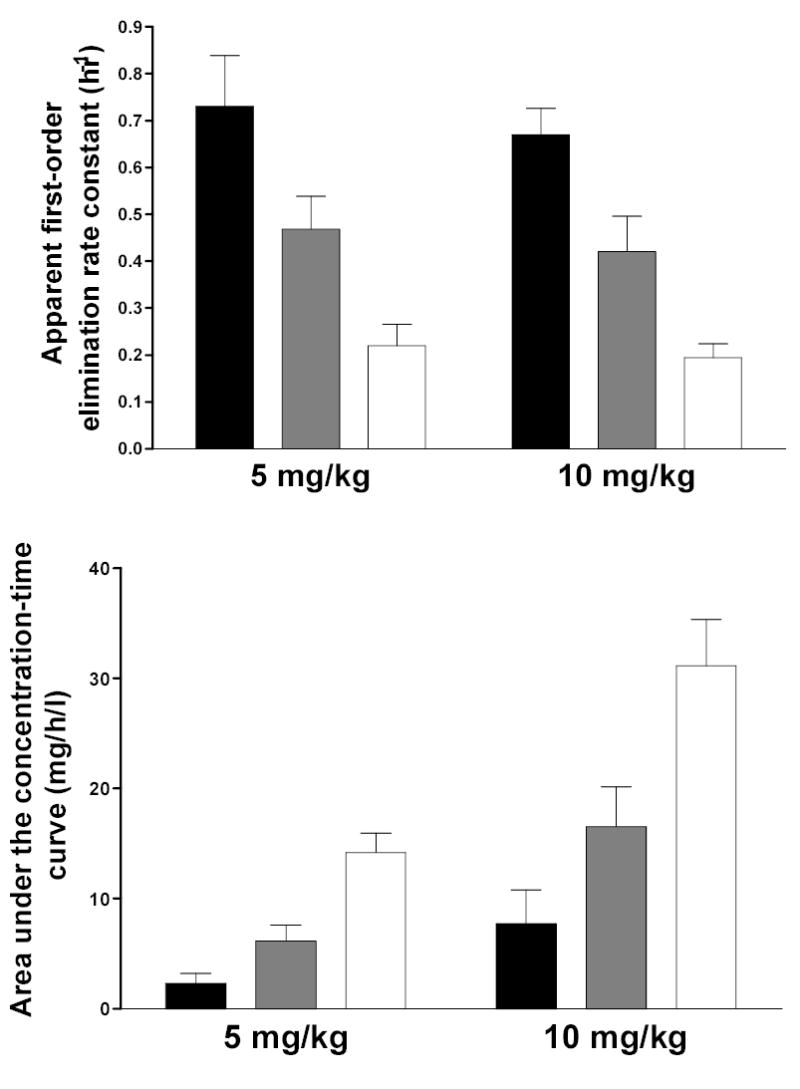
Each bar represents Mean ± SD for the isoniazid elimination rate constant (top) or the area under the concentration-time curve (bottom) following a single oral dose of 5 or 10 mg/kg isoniazid in individuals with homozygous rapid acetylator genotype (black), heterozygous acetylator genotype (gray) or homozygous slow acetylator genotype (white). NAT2 genotypes and phenotypes were 100% concordant and differences among the genotypes were highly significant (p<0.0001) following one way analysis of variance. Adapted from Parkin et al., 1997.
A widely held hypothesis is that human NAT2 is expressed primarily in liver and gastrointestinal tract whereas human NAT1 has widespread tissue distribution. This hypothesis derives from studies in the rabbit model where N-acetyltransferase activities reflected the NAT2 genetic polymorphism in liver and gut, but not in other tissue cytosols suggesting either absence or a much smaller contribution of rabbit NAT2 in these other tissues (Hearse and Weber, 1973). Furthermore, a subsequent study reported that the rapid/slow NAT2 ratio for both sulfamethazine N-acetyltransferase and N-hydroxy-ABP O-acetyltransferase activities were much higher in rabbit liver than small and large intestine (Ilett et al., 1991). Thus, these studies in rabbit suggested that NAT2 genotype-dependent differences are expressed primarily in liver and to a lesser extent the gastrointestinal tract, and therefore suggest that NAT2-genotype dependent differences in carcinogenesis following exposures to carcinogens primarily reflect NAT2 genotype-dependent hepatic versus extrahepatic metabolism of the carcinogen and/or its metabolites.
Studies had shown expression of both NAT1 and NAT2 in human colon (Turesky et al., 1991; Kirlin et al., 1991; Ilett et al., 1994), intestine (Hickman et al., 1998) and widespread tissue distribution of human NAT1 and NAT2 mRNA (Windmill et al., 2000; Boukouvala and Sim, 2005). Although extrahepatic expression of N-acetyltransferase activities have been reported in humans (Pacifici et al., 1986), rat (Hein et al., 1991a), mouse (Chung et al., 1993; Stanley et al., 1997; Sugamori et al., 2003), and Syrian hamster (Hein et al., 1991b; 1994a) models, substrates were not selective for NAT1 and NAT2. NAT2-dependent ABP- and BNA N-acetyltransferase activities have been reported in human urinary bladder (Kirlin et al., 1989; Frederickson et al., 1992; 1994; Pink et al., 1992; Badawi et al., 1995). p-Aminobenzoic acid N-acetyltransferase (selective for NAT1) and N-hydroxy-ABP O-acetyltransferase activities (not selective for NAT1 or NAT2) in human urinary bladder cytosols did not correlate, consistent with catalysis by both NAT1 and NAT2 (Badawi et al., 1995). Other studies are consistent with N-acetylation of ABP and O-acetylation of N-hydroxy-ABP predominantly by NAT1 in urinary bladder (Frederickson et al., 1994). Recent studies with substrates selective for NAT1 versus NAT2 reported widespread distribution of both NAT1 and NAT2 catalytic activities in the rapid and slow acetylator congenic hamster (Hein et al., 2006). NAT2-dependent N-acetylation (Figure 1) and O-acetylation (Figure 4) have been reported in urinary bladder cytosol from rapid and slow acetylator Syrian hamsters congenic at the NAT2 locus. Since both NAT1 and NAT2 catalyze the metabolism of aromatic amine carcinogens (Minchin et al., 1992; Hein et al., 1993b; 1994b; 1995), genetic polymorphism in NAT1 and/or NAT2 may modify cancer risk related to exposures to these carcinogens.
Figure 4.
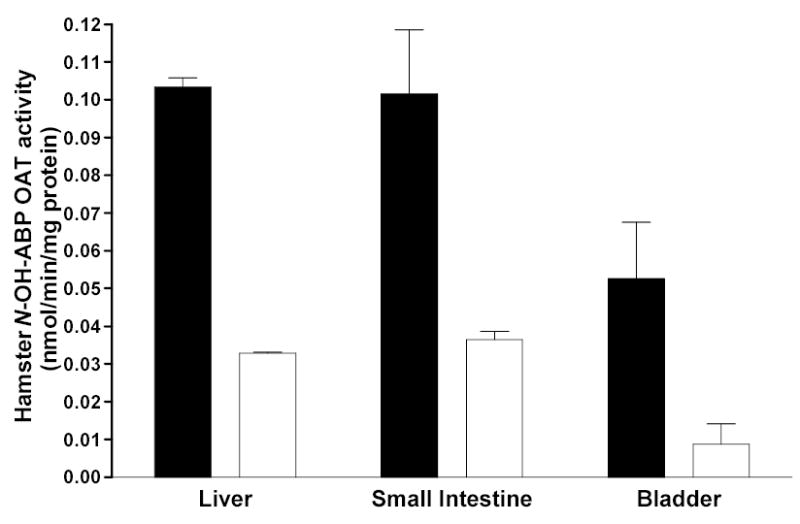
Each bar represents Mean ± SE for cytosolic O-acetyltransferase activities towards N-hydroxy-4-aminobiphenyl (N-OH-ABP) in congenic Syrian hamsters with homozygous rapid acetylator genotype (black) or homozygous slow acetylator genotype (white). Differences between rapid and slow acetylators were significant in each tissue. Modified from Hein et al., 2006.
Molecular genetics
NAT1 and NAT2 are products of single, intronless exons containing single 870 base pair open reading frames encoding 290 amino acids (Blum et al., 1991; Vatsis et al., 1991). NAT1, NAT2, and a pseudogene NATP, are located on the short arm of human chromosome 8 (Blum et al., 1990; Hickman et al., 1994) in the orientation NAT1:NATP:NAT2 (Matsas et al., 1997). NAT1 and NAT2 share 87% nucleotide homology in the coding region, yielding 55 amino acid differences. Human NAT2 transcripts have been identified in many human tissues and derive from the protein-coding exon and a second non-coding exon of 100 base pairs located about 8 kilobases upstream of the translation start site (Blum et al., 1990; Ebisawa and Deguchi, 1991; Boukouvala and Sim, 2005).
A number of single nucleotide polymorphisms (SNPs) have been reported in the NAT2 coding exon. Those that cause amino acid changes include 111T>C (R197Q), 190C>T (R64W), 191G>A (R64Q), 341T>C (I114T), 364G>A (D122N), 411A>T (L137F), 434A>C (Q145P), 590G>A (R197Q), 803A>G (K268R), 845A>C (K282T), 857G>A (K268R) and 859T>C (I287T). SNPs that do not change amino acids include 282C>T, 481C>T and 759C>T. 859T deletion also has been reported resulting in a frame shift at amino acid 287. Various combinations of SNPs are identified as NAT2 alleles (Vatsis et al., 1995) or haplotypes. NAT2*4 is considered the “wild-type” allele or haplotype because of the absence of any SNPs. Variant NAT2 alleles or haplotypes possessing combinations of SNPs are segregated into clusters possessing a signature SNP either alone or in combination with others. The more common NAT2 alleles or haplotypes are illustrated in Table 1. The frequency of NAT2 alleles varies widely across various ethnic groups and NAT2*4 is not the most common in most ethnic groups, including Caucasians and Africans (Figures 5 and 6). NAT2 alleles containing the 191G>A (R64Q), 341T>C (I114T), 590G>A (R197Q), or 857G>A (K268R) SNPs are associated with slow acetylator NAT2 alleles (Table 1). Striking ethnic differences in the frequencies of SNPs and genotypes (http://snp500cancer.nci.nih.gov) are responsible for the corresponding ethnic differences in frequency of rapid (Figure 5) and slow (Figure 6) acetylator NAT2 alleles or haplotypes and therefore phenotypes. For example, the 191G>A (R64Q) SNP common to the NAT2*14 allele cluster is frequent in Africans and African-Americans, but virtually absent in Caucasian, Indian, and Korean populations (Figure 6). Similarly, the NAT2*7 cluster possessing the 857G>A (K268R) SNP is much more frequent in South India and Korea than other populations while the NAT2*5 cluster containing the 341T>C (I114T) SNP is much less frequent in Korea than in Europe, North America, India and Africa (Figure 6). Deduction of NAT2 phenotypes is assigned based on co-dominant expression of rapid and slow acetylator NAT2 alleles or haplotypes as clearly documented in animals (Figures 1–2) and humans (Figure 3). Individuals homozygous for rapid NAT2 acetylator alleles are deduced as rapid acetylators, individuals homozygous for slow acetylator NAT2 alleles are deduced as slow acetylators, and individuals possessing one rapid and one slow NAT2 allele are deduced as intermediate acetylators.
Table 1.
Common human NAT2 alleles (haplotypes)
| Allele (haplotype)a | Nucleotide Change(s)b | Amino Acid Change(s)c | Catalytic activityd |
|---|---|---|---|
| NAT2*4 | None | None | High |
| NAT2*5A | 341T>C; 481C>T | I114T | Low |
| NAT2*5B | 341T>C; 481C>T; 803A>G | I114T; K268R | Low |
| NAT2*5C | 341T>C; 803A>G | I114T; K268R | Low |
| NAT2*6A | 282C>T ; 590G>A | R197Q | Low |
| NAT2*6B | 590G>A | R197Q | Low |
| NAT2*7A | 857G>A | G286E | Low |
| NAT2*7B | 282C>T; 857G>A | G286E | Low |
| NAT2*12A | 803A>G | K268R | High |
| NAT2*12B | 282C>T; 803A>G | K268R | High |
| NAT2*12C | 481C>T; 803A>G | K268R | High |
| NAT2*13 | 282C>T | None | High |
| NAT2*14A | 191G>A | R64Q | Low |
| NAT2*14B | 191G>A; 282C>T | R64Q | Low |
Common NAT2 alleles (haplotypes) associated with low catalytic activityd and slow acetylator phenotype are bolded. Individuals homozygous for these alleles are slow acetylators.
Signature SNP for each allele cluster is bolded.
Amino acid substitutions that confer reduced NAT2 activities are underlined.
Figure 5.
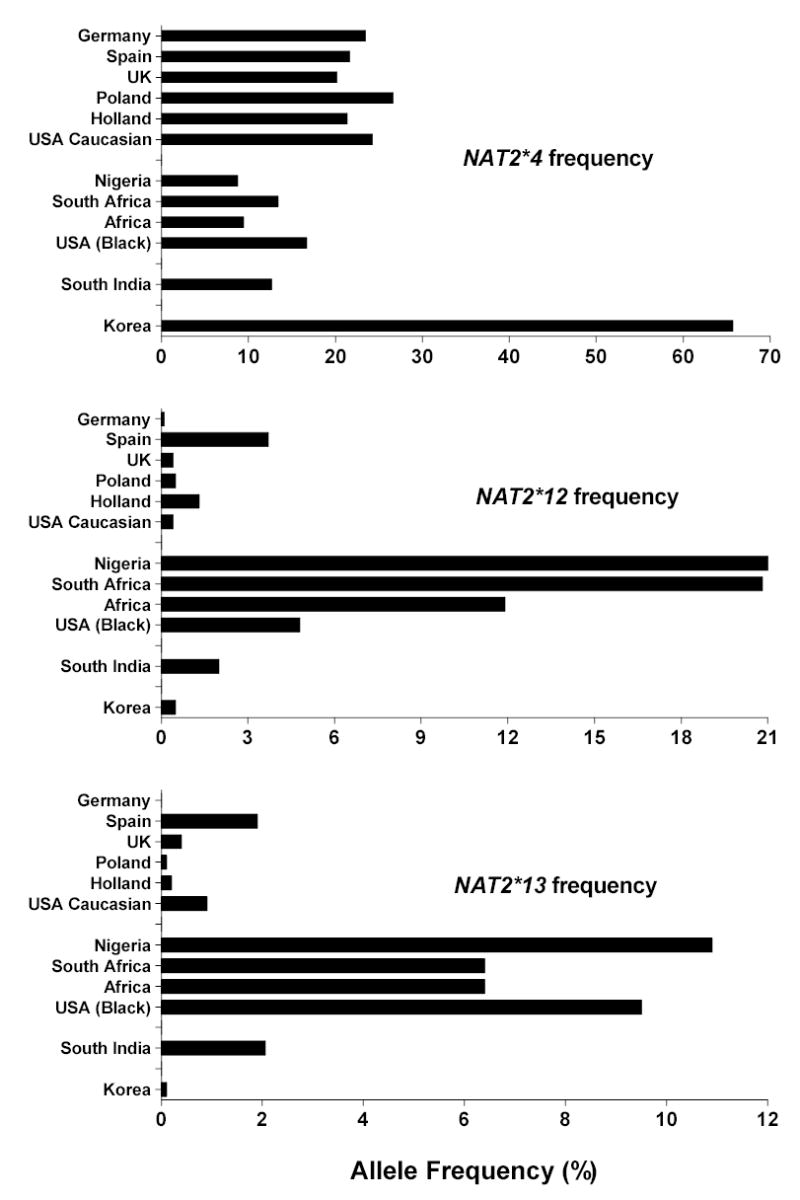
Rapid acetylator NAT2 allelic (haplotype) frequencies reported in various populations. Data for each population was derived from the following sources: Germany (Cascorbi et al., 1999); Spain (Agundez et al., 1996); United Kingdom (UK); (Loktionov et al., 2002); Poland (Lan et al., 2003); Holland (van der Hel et al., 2003); USA Caucasian (Deitz et al., 2000); Nigeria (unpublished data from author’s laboratory); South Africa (Loktionov et al., 2002); Africa (Delomenie et al., 1996); USA Black (O’Neill et al., 2000); South India (Anitha and Banerjee, 2003) and Korea (Lee et al., 2002).
Figure 6.
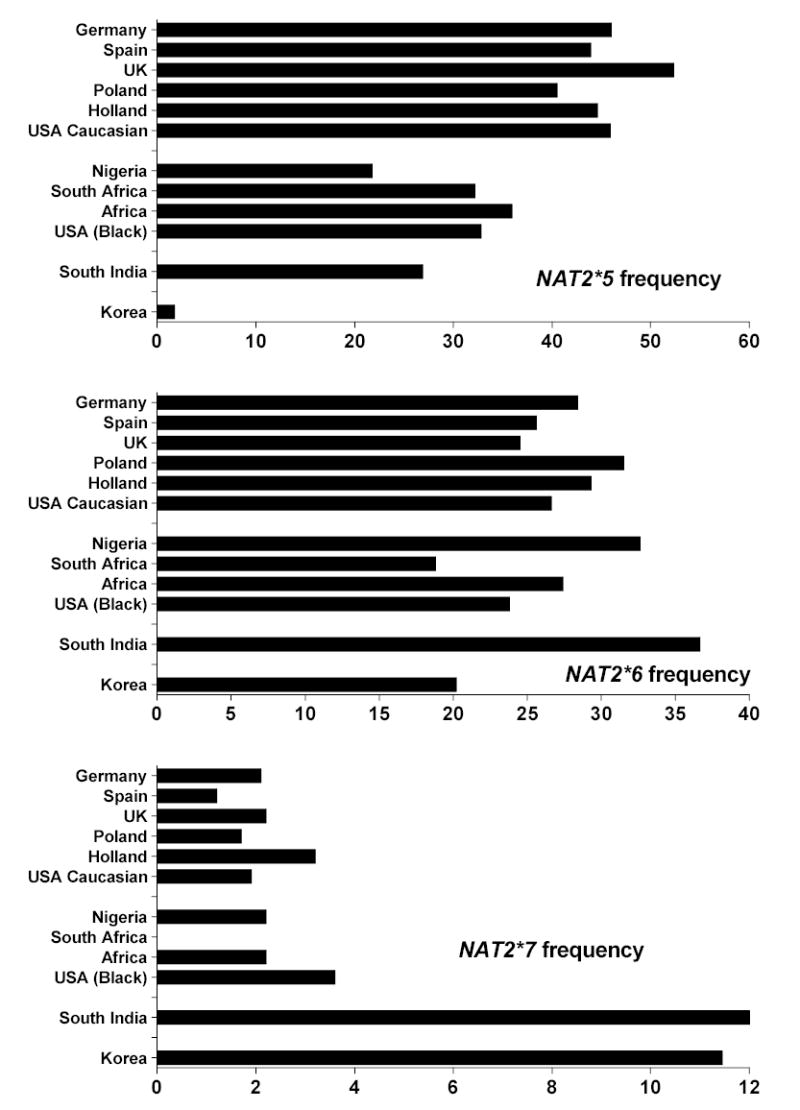
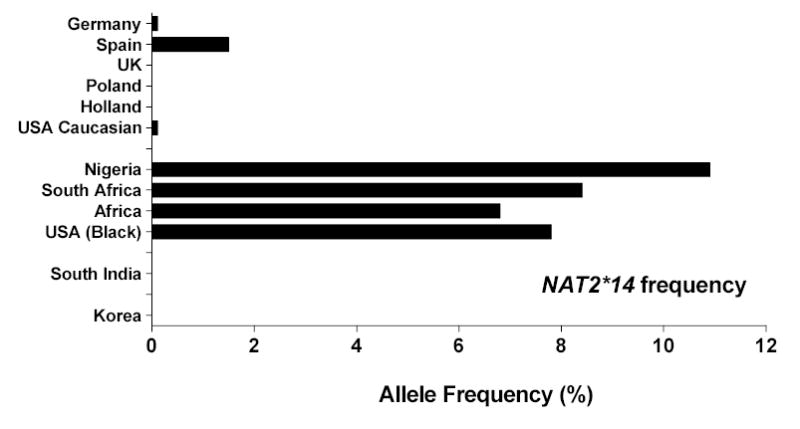
Slow acetylator NAT2 allelic (haplotype) frequencies reported in various populations. Data for each population was derived from the same sources listed in Figure 5.
Over 35 NAT2 alleles or haplotypes have been identified in human populations. A consensus NAT nomenclature was first published in 1995 (Vatsis et al., 1995). An international nomenclature committee publishes an internet accessible website for allele updates at www.louisville.edu/medschool/pharmacology/NAT.html.
Molecular basis for altered function of NAT2 polymorphic variants
Reductions in the amount of NAT2 protein expressed in human liver from individuals with slow acetylator phenotype have been reported (Grant et al., 1990; Deguchi et al., 1990; Deguchi, 1992). Slow acetylator NAT2 alleles recombinantly expressed in COS-1 cells (Blum et al., 1991; Zang et al., 2004), Chinese hamster ovary cells (Abe et al., 1993), and yeast (Leff et al., 1999; Fretland et al., 2001) show reduced levels of NAT2 protein when compared with NAT2*4. These data suggest that slow acetylator phenotype is conferred, at least for some NAT2 alleles, by reduction(s) in NAT2 protein. Recent studies in COS-1 cells also show that the reduction in protein in slow acetylators is the result of increased degradation for SNPs such as 341T>C (I114T) (Zang et al., 2004).
The effects of NAT2 SNPs on catalytic activities have been investigated primarily in recombinant expression systems (reviewed in Hein et al., 2000; Hein, 2002). Nucleotide substitutions identified in human NAT2 allelic variants yield reductions in substrate affinity, catalytic activity and/or protein stability of the recombinant N-acetyltransferase allozymes. Recombinant human NAT2 5, NAT2 6, NAT2 7, and NAT2 14 clusters yield variable reductions in catalytic activity associated with slow acetylator phenotype, while recombinant human NAT2 12 and NAT2 13 clusters catalyze N-, O-, and N,O-acetyltransferase activities at levels comparable to the rapid acetylator NAT2 4 (Hein et al., 1995). Recently, some controversy has arisen regarding the assignment of NAT2*12 and NAT2*13 as rapid acetylator alleles (Bolt et al., 2005). As shown in Table 1, NAT2*12 allele clusters possess the signature 803A>G (K268R) SNP whereas the NAT2*13 allele possesses the 282C>T SNP that does not change the amino acid. These two SNPs and identification of NAT2*12 and NAT2*13 alleles often are not determined in epidemiological studies since they are considered rare. However, as shown in Figure 5, their frequency is not rare in many ethnic groups and it is important both to assess their frequency and to correctly assign them as rapid or slow acetylator status in order to deduce acetylator phenotype. Previous studies have clearly shown that the 803A>G (K268R) SNP characteristic of NAT2*12 alleles and the 282C>T SNP characteristic of NAT2*13 alleles do not alter NAT2 catalytic activity (Hein et al., 1994b; 1995; Fretland et al., 2001; Zang et al., 2005). Three studies using caffeine as a phenotype probe suggested that NAT2*12 and NAT2*13 were associated with slow acetylation phenotype (Cascorbi et al., 1995; Gross et al., 1999; Bolt et al., 2005). Cascorbi et al 1995 initially reported this for NAT2*13 but later reported that it was related to an NAT2 genotyping artifact (Cascorbi et al., 1996; Cascorbi and Roots, 1999). The other studies did not distinguish the NAT2*12 or the NAT2*13 allele from NAT2 genotypes that do not possess NAT2*12 or NAT2*13. Nevertheless, verification of NAT2*12 and NAT2*13 as rapid acetylator alleles has been provided in vivo (Cascorbi et al., 1996; Parkin et al., 1997). The latter study (Parkin et al., 1997) included 5 subjects possessing the NAT2*13 allele and 20 subjects possessing the NAT2*12A allele that consistently confirmed rapid acetylator status based upon measured phenotypes in several people.
Recombinant NAT2 proteins differ in heat stability (Ferguson et al., 1994; Hein et al., 1994b; Grant et al., 1997; Leff et al., 1999; Fretland et al., 2001). The NAT2 7B allozyme has altered affinity for some but not other substrates (Hein et al., 1994b; Hickman et al., 1995) suggesting that expression of acetylator phenotype is dependent upon substrate. Some, but not all of the SNPs in human NAT2 yield reductions in quantity of recombinant NAT2 protein in eukaryotic expression systems (Deguchi, 1992; Blum et al., 1991; Abe et al., 1993; Leff et al., 1999; Fretland et al., 2001). Clearly, more data from tissues is needed to investigate tissue-specific and other regulatory factors.
Since multiple mechanisms for reductions in NAT2 activity are associated with various combinations of SNPs that make up NAT2 alleles, the ability to distinguish among multiple acetylator phenotypes is complex and a function of the sensitivity and specificity of the phenotyping method. Phenotype is influenced by a number of factors including diet, disease, and drug therapy. Depending upon the probe drug and analytical method used, acetylation phenotypes often exhibit overlap due to numerous genetic and/or environmental factors, including the large number and diversity of NAT2 genotypes present in human populations. The relative specificity of the substrate for NAT2 versus NAT1 at the concentrations obtained in vivo will also affect acetylator phenotype. Caffeine is commonly used as a probe drug for NAT2 phenotype determinations because it is relatively non-invasive and excellent NAT2 genotype/phenotype correlations have been reported (Cascorbi et al., 1995; Grant et al., 1997; 2004). Genetic and/or environmental effects on a number of enzyme systems (e.g., cytochrome P450, xanthine oxidase, NAT1) may affect metabolite levels used to assess phenotype. Other potential artifacts in the use of caffeine to determine acetylation phenotype also have been reported (Cribb et al., 1994; Lorenzo and Reidenberg, 1989; O’Neil et al., 2000; Svensson and Hein, 2004).
NAT2 polymorphism and urinary bladder cancer risk
Human epidemiological studies have investigated the role of the NAT2 polymorphism in many cancers. Urinary bladder is a textbook example since individuals are frequently exposed to aromatic amine urinary bladder carcinogens such as ABP and BNA in cigarette smoke (Luceri et al., 1993; Stabbert et al., 2003). These aromatic amine carcinogens require metabolic activation in order to mutate DNA and initiate carcinogenesis. Following N-oxidation, the N-hydroxyaromatic and N-hydroxyheterocyclic amines are further activated (via O-acetylation) by N-acetyltransferases to acetoxy intermediates which react spontaneously with DNA to form DNA adducts (Hein, 1988). Thus, biological plausability for relationships between the NAT2 acetylation polymorphisms are strongest for cancers related to aromatic amine exposures.
The role of rapid versus slow acetylator genotype in cancer predisposition differs between organ sites as might be expected with tissue-specific expression of NAT2. Although reports of associations between NAT2 polymorphism and a number of cancers have been reported, a focus on urinary bladder cancer is useful for illustrating the divergent effects of carcinogenic agent and NAT2 haplotype on individual risk.
The first association between slow acetylator phenotype and urinary bladder cancer was reported over 25 years ago (Lower et al., 1979). The hypothetical mechanism for this association is slow NAT2 acetylation of aromatic amine carcinogens competes poorly with metabolic activation via cytochrome P450(s) and/or prostaglandin H-synthases, thus accounting for higher risk in the slow NAT2 phenotype(s). In a landmark study, English chemical dye workers with documented exposure to aromatic amine carcinogens showed a striking association (OR = 16.7; P=0.00005) between urinary bladder cancer and slow acetylator phenotypes (Cartwright et al., 1982). The population studied had documented exposures to aromatic amines and NAT2 acetylator phenotype was assessed by measurement of plasma monoacetyl-dapsone to dapsone metabolic ratios. Dapsone is an NAT2 selective substrate (more so than caffeine) and measurement of monoacetyl-dapsone to dapsone metabolic ratios in plasma (rather than urine) is a more direct assessment of NAT2 phenotype. Interestingly, the NAT2 phenotype data was not separated into two phenotypes (rapid and slow), but rather into eight ranges of metabolic ratios. Five of these ratios (0.3 and greater) correspond to rapid acetylators, and the other three (0.01 to 0.09; 0.1–0.19; and 0.2 –0.29) correspond to different levels of slow acetylator phenotype. As reviewed previously (Hein, 2002), urinary bladder cancer risk increased as NAT2 metabolic ratio (phenotype) decreased (Ptrend = 0.0006). The risk was markedly increased in the slowest NAT2 phenotype (OR, 20.8; 95%CI, 2.63–164). Four studies found that urinary bladder cancer risk was highest in individuals possessing NAT2*5 haplotypes (Brockmoller et al., 1996; Okkels et al., 1997; Filiadis et al., 1999; El Desoky et al., 2005). The 341T>C (I114T) SNP associated with NAT2*5 alleles or haplotypes yields very large reductions in NAT2 protein and activity (Hein et al., 1994b; 1995; Fretland et al., 2001) resulting from protein degradation (Zang et al., 2004). Recently, NAT2*5 alleles also were associated with increased risk for breast cancer in women smokers (van der Hel et al., 2003). These results suggest that NAT2 slow acetylator phenotype is not homogeneous, but rather that multiple slow acetylator phenotypes exist resulting from different mechanisms inferred by various SNPs and haplotypes.
Among smokers, NAT2 slow acetylators have higher levels of 4-aminobiphenyl hemoglobin adducts (Vineis et al., 1994; Yu et al., 1994; Probst-Hensch et al., 2000). Furthermore, ABP-DNA adducts in higher grade bladder tumors are found at higher levels in smokers who are slow NAT2 acetylators (Airoldi et al., 2002; Hao et al., 2004). A previous review of 21 published case control studies reported that experimental evidence was not sufficient to conclude a real increase in risk for urinary bladder cancer in slow NAT2 acetylators (Green et al., 2000). However, subsequent studies carried out in Europe (Vineis et al., 2001), Japan (Tsukino et al., 2004), the United States (Gu et al., 2005), and Spain (Garcia-Closas et al., 2005) each reported that NAT2 slow acetylators had a significantly increased risk of urinary bladder cancer that was stronger in smokers, particularly heavy or long term smokers. Because of the high frequency of homozygous rapid acetylators in Japan, that study also noted a higher risk in intermediate acetylators compared to homozygous rapid acetylators (Tsukino et al., 2004). Meta-analysis of these and all previous studies show that the overall association with slow NAT2 genotype in the published literature is robust (Figure 7) providing compelling evidence for a role of NAT2 acetylator genotype in urinary bladder cancer associated with aromatic amines in cigarette smoke.
Figure 7.
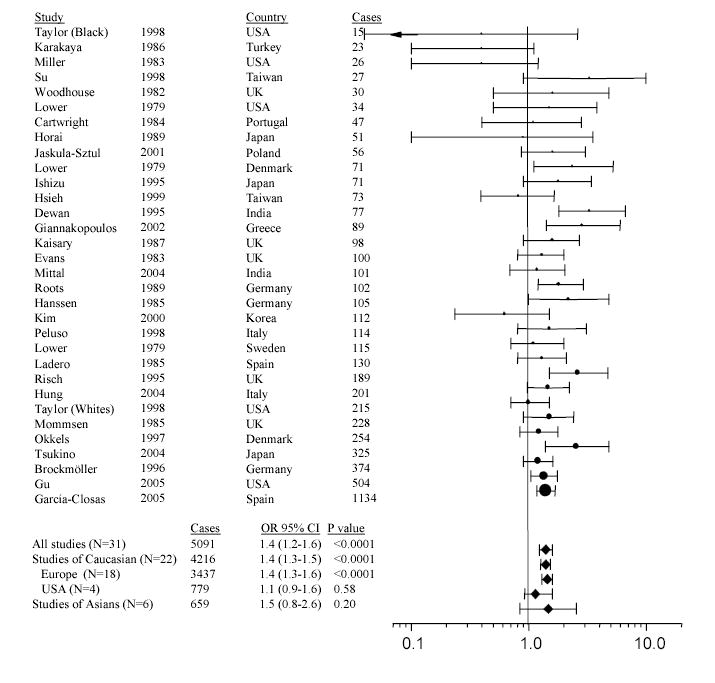
Meta-analysis of NAT2 slow acetylator genotype and bladder cancer risk. Odds ratios (circles) with 95% confidence limits (bars) represent the association of slow NAT2 acetylator phenotype/genotype with urinary bladder cancer reported in various studies throughout the world. Studies carried out in various countries are listed in ascending order of case size which is represented visually by circle size. Group analyses of the world total, and of European, American, and Asian subgroups are shown. Modified with permission from Garcia-Closas et al., 2005.
Since ethnic differences in NAT2 allele frequencies are quite striking (Figures 5 and 6), it has been suggested that that the role of NAT2 polymorphism on urinary cancer risk may differ with ethnic group (Golka et al., 2002). However, strong evidence has been provided by Carreon et al (2006) that this is not the case, but rather that the role of NAT2 polymorphism on urinary bladder cancer risk differs with carcinogenic agent. Although one study found no difference in urinary bladder cancer risk between rapid and slow NAT2 acetylator Chinese workers (Ma et al., 2004), another study of Chinese workers exposed to benzidine (Hayes et al., 1993) subsequently confirmed in a follow-up study (Carreon et al., 2006) reported that slow NAT2 acetylator genotype was associated with decreased risk to urinary bladder (relative to rapid NAT2 acetylators). As noted above and in Figure 7, the effect of NAT2 polymorphism on urinary bladder cancer risk for smokers is not dependent upon ethnic group. Rather than an ethnic difference, these findings are explained by the observation that since benzidine is a aryldiamine, the N-acetylation of one aromatic amine moiety is not a deactivation step and may enhance metabolic activation and/or transport to the urinary bladder. Urinary bladder DNA adducts following benzidine exposures in humans derive from N-acetylated metabolite(s) (Rothman et al., 1996). Support for this hypothesis also derives from the effect of NAT2 polymorphism on hepatoxicity from the aryldiamine 4,4’-methylenedianiline in the rat (Zhang et al., 2006).
NAT2 rapid acetylators potentially would be at decreased risk of urinary bladder cancer following dermal exposures to aromatic amine carcinogens if NAT2 were highly expressed in skin, since a rapid NAT2 acetylator phenotype would have higher capacity to deactivate the carcinogen prior to systemic absorption. The lack of NAT2 expression in human kerotinocytes (Reilly et al., 2000) is consistent with increased risk of urinary bladder cancer in NAT2 slow acetylators following dermal exposures (Gago-Dominguez et al., 2003). Thus following both inhalation and dermal exposures to aromatic monoarylamines, N-acetylation competes with N-hydroxylation conferring higher risk to the slow NAT2 acetylator phenotype(s). Furthermore, local N- or O-acetylation of arylamines or their N-hydroxylated metabolites resulting from dermal exposures would not be modified by NAT2 acetylation polymorphism, except indirectly to the extent that there is linkage disequilibrium between NAT1 and NAT2 alleles.
The effect of NAT2 acetylator polymorphism on urinary bladder cancer susceptibility is dependent upon accuracy of the exposure and genotype assessments (Rothman et al., 1993; Deitz et al., 2004). Indeed, reports suggest that ABP is present in higher yields in sidestream versus mainstream cigarette smoke and aromatic amines are present in indoor environments exposed to side stream cigarette smoke (Luceri et al., 1993; Palmiotto et al., 2001). Since exposure to passive smoking may also increase urinary bladder cancer risk, studies that use controls not exposed to passive cigarette smoke may yield more robust findings. NAT2 genotyping methods resulting in misclassification may confound relationships between NAT2 acetylator polymorphism and urinary bladder cancer risk (Deitz et al., 2004). Functional understanding of the effects of NAT2 genetic polymorphisms on metabolism and genotoxicity, tissue-specific expression, and the molecular mechanisms responsible for these effects are critical for the interpretation of previous and future human molecular epidemiology studies.
Acknowledgments
The author gratefully acknowledges the assistance of Dr. Montserrat Garcia-Closas for providing Figure 7 as an update from that recently published in Lancet (Garcia-Closas et al., 2005). Thanks also to faculty, staff and students at the University of Louisville who reviewed and improved the manuscript. Studies and hypotheses from the author’s laboratory described in this paper have been supported by over twenty years of research funding from the National Cancer Institute (R01-CA34627) and more recently by Philip Morris USA and the Kentucky Lung Cancer Research Program.
References
- Abe M, Deguchi T, Suzuki T. Biochem Biophys Res Commun. 1993;191:811–6. doi: 10.1006/bbrc.1993.1289. [DOI] [PubMed] [Google Scholar]
- Agundez JA, Menaya JG, Tejeda R, Lago F, Chavez M, Benitez J. Pharmacogenetics. 1996;6:465–72. doi: 10.1097/00008571-199610000-00011. [DOI] [PubMed] [Google Scholar]
- Airoldi L, Orsi F, Magagnotti C, Coda R, Randone D, Casetta G, Peluso M, Hautefeuille A, Malaveille C, Vineis P. Carcinogenesis. 2002;23:861–6. doi: 10.1093/carcin/23.5.861. [DOI] [PubMed] [Google Scholar]
- Anitha A, Banerjee M. Int J Mol Med. 2003;11:125–31. [PubMed] [Google Scholar]
- Badawi AF, Hirvonen A, Bell DA, Lang NP, Kadlubar FF. Cancer Res. 1995;55:5230–7. [PubMed] [Google Scholar]
- Blum M, Demierre A, Grant DM, Heim M, Meyer UA. Proc Natl Acad Sci U S A. 1991;88:5237–41. doi: 10.1073/pnas.88.12.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum M, Grant DM, Demierre A, Meyer UA. Proc Natl Acad Sci U S A. 1989;86:9554–7. doi: 10.1073/pnas.86.23.9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum M, Grant DM, McBride W, Heim M, Meyer UA. DNA Cell Biol. 1990;9:193–203. doi: 10.1089/dna.1990.9.193. [DOI] [PubMed] [Google Scholar]
- Bolt HM, Selinski S, Dannappel D, Blaszkewicz M, Golka K. Arch Toxicol. 2005;79:196–200. doi: 10.1007/s00204-004-0622-8. [DOI] [PubMed] [Google Scholar]
- Boukouvala S, Sim E. Basic Clin Pharmacol Toxicol. 2005;96:343–51. doi: 10.1111/j.1742-7843.2005.pto_02.x. [DOI] [PubMed] [Google Scholar]
- Brockmoller J, Cascorbi I, Kerb R, Roots I. Cancer Res. 1996;56:3915–25. [PubMed] [Google Scholar]
- Carreon T, Ruder AM, Schulte PA, Hayes RB, Rothman N, Waters M, Grant DJ, Boissy R, Bell DA, Kadlubar FF, Hemstreet GP, 3rd, Yin S, Lemasters GK. Int J Cancer. 2006;118:161–168. doi: 10.1002/ijc.21308. [DOI] [PubMed] [Google Scholar]
- Cartwright RA, Glashan RW, Rogers HJ, Ahmad RA, Barham-Hall D, Higgins E, Kahn MA. Lancet. 1982;2:842–5. doi: 10.1016/s0140-6736(82)90810-8. [DOI] [PubMed] [Google Scholar]
- Cascorbi I, Brockmoller J, Bauer S, Reum T, Roots I. Pharmacogenetics. 1996;6:257–9. doi: 10.1097/00008571-199606000-00009. [DOI] [PubMed] [Google Scholar]
- Cascorbi I, Brockmoller J, Mrozikiewicz PM, Muller A, Roots I. Drug Metab Rev. 1999;31:489–502. doi: 10.1081/dmr-100101932. [DOI] [PubMed] [Google Scholar]
- Cascorbi I, Drakoulis N, Brockmoller J, Maurer A, Sperling K, Roots I. Am J Hum Genet. 1995;57:581–92. [PMC free article] [PubMed] [Google Scholar]
- Cascorbi I, Roots I. Pharmacogenetics. 1999;9:123–7. doi: 10.1097/00008571-199902000-00016. [DOI] [PubMed] [Google Scholar]
- Chapron DJ, Kramer PA, Mercik SA. Clin Pharmacol Ther. 1980;27:104–13. doi: 10.1038/clpt.1980.16. [DOI] [PubMed] [Google Scholar]
- Chung JG, Levy GN, Weber WW. Drug Metab Dispos. 1993;21:1057–63. [PubMed] [Google Scholar]
- Cribb AE, Isbrucker R, Levatte T, Tsui B, Gillespie CT, Renton KW. Pharmacogenetics. 1994;4:166–70. [PubMed] [Google Scholar]
- Deguchi T. J Biol Chem. 1992;267:18140–7. [PubMed] [Google Scholar]
- Deguchi T, Mashimo M, Suzuki T. J Biol Chem. 1990;265:12757–60. [PubMed] [Google Scholar]
- Deitz AC, Rothman N, Rebbeck TR, Hayes RB, Chow WH, Zheng W, Hein DW, Garcia-Closas M. Cancer Epidemiol Biomarkers Prev. 2004;13:1543–6. [PubMed] [Google Scholar]
- Deitz AC, Zheng W, Leff MA, Gross M, Wen WQ, Doll MA, Xiao GH, Folsom AR, Hein DW. Cancer Epidemiol Biomarkers Prev. 2000;9:905–10. [PubMed] [Google Scholar]
- Delomenie C, Sica L, Grant DM, Krishnamoorthy R, Dupret JM. Pharmacogenetics. 1996;6:177–85. doi: 10.1097/00008571-199604000-00004. [DOI] [PubMed] [Google Scholar]
- Dewan A, Chattopadhyay P, Kulkarni PK. Indian J Cancer. 1995;32:15–9. [PubMed] [Google Scholar]
- Doll MA, Hein DW. Pharmacogenetics. 1995;5:247–51. doi: 10.1097/00008571-199508000-00009. [DOI] [PubMed] [Google Scholar]
- Dupret JM, Grant DM. J Biol Chem. 1992;267:7381–5. [PubMed] [Google Scholar]
- Dupret JM, Rodrigues-Lima F. Curr Med Chem. 2005;12:311–8. doi: 10.2174/0929867053363289. [DOI] [PubMed] [Google Scholar]
- Ebisawa T, Deguchi T. Biochem Biophys Res Commun. 1991;177:1252–7. doi: 10.1016/0006-291x(91)90676-x. [DOI] [PubMed] [Google Scholar]
- El Desoky ES, AbdelSalam YM, Salama RH, El Akkad MA, Atanasova S, von Ahsen N, Armstrong VW, Oellerich M. Ther Drug Monit. 2005;27:297–304. doi: 10.1097/01.ftd.0000164197.95494.aa. [DOI] [PubMed] [Google Scholar]
- Ferguson RJ, Doll MA, Rustan TD, Gray K, Hein DW. Drug Metab Dispos. 1994;22:371–6. [PubMed] [Google Scholar]
- Ferguson RJ, Doll MA, Rustan TD, Hein DW. Pharmacogenetics. 1996;6:55–66. doi: 10.1097/00008571-199602000-00004. [DOI] [PubMed] [Google Scholar]
- Filiadis IF, Georgiou I, Alamanos Y, Kranas V, Giannakopoulos X, Lolis D. J Urol. 1999;161:1672–5. [PubMed] [Google Scholar]
- Fretland AJ, Doll MA, Leff MA, Hein DW. Pharmacogenetics. 2001;11:511–20. doi: 10.1097/00008571-200108000-00006. [DOI] [PubMed] [Google Scholar]
- Frederickson SM, Hatcher JF, Reznikoff CA, Swaminathan S. Carcinogenesis. 1992;13:955–61. doi: 10.1093/carcin/13.6.955. [DOI] [PubMed] [Google Scholar]
- Frederickson SM, Messing EM, Reznikoff CA, Swaminathan S. Cancer Epidemiol Biomarkers Prev. 1994;3:25–32. [PubMed] [Google Scholar]
- Gago-Dominguez M, Bell DA, Watson MA, Yuan JM, Castelao JE, Hein DW, Chan KK, Coetzee GA, Ross RK, Yu MC. Carcinogenesis. 2003;24:483–9. doi: 10.1093/carcin/24.3.483. [DOI] [PubMed] [Google Scholar]
- Garcia-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, Tardon A, Serra C, Carrato A, Garcia-Closas R, Lloreta J, Castano-Vinyals G, Yeager M, Welch R, Chanock S, Chatterjee N, Wacholder S, Samanic C, Tora M, Fernandez F, Real FX, Rothman N. Lancet. 2005;366:649–59. doi: 10.1016/S0140-6736(05)67137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos X, Charalabopoulos K, Baltogiannis D, Chatzikiriakidou A, Alamanos Y, Georgiou I, Evangelou A, Agnantis N, Sofikitis N. Anticancer Res. 2002;22:3801–4. [PubMed] [Google Scholar]
- Green J, Banks E, Berrington A, Darby S, Deo H, Newton R. Br J Cancer. 2000;83:412–7. doi: 10.1054/bjoc.2000.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golka K, Prior V, Blaszkewicz M, Bolt HM. Toxicol Lett. 2002;128:229–41. doi: 10.1016/s0378-4274(01)00544-6. [DOI] [PubMed] [Google Scholar]
- Grant DM, Hughes NC, Janezic SA, Goodfellow GH, Chen HJ, Gaedigk A, Yu VL, Grewal R. Mutat Res. 1997;376:61–70. doi: 10.1016/s0027-5107(97)00026-2. [DOI] [PubMed] [Google Scholar]
- Grant DM, Morike K, Eichelbaum M, Meyer UA. J Clin Invest. 1990;85:968–72. doi: 10.1172/JCI114527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant DM, Tang BK, Kalow W. Br J Clin Pharmacol. 2004;58:S788–93. doi: 10.1111/j.1365-2125.2004.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M, Kruisselbrink T, Anderson K, Lang N, McGovern P, Delongchamp R, Kadlubar F. Cancer Epidemiol Biomarkers Prev. 1999;8:683–92. [PubMed] [Google Scholar]
- Gu J, Liang D, Wang Y, Lu C, Wu X. Mutat Res. 2005;581:97–104. doi: 10.1016/j.mrgentox.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Hanssen HP, Agarwal DP, Goedde HW, Bucher H, Huland H, Brachmann W, Ovenbeck R. Eur Urol. 1985;11:263–6. doi: 10.1159/000472511. [DOI] [PubMed] [Google Scholar]
- Hao GY, Zhang WD, Chen YH, Zhang DX, Zhang YH. Zhonghua Zhong Liu Za Zhi. 2004;26:283–6. [PubMed] [Google Scholar]
- Hayes RB, Bi W, Rothman N, Broly F, Caporaso N, Feng P, You X, Yin S, Woosley RL, Meyer UA. Carcinogenesis. 1993;14:675–8. doi: 10.1093/carcin/14.4.675. [DOI] [PubMed] [Google Scholar]
- Hearse DJ, Weber WW. Biochem J. 1973;132:519–26. doi: 10.1042/bj1320519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein DW. Biochim Biophys Acta. 1988;948:37–66. doi: 10.1016/0304-419x(88)90004-2. [DOI] [PubMed] [Google Scholar]
- Hein DW. (2002). Mutat Res, 506–507, 65–77. [DOI] [PubMed]
- Hein DW, Doll MA, Fretland AJ, Gray K, Deitz AC, Feng Y, Jiang W, Rustan TD, Satran SL, Wilkie TR., Sr Mutat Res. 1997;376:101–6. doi: 10.1016/s0027-5107(97)00031-6. [DOI] [PubMed] [Google Scholar]
- Hein DW, Doll MA, Fretland AJ, Leff MA, Webb SJ, Xiao GH, Devanaboyina US, Nangju NA, Feng Y. Cancer Epidemiol Biomarkers Prev. 2000;9:29–42. [PubMed] [Google Scholar]
- Hein DW, Doll MA, Gray K, Rustan TD, Ferguson RJ. Cancer Res. 1993a;53:509–14. [PubMed] [Google Scholar]
- Hein DW, Doll MA, Nerland DE, Fretland AJ. Mol Carcinog. 2006;45:230–8. doi: 10.1002/mc.20164. [DOI] [PubMed] [Google Scholar]
- Hein DW, Doll MA, Rustan TD, Ferguson RJ. Cancer Res. 1995;55:3531–6. [PubMed] [Google Scholar]
- Hein DW, Doll MA, Rustan TD, Gray K, Feng Y, Ferguson RJ, Grant DM. Carcinogenesis. 1993b;14:1633–8. doi: 10.1093/carcin/14.8.1633. [DOI] [PubMed] [Google Scholar]
- Hein DW, Doll MA, Rustan TD, Gray K, Ferguson RJ, Feng Y. Toxicol Appl Pharmacol. 1994a;124:16–24. doi: 10.1006/taap.1994.1003. [DOI] [PubMed] [Google Scholar]
- Hein DW, Doll MA, Xiao GH, Feng Y. Pharmacogenetics. 2003;13:159–67. doi: 10.1097/00008571-200303000-00005. [DOI] [PubMed] [Google Scholar]
- Hein DW, Ferguson RJ, Doll MA, Rustan TD, Gray K. Hum Mol Genet. 1994b;3:729–34. doi: 10.1093/hmg/3.5.729. [DOI] [PubMed] [Google Scholar]
- Hein DW, Kirlin WG, Ferguson RJ, Weber WW. J Pharmacol Exp Ther. 1985;234:358–64. [PubMed] [Google Scholar]
- Hein DW, Rustan TD, Bucher KD, Martin WJ, Furman EJ. Drug Metab Dispos. 1991a;19:933–7. [PubMed] [Google Scholar]
- Hein DW, Rustan TD, Bucher KD, Miller LS. J Pharmacol Exp Ther. 1991b;259:699–704. [PubMed] [Google Scholar]
- Hein DW, Trinidad A, Yerokun T, Ferguson RJ, Kirlin WG, Weber WW. Drug Metab Dispos. 1988;16:341–7. [PubMed] [Google Scholar]
- Hickman D, Palamanda JR, Unadkat JD, Sim E. Biochem Pharmacol. 1995;50:697–703. doi: 10.1016/0006-2952(95)00182-y. [DOI] [PubMed] [Google Scholar]
- Hickman D, Pope J, Patil SD, Fakis G, Smelt V, Stanley LA, Payton M, Unadkat JD, Sim E. Gut. 1998;42:402–9. doi: 10.1136/gut.42.3.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman D, Risch A, Buckle V, Spurr NK, Jeremiah SJ, McCarthy A, Sim E. Biochem J. 1994;297 ( Pt 3):441–5. doi: 10.1042/bj2970441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horai Y, Fujita K, Ishizaki T. Eur J Clin Pharmacol. 1989;37:581–7. doi: 10.1007/BF00562549. [DOI] [PubMed] [Google Scholar]
- Hsieh FI, Pu YS, Chern HD, Hsu LI, Chiou HY, Chen CJ. Br J Cancer. 1999;81:537–41. doi: 10.1038/sj.bjc.6690727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RJ, Boffetta P, Brennan P, Malaveille C, Hautefeuille A, Donato F, Gelatti U, Spaliviero M, Placidi D, Carta A, Scotto di Carlo A, Porru S. Int J Cancer. 2004;110:598–604. doi: 10.1002/ijc.20157. [DOI] [PubMed] [Google Scholar]
- Hughes HB, Biehl JP, Jones AP, Scmidt LH. Am Rev Res Dis. 1954;70:266–73. doi: 10.1164/art.1954.70.2.266. [DOI] [PubMed] [Google Scholar]
- Ilett KF, Ingram DM, Carpenter DS, Teitel CH, Lang NP, Kadlubar FF, Minchin RF. Biochem Pharmacol. 1994;47:914–7. doi: 10.1016/0006-2952(94)90493-6. [DOI] [PubMed] [Google Scholar]
- Ilett KF, Reeves PT, Minchin RF, Kinnear BF, Watson HF, Kadlubar FF. Carcinogenesis. 1991;12:1465–9. doi: 10.1093/carcin/12.8.1465. [DOI] [PubMed] [Google Scholar]
- Ishizu S, Hashida C, Hanaoka T, Maeda K, Ohishi Y. Jpn J Cancer Res. 1995;86:1179–81. doi: 10.1111/j.1349-7006.1995.tb03312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskula-Sztul R, Sokolowski W, Gajecka M, Szyfter K. J Appl Genet. 2001;42:223–31. [PubMed] [Google Scholar]
- Jenne JW. J Clin Invest. 1965;44:1992–2002. doi: 10.1172/JCI105306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisary A, Smith P, Jaczq E, McAllister CB, Wilkinson GR, Ray WA, Branch RA. Cancer Res. 1987;47:5488–93. [PubMed] [Google Scholar]
- Karakaya AE, Cok I, Sardas S, Gogus O, Sardas OS. Hum Toxicol. 1986;5:333–5. doi: 10.1177/096032718600500507. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Lee HL, Lee SC, Kim YT, Kim H. J Urol. 2000;164:209–13. [PubMed] [Google Scholar]
- Kirlin WG, Ogolla F, Andrews AF, Trinidad A, Ferguson RJ, Yerokun T, Mpezo M, Hein DW. Cancer Res. 1991;51:549–55. [PubMed] [Google Scholar]
- Kirlin WG, Trinidad A, Yerokun T, Ogolla F, Ferguson RJ, Andrews AF, Brady PK, Hein DW. Cancer Res. 1989;49:2448–54. [PubMed] [Google Scholar]
- Ladero JM, Kwok CK, Jara C, Fernandez L, Silmi AM, Tapia D, Uson AC. Ann Clin Res. 1985;17:96–9. [PubMed] [Google Scholar]
- Lan Q, Rothman N, Chow WH, Lissowska J, Doll MA, Xiao GH, Zatonski W, Hein DW. Cancer Epidemiol Biomarkers Prev. 2003;12:384–6. [PubMed] [Google Scholar]
- Lee EJ, Lee LK. Br J Clin Pharmacol. 1982;13:375–8. doi: 10.1111/j.1365-2125.1982.tb01388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Lee KA, Ki CS, Kwon OJ, Kim HJ, Chung MP, Suh GY, Kim JW. Clin Chem. 2002;48:775–7. [PubMed] [Google Scholar]
- Leff MA, Fretland AJ, Doll MA, Hein DW. J Biol Chem. 1999;274:34519–22. doi: 10.1074/jbc.274.49.34519. [DOI] [PubMed] [Google Scholar]
- Lorenzo B, Reidenberg MM. Br J Clin Pharmacol. 1989;28:207–8. doi: 10.1111/j.1365-2125.1989.tb05420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktionov A, Moore W, Spencer SP, Vorster H, Nell T, O’Neill IK, Bingham SA, Cummings JH. Cancer Detect Prev. 2002;26:15–22. doi: 10.1016/s0361-090x(02)00010-7. [DOI] [PubMed] [Google Scholar]
- Lower GM, Jr, Nilsson T, Nelson CE, Wolf H, Gamsky TE, Bryan GT. Environ Health Perspect. 1979;29:71–9. doi: 10.1289/ehp.792971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luceri F, Pieraccini G, Moneti G, Dolara P. Toxicol Ind Health. 1993;9:405–13. doi: 10.1177/074823379300900302. [DOI] [PubMed] [Google Scholar]
- Ma QW, Lin GF, Chen JG, Xiang CQ, Guo WC, Golka K, Shen JH. Biomed Environ Sci. 2004;17:291–8. [PubMed] [Google Scholar]
- Martell KJ, Vatsis KP, Weber WW. Mol Pharmacol. 1991;40:218–27. [PubMed] [Google Scholar]
- Matas N, Thygesen P, Stacey M, Risch A, Sim E. Cytogenet Cell Genet. 1997;77:290–5. doi: 10.1159/000134601. [DOI] [PubMed] [Google Scholar]
- Miller ME, Cosgriff JM. J Urol. 1983;130:65–6. doi: 10.1016/s0022-5347(17)50956-8. [DOI] [PubMed] [Google Scholar]
- Minchin RF, Reeves PT, Teitel CH, McManus ME, Mojarrabi B, Ilett KF, Kadlubar FF. Biochem Biophys Res Commun. 1992;185:839–44. doi: 10.1016/0006-291x(92)91703-s. [DOI] [PubMed] [Google Scholar]
- Mittal RD, Srivastava DS, Mandhani A. Int Braz J Urol. 2004;30:279–85. doi: 10.1590/s1677-55382004000400003. discussion 285–8. [DOI] [PubMed] [Google Scholar]
- Mommsen S, Wolf H. Scand J Urol Nephrol. 1985;19:203–4. doi: 10.3109/00365598509180254. [DOI] [PubMed] [Google Scholar]
- Nagata K, Ozawa S, Miyata M, Shimada M, Yamazoe Y, Kato R. Pharmacogenetics. 1994;4:91–100. doi: 10.1097/00008571-199404000-00006. [DOI] [PubMed] [Google Scholar]
- Okkels H, Sigsgaard T, Wolf H, Autrup H. Cancer Epidemiol Biomarkers Prev. 1997;6:225–31. [PubMed] [Google Scholar]
- O’Neil WM, Drobitch RK, MacArthur RD, Farrough MJ, Doll MA, Fretland AJ, Hein DW, Crane LR, Svensson CK. Pharmacogenetics. 2000;10:171–82. doi: 10.1097/00008571-200003000-00009. [DOI] [PubMed] [Google Scholar]
- Ozawa S, Abu-Zeid M, Kawakubo Y, Toyama S, Yamazoe Y, Kato R. Carcinogenesis. 1990;11:2137–44. doi: 10.1093/carcin/11.12.2137. [DOI] [PubMed] [Google Scholar]
- Pacifici GM, Bencini C, Rane A. Pharmacology. 1986;32:283–91. doi: 10.1159/000138181. [DOI] [PubMed] [Google Scholar]
- Palmiotto G, Pieraccini G, Moneti G, Dolara P. Chemosphere. 2001;43:355–61. doi: 10.1016/s0045-6535(00)00109-0. [DOI] [PubMed] [Google Scholar]
- Parkin DP, Vandenplas S, Botha FJ, Vandenplas ML, Seifart HI, van Helden PD, van der Walt BJ, Donald PR, van Jaarsveld PP. Am J Respir Crit Care Med. 1997;155:1717–22. doi: 10.1164/ajrccm.155.5.9154882. [DOI] [PubMed] [Google Scholar]
- Peluso M, Airoldi L, Armelle M, Martone T, Coda R, Malaveille C, Giacomelli G, Terrone C, Casetta G, Vineis P. Cancer Epidemiol Biomarkers Prev. 1998;7:341–6. [PubMed] [Google Scholar]
- Pink JC, Messing EM, Reznikoff CA, Bryan GT, Swaminathan S. Drug Metab Dispos. 1992;20:559–65. [PubMed] [Google Scholar]
- Probst-Hensch NM, Bell DA, Watson MA, Skipper PL, Tannenbaum SR, Chan KK, Ross RK, Yu MC. Cancer Epidemiol Biomarkers Prev. 2000;9:619–23. [PubMed] [Google Scholar]
- Reilly TP, Lash LH, Doll MA, Hein DW, Woster PM, Svensson CK. J Invest Dermatol. 2000;114:1164–73. doi: 10.1046/j.1523-1747.2000.00985.x. [DOI] [PubMed] [Google Scholar]
- Risch A, Wallace DM, Bathers S, Sim E. Hum Mol Genet. 1995;4:231–6. doi: 10.1093/hmg/4.2.231. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Lima F, Delomenie C, Goodfellow GH, Grant DM, Dupret JM. Biochem J. 2001;356:327–34. doi: 10.1042/0264-6021:3560327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues-Lima F, Dupret JM. Biochem Biophys Res Commun. 2002;291:116–23. doi: 10.1006/bbrc.2002.6414. [DOI] [PubMed] [Google Scholar]
- Rothman N, Bhatnagar VK, Hayes RB, Zenser TV, Kashyap SK, Butler MA, Bell DA, Lakshmi V, Jaeger M, Kashyap R, Hirvonen A, Schulte PA, Dosemeci M, Hsu F, Parikh DJ, Davis BB, Talaska G. Proc Natl Acad Sci U S A. 1996;93:5084–9. doi: 10.1073/pnas.93.10.5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman N, Stewart WF, Caporaso NE, Hayes RB. Cancer Epidemiol Biomarkers Prev. 1993;2:299–303. [PubMed] [Google Scholar]
- Sandy J, Mushtaq A, Kawamura A, Sinclair J, Sim E, Noble M. J Mol Biol. 2002;318:1071–83. doi: 10.1016/S0022-2836(02)00141-9. [DOI] [PubMed] [Google Scholar]
- Sinclair JC, Sandy J, Delgoda R, Sim E, Noble ME. Nat Struct Biol. 2000;7:560–4. doi: 10.1038/76783. [DOI] [PubMed] [Google Scholar]
- Smith TJ, Hanna PE. Carcinogenesis. 1986;7:697–702. doi: 10.1093/carcin/7.5.697. [DOI] [PubMed] [Google Scholar]
- Smith CA, Wadelius M, Gough AC, Harrison DJ, Wolf CR, Rane A. J Med Genet. 1997;34:758–60. doi: 10.1136/jmg.34.9.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabbert R, Schafer KH, Biefel C, Rustemeier K. Rapid Commun Mass Spectrom. 2003;17:2125–32. doi: 10.1002/rcm.1161. [DOI] [PubMed] [Google Scholar]
- Stanley LA, Mills IG, Sim E. Pharmacogenetics. 1997;7:121–30. doi: 10.1097/00008571-199704000-00005. [DOI] [PubMed] [Google Scholar]
- Su HJ, Guo YL, Lai MD, Huang JD, Cheng Y, Christiani DC. Pharmacogenetics. 1998;8:187–90. doi: 10.1097/00008571-199804000-00011. [DOI] [PubMed] [Google Scholar]
- Sugamori KS, Wong S, Gaedigk A, Yu V, Abramovici H, Rozmahel R, Grant DM. Mol Pharmacol. 2003;64:170–9. doi: 10.1124/mol.64.1.170. [DOI] [PubMed] [Google Scholar]
- Svensson CK and Hein DW. (2004). Drug Metabolism and Transport: Molecular Methods and Mechanisms: Methods in Pharmacology and Toxicology. Lash LH (ed.). Humana Press: Totowa, NJ, pp 173–195.
- Taylor JA, Umbach DM, Stephens E, Castranio T, Paulson D, Robertson C, Mohler JL, Bell DA. Cancer Res. 1998;58:3603–10. [PubMed] [Google Scholar]
- Trinidad A, Kirlin WG, Ogolla F, Andrews AF, Yerokun T, Ferguson RJ, Brady PK, Hein DW. Drug Metab Dispos. 1989;17:238–47. [PubMed] [Google Scholar]
- Tsukino H, Nakao H, Kuroda Y, Imai H, Inatomi H, Osada Y, Katoh T. Eur J Cancer Prev. 2004;13:509–14. doi: 10.1097/00008469-200412000-00008. [DOI] [PubMed] [Google Scholar]
- Turesky RJ, Lang NP, Butler MA, Teitel CH, Kadlubar FF. Carcinogenesis. 1991;12:1839–45. doi: 10.1093/carcin/12.10.1839. [DOI] [PubMed] [Google Scholar]
- van der Hel OL, Peeters PH, Hein DW, Doll MA, Grobbee DE, Kromhout D, Bueno de Mesquita HB. Pharmacogenetics. 2003;13:399–407. doi: 10.1097/00008571-200307000-00005. [DOI] [PubMed] [Google Scholar]
- Vatsis KP, Martell KJ, Weber WW. Proc Natl Acad Sci U S A. 1991;88:6333–7. doi: 10.1073/pnas.88.14.6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatsis KP, Weber WW, Bell DA, Dupret JM, Evans DA, Grant DM, Hein DW, Lin HJ, Meyer UA, Relling MV, et al. Pharmacogenetics. 1995;5:1–17. doi: 10.1097/00008571-199502000-00001. [DOI] [PubMed] [Google Scholar]
- Vineis P, Alavanja M, Garte S. Int J Cancer. 2004;108:2–7. doi: 10.1002/ijc.11467. [DOI] [PubMed] [Google Scholar]
- Vineis P, Bartsch H, Caporaso N, Harrington AM, Kadlubar FF, Landi MT, Malaveille C, Shields PG, Skipper P, Talaska G, et al. Nature. 1994;369:154–6. doi: 10.1038/369154a0. [DOI] [PubMed] [Google Scholar]
- Vineis P, Marinelli D, Autrup H, Brockmoller J, Cascorbi I, Daly AK, Golka K, Okkels H, Risch A, Rothman N, Sim E, Taioli E. Cancer Epidemiol Biomarkers Prev. 2001;10:1249–52. [PubMed] [Google Scholar]
- Weber WW, Hein DW. Pharmacol Rev. 1985;37:25–79. [PubMed] [Google Scholar]
- Windmill KF, Gaedigk A, Hall PM, Samaratunga H, Grant DM, McManus ME. Toxicol Sci. 2000;54:19–29. doi: 10.1093/toxsci/54.1.19. [DOI] [PubMed] [Google Scholar]
- Woodhouse KW, Adams PC, Clothier A, Mucklow JC, Rawlins MD. Hum Toxicol. 1982;1:443–5. doi: 10.1177/096032718200100411. [DOI] [PubMed] [Google Scholar]
- Yerokun T, Kirlin WG, Trinidad A, Ferguson RJ, Ogolla F, Andrews AF, Brady PK, Hein DW. Drug Metab Dispos. 1989;17:231–7. [PubMed] [Google Scholar]
- Yu MC, Skipper PL, Taghizadeh K, Tannenbaum SR, Chan KK, Henderson BE, Ross RK. J Natl Cancer Inst. 1994;86:712–6. doi: 10.1093/jnci/86.9.712. [DOI] [PubMed] [Google Scholar]
- Zang Y, Zhao S, Doll MA and Hein DW. (2005). Proc Amer Assoc Cancer Res 46, Abstract #4060.
- Zang Y, Zhao S, Doll MA, States JC, Hein DW. Pharmacogenetics. 2004;14:717–23. doi: 10.1097/00008571-200411000-00002. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lambert JC, Doll MA, Walraven JM, Arteel GE, Hein DW. J Pharmacol Exp Ther. 2006;316:289–294. doi: 10.1124/jpet.105.093302. [DOI] [PubMed] [Google Scholar]