

Abstract
Objectives
To review the clinical and laboratory features of twelve cases of nephrogenic fibrosing dermopathy (NFD) studied at our institution and of 70 previously described cases in the literature.
Methods
Clinical evaluation and laboratory studies of twelve patients with NFD associated with chronic hemodialysis or peritoneal dialysis for end-stage renal disease and a review of 23 previous publications describing 70 patients with this disease.
Results
Eleven patients undergoing chronic hemodialysis and one patient undergoing chronic peritoneal dialysis for end-stage renal failure developed a severe and progressive cutaneous fibrotic process with woody induration of legs, thighs, hands and forearms, and severe loss of motion and flexion contractures in multiple joints. Several patients displayed systemic involvement including fibrosis of muscles, myocardium and lungs and marked elevations of the erythrocyte sedimentation rate and/or C reactive protein. Three patients died within two years of onset of symptoms. A review of previously published reports of this disorder confirmed the presence of systemic involvement and a poor prognosis with a high rate of mortality.
Conclusions
The analysis of twelve cases and a review of 70 previously described cases indicate that NFD is a severe and usually progressive systemic fibrotic disease affecting the dermis, subcutaneous fascia and striated muscles. It also appears that the disease can cause fibrosis of lungs, myocardium and other organs.
Keywords: Nephrogenic Fibrosing Dermopathy, Scleroderma, Fibrosis, TGFβ
INTRODUCTION
In 2000, Cowper et al. published the first report of 15 patients with scleromyxedema-like skin lesions in renal-dialysis patients (1). These patients had extensive thickening and hardening of the skin associated with browny hyperpigmentation, papules and subcutaneous nodules. Histopathologically the affected skin resembled scleromyxedema. However, the absence of systemic involvement and monoclonal paraproteinemia, and the unique histopathological findings indicated that the disorder was novel and distinct. A more detailed histopathological study was published subsequently and the term Nephrogenic Fibrosing Dermopathy (NFD) was used to refer to this novel clinical entity (2).
Following the initial identification of this disorder, a registry was established and by June 2005 the registry indicates that 170 cases had been published or reported (3). In contrast with the original description of a purely cutaneous disorder, recent reports indicate that this disorder displays evidence of systemic involvement (4–6). We recently published a brief description of nine cases with this disorder (5) and emphasized the occurrence of a severe inflammatory and systemic fibrotic process associated with chronic hemodialysis or peritoneal dialysis in the setting of end-stage renal disease.
Despite extensive epidemiologic and histopathological studies, the etiology of this disorder remains elusive. Renal failure is present in all cases and hemo- or peritoneal dialyses is a common factor present essentially in all cases described, although a few patients who never received dialysis have been reported (2, 8–9). A recent paper has reviewed briefly the first six years’ experience with NFD (10).
We describe here the clinical and histopathological features and the results of laboratory and ancillary tests in nine previously described and three new patients evaluated at our institution, and we review the salient manifestations of 70 patients with NFD described in 23 English language publications between the years 2000 and 2004.
METHODS
Clinical, laboratory and histopathological studies were performed in ten patients evaluated at the Scleroderma Center of Thomas Jefferson University between the years 2000 and 2005, in one patient followed at the Veterans Administration Hospital of St. Louis, MO, and in one patient followed at the Robert Wood Johnson Medical School (Camden, NJ). This evaluation included a detailed assessment for the presence of systemic involvement, including a review of systems and physical examination for cardiovascular, respiratory, gastrointestinal, genitourinary, neurologic and musculoskeletal and articular systems, as well as hematological, biochemical and serological laboratory analysis, electrocardiograms, echocardiograms and pulmonary function tests with diffusion capacity and in most patients computerized axial tomography of the chest. Full thickness skin biopsies including fascia (11 patients), skin punch biopsies (1 patient), muscle biopsies (6 patients), and several tissues from one deceased patient whose autopsy samples were available were examined by histopathology. Discarded tissues remaining from samples employed for diagnostic purposes and the autopsy specimens were stained with hematoxylin and eosin and Masson’s trichrome after fixation in formaldehyde. Some samples were stained with PAS, Giemsa, alcian blue, colloidal iron and Verhoeff–Van Gieson stains. Seventy cases described in 23 English language publications were also reviewed. These cases were identified searching the PubMed database with the following identifiers: Nephrogenic Fibrosing Dermopathy; Renal Failure AND Fibrosis; Dialysis AND Fibrosis; and Chronic Renal Disease AND Fibrosis. From all the cases identified those providing sufficient clinical and laboratory studies information were included for review.
RESULTS
Demographical and epidemiological characteristics
The details of the epidemiologic and demographic data of the twelve patients we studied are shown on Table 1. Ten patients received hemodialysis (HD), one patient received peritoneal dialysis (PD), and another patient was on HD at the time of diagnosis but had received PD previously. Six of twelve patients had renal transplantation which failed in all and required removal. The cause of the renal failure was diverse, as well as the duration of dialysis (Table 1).
Table 1.
Demographic data and relevant renal disease history.
| Age | Sex | Renal Disease | Dialysis (duration to onset) | Renal Transplant | |
|---|---|---|---|---|---|
| Case 1 | 55 | F | Polycystic kidneys | Hemodialysis (2–3 mos) | Yes* |
| Case 2 | 52 | M | Chronic glomerulosclerosis | Hemodialysis (16 mos)** | No |
| Case 3 | 74 | M | Diabetic nephrosclerosis | Hemodialysis (14 mos) | No |
| Case 4 | 47 | M | Polycystic kidneys | Hemodialysis (2 mos) | No |
| Case 5 | 55 | M | Diabetic nephrosclerosis | Hemodialysis (3 yrs) | No |
| Case 6 | 52 | F | Diabetic nephrosclerosis | Hemodialysis (6 mos)*** | Yes (x2)* |
| Case 7 | 53 | F | Chronic glomerulonephritis | Hemodialysis (22 yrs) | Yes (x3)* |
| Case 8 | 42 | F | Chronic glomerulonephritis | Peritoneal dialysis (10 yrs) | Yes (x2)* |
| Case 9 | 33 | M | Chronic nephritis | Hemodialysis (17 yrs) | No |
| Case 10 | 41 | F | Polycystic kidneys | Hemodialysis (8 yrs) | Yes (x2) |
| Case 11 | 55 | M | Focal segmental glomerulosclerosis**** | Hemodialysis (9 yrs) | Yes (x3) |
| Case 12 | 65 | M | Diabetic nephrosclerosis | Hemodialysis (2 mos) | No |
Renal transplant(s) failed and required removal.
Started on “hemofiltration” 3 months prior to onset.
Had been on peritoneal dialysis for 3 years; started hemodialysis 6 months prior to onset of skin tightness.
Had been on peritoneal dialysis intermittently for 8 years; started hemodialysis 2 years prior to onset of skin tightness
Clinical Features
The clinical features of the twelve patients we studied are summarized in Table 2 and are briefly discussed below within the context of the clinical manifestations of the other patients from the literature.
Table 2.
Clinical features and mortality.
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | Case 11 | Case 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Skin Sclerosis/Puckering | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes | Yes/Yes |
| Pruritus and Burning | Yes | Yes | Yes | No | No | No | No | No | No | Yes | No | No |
| Extremity Swelling | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Raynaud Phenomenon | No | No | No | No | No | No | No | No | No | No | No | No |
| Gastrointestinal Involvement | No | No | No | No | Yes* | Yes* | Yes* | No | No | Yes* | No | No |
| Lung Involvement (TLC/DLCO)** | No (N.D.) | Yes (65%/75%) | No (N.D.) | Yes (64%/66%) | Yes (86%/59.5%) | Yes (71%/49%) | Yes (85%/64%) | Yes (81%/60%) | No (N.D.) | No (N.D.) | Yes (85%/62%) | No (N.D.) |
| Muscle induration | Yes | No | Yes | Yes | Yes | Yes | Yes | No | No | Yes | Yes | Yes |
| Joint flexion contractures | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Neuropathy | Yes | No | No | No | No | Yes | No | No | No | Yes | No | Yes |
| Thrombotic events | Yes (peripheral vascular graft access) | No | Yes (peripheral vascular graft access) | No | No | No | No | Yes (TIA) | Yes (brain) | No | No | No |
| Mortality | Deceased | Alive | Deceased | Alive | Deceased | Alive | Alive | Alive | Alive | Alive | Alive | Alive |
Difficulty swallowing solid foods.
Shown as % of predicted values.
ND: Not done
Cutaneous Manifestations
The CDC defined the clinical characteristic of the disorder as “large areas of hardened skin with slightly raised plaques or confluent papules, with or without pigment alteration” in their case definition (3). The lesions are usually erythematous papules, but usually progress to diffuse browny induration or display the typical “peau d’orange” appearance described in cases of eosinophillic fasciitis. The appearance of affected skin in some of the patients we studied is illustrated in Figure 1.
Figure 1. Cutaneous involvement.

Note marked skin thickening including sclerodactyly and involvement of the dorsum of the hands (A, B) and “peau de orange” appearance in several affected areas (C, D). Severe fixed flexion contractures in fingers and wrist joints are apparent (C). Involvement of the lower legs (E) and a marked hypertrophic and neovascularized scar at a site of a minor dermal injury are illustrated (F).
In our series, all patients developed swelling of the feet and legs followed by severe and progressive skin induration. Many patients also noted pruritus and burning sensation in the affected skin. The clinical features of well established cutaneous lesions resembled the appearance of the cutaneous involvement of both systemic sclerosis and diffuse fasciitis. There was sclerodactyly, thickening and induration of the dorsum of the hands and lower legs, but the face was spared. The affected skin in arms, legs, thighs and abdomen displayed a browny induration and thickening, “peau d’orange” appearance, deep furrowing and loss of skin appendages (Figure 1). The cutaneous involvement initially affected the feet, lower legs and hands and in all our cases it progressed proximally to involve the upper legs, thighs, and forearms. The trunk and/or the abdomen were involved in six cases (Figure 2). Two patients noted localized inflammatory changes in the affected skin with erythema and increased skin temperature and one patient developed a remarkable fibroproliferative reaction at the site of a minor dermal abrasion with a large hypertrophic and neovascularized scar (Figure 1). One patient noticed coldness in the affected extremities but none of the patients had symptoms of Raynaud’s phenomenon.
Figure 2.

Diagram illustrating the pattern of cutaneous involvement in the patients of the cohort we studied.
From a review of the published cases (1, 2, 4–6, 8, 9, 11–26), it is apparent that skin lesions affect the lower extremities in almost all patients (97%) (55/57). Ankles, shins and lower thighs are the most commonly affected areas. Upper extremities are the second most common area of involvement being affected in 77% (44/57) of the patients, especially in the hands and wrists. The trunk was involved in 30% (17/57) whereas the face and neck were not involved.
Non-Cutaneous Manifestations
In initials reports, systemic involvement was not described and the absence of systemic manifestations was proposed as one of the important features and criteria in the differential diagnosis (1, 2). However, several subsequent studies have described prominent systemic involvement in these patients (4, 5, 6, 9). The first report of systemic involvement in this disorder described a patient whose autopsy revealed fibrosis and calcification of the diaphragm and psoas muscle, and fibrosis of the renal tubules and rete testis (4). Subsequently, Jimenez et al. (5) and Levine et al. (6) reported frequent systemic involvement including involvement of skeletal muscles, lung and myocardium, and Swartz et al. (9) described visceral involvement in 4 of the 13 cases they reported including cardiomyopathy and pulmonary fibrosis, although the authors considered these two alterations to represent co-morbidities rather than being caused by the disorder.
Joints and tendons
All patients in our series had severe involvement of multiple joints with loss of motion and flexion contractures leading to wheelchair confinement in four cases. The joints did not show clinical evidence of synovitis or arthritis and the severe limitation of motion appeared to be related to thickening of the tendons and periarticular tissues. A remarkable thickening and shortening of the digital flexor tendons and thickening of the palmar fascia were observed in four patients. Decreased range of motion of affected joints which progress to severe joint flexion contractures and severe disability have been described in numerous other cases in the literature (10).
Muscles and peripheral nerves
Eight patients in our series displayed a severe woody induration of muscles in the legs, thighs and forearms but true muscle weakness was not observed. Two patients had clinical evidence of sensory and motor neuropathy. Two patients had muscle and nerve electrophysiological studies. One of them had mild sensory neuropathy and the other had a moderate sensory motor neuropathy. A recent publication from our group described a detailed study of skeletal muscle and peripheral nerve involvement in three of previously described patients and in two additional cases not reported previously (6). In this study, peripheral nerve involvement evaluation by electromyography showed a sensory-motor polyneuropathy in 4/5 patients although it is not clear whether the nerve involvement was exclusively related to the disorder or was caused by the end-stage renal disease (6). Clinically, it is important to differentiate neuropathic pain, usually referred as paresthesias or burning sensation from myopathic pain, presented as “aching or cramping”; several of our patients reported the occurrence of both types of pain indicating that the process involved both muscles and nerves in these patients.
Cardiovascular
Heart involvement was not studied with specific tests, however, histopathological study in the autopsy specimens from one of our patients showed fibrotic changes in the myocardium as well as perivascular fibrosis in small coronary arterioles. Similar perivascular fibrosis in the myocardium was observed in the autopsy specimens of the case described by Ting et al (4). It is of interest that in the series of Swartz et al. (9), four patients had evidence of cardiomyopathy, however, it was considered to be an unrelated co-morbidity. In our cohort, two patients developed pulmonary arterial hypertension as detected by echocardiography. In one case the estimated pulmonary artery pressure was 60 mm Hg whereas in the other it was 80 mm Hg. None of these patients had symptoms or signs of right heart failure. No cases of pulmonary hypertension were reported in any of the published series reviewed.
Pulmonary
Six of seven of our patients in whom CO diffusion capacity was examined had a reduction of CO diffusion capacity on pulmonary function tests. The review of published cases in the literature did not disclose any additional patients in whom pulmonary function tests were reported. Radiographic interstitial lung disease was present in one case in the series of Swartz et al. (9), however, in this report the radiographic findings were considered to represent a co-morbid condition without direct association with the disease.
Other systemic events
Four patients in our series had thrombotic events including repeated occlusion of the dialysis graft, peripheral vascular occlusion, transient ischemic attacks and multiple brain infarcts. Similar thrombotic events were described in several other patients in the literature (9, 10, 25–27).
Laboratory Findings
The results of relevant laboratory test in our patients are summarized in Table 3. There are no specific laboratory findings associated with this disorder. However, very high erythrocyte sedimentation rates and/or C-reactive protein levels were observed in nine patients in our series. Elevation of the erythrocyte sedimentation rate was also common in the literature review with 16/20 reported patients with abnormal values (5, 9, 23). Creatine kinase (CK) has been normal in all cases in whom the test was performed (8/8) (5, 9). However, it is interesting to notice than CK values below the normal reference range were observed in 88% of these cases (7/8). Paraproteinemia was not found in any of the cases in which it was examined (0/57), constituting one of the most important differences with scleromyxedema (2, 9). Antinuclear antibodies (ANA) examined by immunofluorescence were present in four patients of our series at low titers (less than 1/360) showing a homogenous pattern. A nucleolar pattern was also observed in the serum of one patient. Anticentromere and Scl70 antibodies were negative in the patients in whom the test was performed. ANA tests were negative in the majority of the published cases, however, 3 series described positive ANA (homogeneous or nucleolar pattern), generally in low titers (5, 12).
Table 3.
Laboratory studies.
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | Case 11 | Case 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CPK/Aldolase* | (N.D.) | 5/4.7 | N.D. | 14/3.4 | 2/N.D. | 22/9.0 | 34/9 | <20/6.8 | N.D. | N.D./3.1 | 2.9/N.D. | N.D |
| Eosinophilia | No | No | No | No | No | No | No | No | No | No | No | No |
| Paraproteinemia | No | No | No | No | No | No | No | No | No | No | No | No |
| ANA titer/pattern | 1:160 Homo-geneous | Neg | 1:320 Homo-geneous | Neg | Neg | 1:80 Homo-geneous | 1:160 Homo-geneous
1:160 Nucleolar |
Neg | Neg | Neg | Neg | Neg |
| Anticentromere/Scl-70 | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg | Neg/Neg |
| Sed Rate/CRP ** | 58/11.3 | 22/1.3 | 110/N.D. | 73/N.D. | 68/N.D. | 20/5 | 67/10.4 | 109/3.5 | 40/4.6 | 33/<0.7 | 65/0.93 | 118/3.7 |
| Antiphospholipid Antibody (level in ug/ml) | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | IgG(24) | IgG(13)
IgM(54) |
N.D. |
CPK: Creatine phosphokinase. Normal values are 25–215 IU/L. Aldolase normal values and 2.0 –7.0 U/L.
Sed Rate: Erythrocyte Sedimentation Rate (Westergreen Method). Normal values <30 mm/hr. CRP: C reactive protein. Normal values <0.8 μg/ml.
ND Not done
Histopathology
Sections from the skin biopsies in our patients demonstrated marked thickening of the dermis with accumulation of thick collagen bundles separated by large clefts in the papillary and deep dermis (Figure 3), and thick fibrous tissue tracts extending across the adipose tissue into the fascia which was severely thickened. The adipose tissue was encroached within the fibrous tracts yielding a microlobular architecture (Figure 3). Markedly thickened and tortuous collagen bundles were present in the affected tissues (Figure 4). The alcian blue or colloidal iron was positive in the dermis and sub-dermal tissues confirming mucin accumulation in the clefts between the collagen bundles. Numerous spindle-shaped and elongated fibroblasts were present throughout the dermis and fascia. Our observations are similar to those described in the extensive and detailed histopathologic study by Cowper et al. (2). These authors also first noted the presence of abundant CD34+ spindle cells immersed in a network of elastic fibers and collagen, and of either CD68+ or XIIIa+ mono- or multi-nucleated cells in skin biopsies from these patients (2). CD34+ spindle fibroblasts and cells staining with both CD68 and factor XIIIa labeled antibodies were present in the samples we examined (Figure 5). These cells are most likely dendritic cells co-expressing both CD68 and factor XIIIa epitopes. CD68+ or factor XIIIa+ cells are a constant finding in early disease (2, 5, 6,12,16, 17, 20). In samples taken later after clinical onset (more than 20 weeks) there is a decrease in CD34+/CD68+ cells and in mucin accumulation (2). CD45RO+/CD34+ cells have been also described in another study of early NFD skin lesions (12). Other inflammatory cell populations such as CD3+ or CD4+ lymphocytes, and smooth muscle actin positive fibroblasts (myofibroblasts), have been described in different series (6, 14, 16). Calcium deposits may be present, especially in the setting of hyperparathyroidism (4), however, calcium deposits have been observed in patients without secondary hyperparathyroidism.
Figure 3. Histopathology of skin and subcutaneous tissues.

(A) Hematoxylineosin and (B) trichrome stains. Note marked thickening of the entire dermis and subcutaneous tissues with severe accumulation of thick collagen bundles separated by numerous clefts. (C) Hematoxylineosin showing thick tracts of fibrous tissue extending into the adipose layer resulting in microlobular architecture and in thickening of the fascia. Magnification 60X.
Figure 4. Morphology of dermal and subdermal collagen fibers.
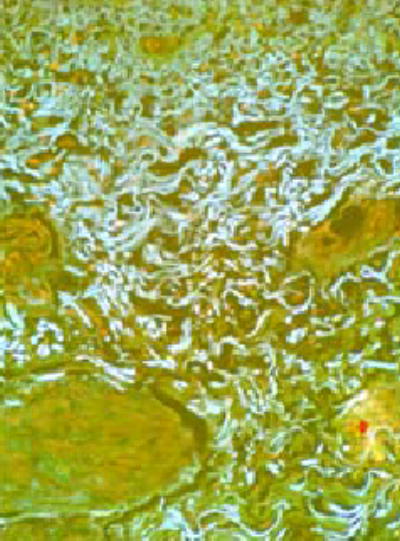
Thick and tortuous collagen bundles in the deep dermis and subcutaneous tissue of affected skin viewed under fluorescence microscopy with a triple-band pass filter. Magnification 400X.
Figure 5. Co-expression of CD68 and FXIIIa in infiltrating cells in skin.
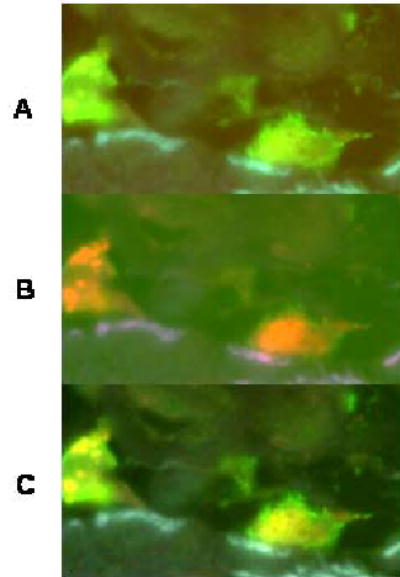
Cells were dual-stained for CD68 (red) and FXIIIa (green). Both antibodies were diluted 1:100 and simultaneously applied to the section for 40 min. Unbound antibodies were removed with 3 washes of 2 min each of PBS. The secondary antibodies; rabbit-anti-mouse Cy3 (1:100) and donkey-anti-goat-FITC (1:100) were applied to the section for 40 min and the unbound proteins washed off. Sections were counterstained with DapI and viewed with an epi-fluorescence microscope. Panel A shows two cells that are positive for FXIIIa. Panel B: the same cells are viewed through the red filter to detect CD68. Panel C: the cells are viewed through the triple bandpass filter to detect the co-expression of FXIIIa and CD68. The cells appear yellow when there is co-localization of the two antibodies. Magnitication 400X.
Histopathology of the muscles in the patients we studied (5, 6) showed pronounced infiltration of the perimysium and endomysium with fibrotic tissue and muscle fiber atrophy (Figure 6). There was no evidence of polymorphonuclear leukocyte or lymphocyte infiltration in the affected muscles although numerous CD68+ cells, most likely macrophages, have been shown within the fibrous tracts of the diaphragm and other affected muscles (4–6). The myocardium was also affected by a similar fibrotic process (Figure 7). The lungs showed patchy interstitial fibrosis with inflammatory cell infiltration (Figure 8). A pronounced increase in the thickness of the adventitial layer of small and medium size lung arterioles with marked accumulation of fibrous tissue was also observed in the autopsy specimen of one of our patients.
Figure 6. Histopathology of muscle.

Trichrome stain. Note remarkable accumulation of fibrotic tissue in the interfascicular septae and surrounding muscle fibers. Magnification 200X.
Figure 7. Histopathology of myocardium.

Trichrome stain. Note thickening of pericardium and accumulation of large amounts of fibrous tissue surrounding and between myocardial fibers. Magnification 200X.
Figure 8. Histopathology of lung tissue.

Trichrome stain. (A) Lung parenchyma showing interstitial fibrosis with severe distortion of alveolar architecture and moderate inflammatory cell infiltrate. (B) Small pulmonary arterioles showing marked thickening and fibrosis of adventitial layer. Interstitial fibrosis is also observed. Magnification 100X.
Etiology and Epidemiology
The etiology of NFD is not known, however, there is a strong and constant epidemiological association with renal failure. In the published literature, the vast majority of patients had end-stage renal disease of various etiologies and received hemodialysis or peritoneal dialysis. A large number of cases had failed renal transplantation (Table 4).
Table 4.
Epidemiological, clinical and laboratory abnormalities in 70 patients described in the literature.
| Number of patients | Total | % of Patients | |
|---|---|---|---|
| Renal disease | |||
| Unspecified GN | 11 | 62 | 18 |
| Diabetic nephropathy | 10 | 62 | 16 |
| HTN nephropathy | 6 | 62 | 10 |
| MPGN | 4 | 62 | 6 |
| Atherosclerosis | 4 | 62 | 6 |
| Polycystic disease | 4 | 62 | 6 |
| Interstitial nephritis | 3 | 62 | 5 |
| Unknown etiology | 3 | 62 | 5 |
| Others | 17 | 62 | 27 |
| Type of Dialysis | |||
| Hemodialysis | 61 | 70 | 87 |
| Peritoneal Dialysis | 6 | 70 | 9 |
| Never dialyzed | 3 | 70 | 4 |
| Renal transplant | 30 | 67 | 44 |
| Extent of Skin Sclerosis | |||
| Lower extremity | 55 | 57 | 96 |
| Upper extremitiy | 44 | 57 | 77 |
| Trunk and abdomen | 17 | 57 | 30 |
| Laboratory | |||
| Paraproteinemia* | 3 | 57 | 5 |
| ANA | 7 | 23 | 30 |
| Increased ESR** | 13 | 20 | 65 |
| Increased CK*** | 0 | 8 | 0 |
| Decreased CK | 7 | 8 | 88 |
2 patients with polyclonal and 1 patient with IgA paraproteinemia
Normal values for ERS; Thomas Jefferson University Hospital: <30 mm/h; University of Michigan Health System: <20 mm/h; Loma Linda University Medical Center (males): <9 mm/h (<50yo)/<20mm/h (50–55yo)
Normal values for CK; Thomas Jefferson University Hospital: 25–215 IU/l; University of Michigan Health System: 39–275 IU/l
HTN: Hypertensive nephropathy
MPGN: Membrano-proliferative glomerulonephritis
GN: Glomerulonephritis
ANA: Antinuclear antibodies (fluorescence)
ESR: Erythrocyte Sedimentation Rate (Westergreen)
CK: Creatine Kinase
The disorder affects male and female patients in approximately equal numbers showing a 1:1 relationship (1, 2, 4–6, 8, 9, 11–26). The mean age of diagnosis was 49.8 years with a range between 8 and 81. Pediatric cases with the same clinical presentation have also been published (22, 27).
The sudden emergency of small clusters of cases after 1997 and its resemblance to other toxin-related fibrotic disorders suggested the possible involvement of a toxic or infectious agent in a susceptible host. However, various putative agents such as the dialysis fluid, the methods of membrane cleaning, and the type of dialysis equipment used have not shown a consistent epidemiological relation. There is no predominance of any specific renal pathology and the cause of the renal failure is heterogeneous (Table 4). Renal transplantation was present in 44% (30/67) of the published cases (1, 2, 4–6, 8, 9, 11–26). However, as the majority of cases did not have a renal transplant it is not likely that there is an etiological relationship.
Many reports have described associated extra-renal diseases. For example, in the series of Mackay-Wiggan et al. (25) a strong association with antiphospholipid antibodies, which were present in higher proportion than in end-stage renal disease patients, was found. Liver insufficiency and hepatitis B and C have been reported in few cases (8, 9, 23), and Cowper et al. emphasized the relationship between the onset of symptoms and an immediately preceding surgical procedure (90%). However, in this latter review, the majority of the surgical procedures were dialysis catheter placement or renal transplantation (10). Therapy with calcineurin inhibitors has been implicated in the initiation of tissue fibrosis owing to their ability to induce increased levels of the profibrotic cytokine, TGFβ; however, in the published cases, there is no consistent association between any particular therapy for the renal disease and the development of this disorder (11).
Clinical Course Treatment and Prognosis
The primary lesion appears in the lower extremities in the majority of the patients followed by upper extremities and trunk involvement (1,2,5,9,10). In many cases, puffiness and swelling of the affected extremities has been described before the appearance of the characteristic lesions. When this edema is present, it gradually resolves leaving erythematous indurated plaques which progress to more extreme brawny indurations and thickening of the affected skin, “peau d’orange” appearance and deep furrowing. Progressively, this induration involves the periarticular tissues, causing limitations of motion and flexion contractures (5, 6, 9, 27). The chronology of the visceral involvement is not clear, but is more common in patients with extensive cutaneous involvement (5, 6).
The severity and rapidity of progression of the cutaneous lesions correlate with poor prognosis and death. In six of our patients a progressive course was evident leading to the demise of three and to wheelchair confinement in three cases.
A review of published reports indicates that there is no proven effective therapy, and the prognosis depends on the extent, severity, and rapidity of cutaneous involvement and in the severity of the systemic involvement. In the published literature, it was found that 28% of the patients died, 28% had no improvement, and only 20% had modest improvement. Only a small percentage of patients improved following recovery of renal function and discontinuation of dialysis (2, 4, 8–10, 14, 15, 22–24).
Plasmapheresis has been the most extensively used treatment; however, the results reported are inconsistent. It is important to note that the patients who had mild to marked improvement were patients who discontinued dialysis because of improvement of their renal function (11, 23, 25). However, complete remission following cessation of dialysis has been observed in less than 40% of the patients (2, 9, 23, 25).
In a few patients, local administration of interferon or treatment with IV IgG have proven to be partially effective but in the majority of the cases with only a transient effect (11, 17). Photophoresis has not shown effectiveness in some of the cases in which was employed (5, 15, 24). Other treatments such as corticosteroids (local and systemic) cyclosporine, and methotrexate have failed to show improvement (5, 11, 22, 24). In our patients we employed a combination of high dose oral corticosteroids and D-penicillamine. Although in some patients there was an improvement in the cutaneous fibrotic process our experience is too limited to allow reaching a conclusion about the therapeutic effects of this regime.
DISCUSSION
NFD is a severe, usually progressive, fibrotic disorder occurring in patients with end-stage renal disease who received, with few exceptions, hemodialysis or peritoneal dialysis. In addition to the severe cutaneous alterations, frequently there is evidence of systemic involvement including: 1) a very marked elevation of the erythrocyte sedimentation rate and C reactive protein, 2) tendinous, peri-articular and striated muscle involvement, and 3) visceral fibrosis of heart, lungs and other organs. Therefore, the presence and extent of systemic involvement should be searched aggressively as it may have a great impact on the course and prognosis of the disease. We previously suggested that a more appropriate denomination of this disorder was “dialysis-associated systemic fibrosis” (5). This suggestion was based on our belief that the term NFD is a misnomer for two reasons. The first reason is that the term NFD implies that kidney failure is the cause of the disease. If chronic renal disease is the only cause of this disorder, it is difficult to explain its rarity and the absence of descriptions prior to 1997 given the extremely large number of patients with renal failure worldwide. The second reason is that the disorder is not only a cutaneous disease as implied in the word dermopathy as several recent reports (4–6) and our data presented here have shown that the fibrotic process is a systemic one affecting muscles, myocardium, lungs, kidneys, testis and very likely other organs as well. However, we have here maintained the NFD nomenclature to avoid confusion until the etiology and pathogenesis of the disease are elucidated.
The sequence of physiopathological events of this severe fibrotic disorder remains unclear and very few studies have attempted to examine the mechanisms involved (5, 12, 13). In our study (5) we found by immunohistochemistry an early dermal infiltration with CD 68+/factor XIIIa+ dendritic cells (Figure 5) that could represent a possible response of the host to noxious stimuli. Furthermore, in situ hybridization studies and immunohistochemistry showed a marked increase in the expression of TGFβ1 mRNA, diffusely distributed in the skin, fascia and affected muscles (Figure 9). Thus, these observations allowed us to postulate the hypothesis that CD68+/Factor XIIIa+ dendritic cells and TGFβ are intimately involved. Dendritic cells are important cellular elements of the immune system participating in numerous immune functions including antigen presentation. Although there is no direct evidence that dendritic cells in the tissue are responsible for the increased expression of the growth factor, this is a likely possibility as expression of TGFβ in dendritic cells has been previously documented (28). On the other hand, recent evidence has shown that TGFβ is involved in the regulation of the complex process of dendritic cell maturation and that it is an essential requirement for the appearance of dendritic cells in the epidermis (29). Thus, it is possible that the causative agent(s) resulted in increased expression of the growth factor as part of the response of the dendritic cells to this noxious agent. The TGFβ produced by these dendritic cells in turn would be responsible for both the fibrotic process and the expansion and enhancement or initiation of antigen presenting functions of further dendritic cells establishing a vicious circle which results in their accumulation in affected tissues and in the severe fibrotic process. This hypothesis is schematically shown in Figure 10.
Figure 9. Expression of TGFβ in NFD skin.

Fixed skin slides were stained with a fluorescein-labeled TGFβ specific antibody (green). The TGFβ antibody was diluted 1:100 and applied to the section for 40 min. The unbound antibody was removed with 3 washes of 2 min each of PBS. The secondary antibody was donkey-anti-goat-FITC (1:100) and it was applied to the section for 40 min and the unbound proteins washed off. Sections were counterstained with DapI and viewed with an epi-fluorescence microscope. Note the abundant expression of TGFβ epitopes throughout the dermis in dermal fibroblasts and infiltrating inflammatory cells.
Figure 10.
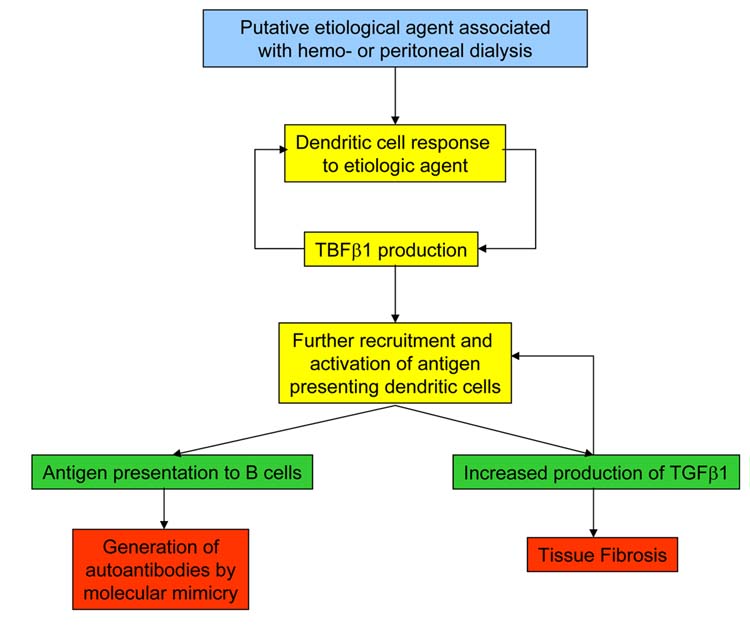
Schematic diagram of NFD pathogenesis through the involvement of dendritic cells and TGFβ.
A second hypothesis proposed for the pathogenesis of this disorder considers the possible early dermal infiltration with bone marrow-derived CD45RO+/CD34+ circulating fibrocytes (12), a novel leukocytic subpopulation that plays a role in the proliferative phase of the normal wound repair (30). This hypothesis is schematically shown in Figure 11.
Figure 11.

Schematic diagram of NFD pathogenesis through the involvement of circulating fibrocytes.
Since the first recognized cases of NFD in 1997, very little progress has been accomplished regarding its etiology and pathogenesis, therefore, this challenging and serious disease with newly recognized systemic involvement requires intensive investigation. Further research on the pathogenesis of this disease should focus, among other topics, on the role of cutaneous dendritic cells or of circulating fibrocytes and on the cellular pathways involved in this particular form of tissue fibrosis.
On the other hand, epidemiological surveillance should be the pillar for the recognition of the putative etiologic agent. Studies of genetic predisposition should be also part of this effort. Therapeutic approaches will remain empirical until the pathophysiology of this severe disorder becomes better understood.
Acknowledgments
Supported by NIH grant AR19161 (to SAJ). Dr. Nora Sandorfi was supported by NIH training grant AR07583. Dr. Sonsoles Piera-Velazquez was supported by a Postdoctoral Fellowship from the Arthritis Foundation. The expert assistance of Kate Salmon in the preparation of this paper is gratefully acknowledged.
Footnotes
Supported by NIH Grant AR19616.
References
- 1.Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous disease in renal-dialysis patients. Lancet. 2000;356:1000–01. doi: 10.1016/S0140-6736(00)02694-5. [DOI] [PubMed] [Google Scholar]
- 2.Cowper SE, Su LD, Bhawan J, Robin HS, LeBoit PE. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23:383–93. doi: 10.1097/00000372-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 3.http://www.icnfdr.org/ Accessed June 9, 2005.
- 4.Ting WW, Stone MS, Madison KC, Kurtz K. Nephrogenic fibrosing dermopathy with systemic involvement. Arch Dermatol. 2003;139:903–06. doi: 10.1001/archderm.139.7.903. [DOI] [PubMed] [Google Scholar]
- 5.Jimenez SA, Arlett CM, Sandorfi N, Derk C, Latinis K, Sawaya H, et al. Dialysis-Associated Systemic Fibrosis (Nephrogenic Fibrosing Dermopathy) Arthritis Rheum. 2004;50:2660–66. doi: 10.1002/art.20362. [DOI] [PubMed] [Google Scholar]
- 6.Levine JM, Taylor RA, Elman LB, Bird SJ, Lavi E, Stolzenberg ED, et al. Involvement of skeletal muscle in dialysis-associated fibrosis (Nephrogenic Fibrosing Dermopathy) Muscle & Nerve. 2004;30:569–77. doi: 10.1002/mus.20153. [DOI] [PubMed] [Google Scholar]
- 7.Hershko K, Hull C, Ettefagh L, Nedorost S, Dyson S, Horn T, et al. A variant of nephrogenic fibrosing dermopathy with osteoclast-like giant cells: a syndrome of dysregulated matrix remodeling? J Cutan Pathol. 2004;31:262–65. doi: 10.1111/j.0303-6987.2004.00177.x. [DOI] [PubMed] [Google Scholar]
- 8.Chiu H, Wells G, Carag H, Canova E, Firpi RJ. Nephrogenic fibrosing dermopathy: a rare entity in patients awaiting liver transplantation. Liver Transpl. 2004;10:465–66. doi: 10.1002/lt.20068. [DOI] [PubMed] [Google Scholar]
- 9.Swartz RD, Crofford LJ, Phan SH, Ike RW, Su LD. Nephrogenic Fibrosing Dermopathy: A novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563–72. doi: 10.1016/s0002-9343(03)00085-8. [DOI] [PubMed] [Google Scholar]
- 10.Cowper SE. Nephrogenic fibrosing dermopathy: the first 6 years. Curr Opin Rheumatol. 2003;15:785–90. doi: 10.1097/00002281-200311000-00017. [DOI] [PubMed] [Google Scholar]
- 11.Hubbard V, Davenport A, Jarmulowicz M, Rustin M. Scleromyxoedema-like changes in four renal dialysis patients. Br J Dermatol. 2003;148:563–68. doi: 10.1046/j.1365-2133.2003.05181.x. [DOI] [PubMed] [Google Scholar]
- 12.Ortonne N, Lipsker D, Chantrel F, Boehm N, Grosshans E, Cribier B. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J Dermatol. 2004;150:1050–52. doi: 10.1111/j.1365-2133.2004.05900.x. [DOI] [PubMed] [Google Scholar]
- 13.McNeil AM, Barr R. Scleromyxedema-like fibromucinosis in a patient undergoing hemodialysis. Int J Dermatol. 2002;41:364–67. [PubMed] [Google Scholar]
- 14.Hauser C, Kaya G, Chizzolini C. Nephrogenic fibrosing dermopathy in a renal transplant recipient with tubulointerstitial nephritis and uveitis. Dermatology. 2004;209:50–52. doi: 10.1159/000078587. [DOI] [PubMed] [Google Scholar]
- 15.Lauchli S, Zortea-Caflisch C, Nestle FO, Burg G, Kempf W. Nephrogenic fibrosing dermopathy treated with extracorporeal photopheresis. Dermatology. 2004;208:278–80. doi: 10.1159/000077321. [DOI] [PubMed] [Google Scholar]
- 16.Gambichler T, Paech V, Kreuter A, Wilmert M, Altmeyer P, Stucker M. Nephrogenic fibrosing dermopathy. Clin Exp Dermatol. 2004;29:258–60. doi: 10.1111/j.0307-6938.2004.01474.x. [DOI] [PubMed] [Google Scholar]
- 17.Chung HJ, Chung KY. Nephrogenic fibrosing dermopathy: response to high-dose intravenous immunoglobulin. Br J Dermatol. 2004;150:596–97. doi: 10.1111/j.1365-2133.2003.05795.x. [DOI] [PubMed] [Google Scholar]
- 18.Edsall LC, English JC, 3rd, Patterson JW. Calciphylaxis and metastatic calcification associated with nephrogenic fibrosing dermopathy. J Cutan Pathol. 2004;31:247–53. doi: 10.1111/j.0303-6987.2004.00169.x. [DOI] [PubMed] [Google Scholar]
- 19.Dawn G, Holmes SC. Scleromyxoedema-like eruption following haemodialysis or nephrogenic fibrosing dermopathy? Br J Dermatol. 2004;150:167–68. doi: 10.1111/j.1365-2133.2004.05693.x. [DOI] [PubMed] [Google Scholar]
- 20.Evenepoel P, Zeegers M, Segaert S, Claes K, Kuypers D, Maes B, et al. Nephrogenic fibrosing dermopathy: a novel, disabling disorder in patients with renal failure. Nephrol Dial Transplant. 2004;19:469–73. doi: 10.1093/ndt/gfg506. [DOI] [PubMed] [Google Scholar]
- 21.Engelen JW, Kooistra MP, Canninga-van Dijk MR, Toonstra J, Sigurdsson V. [Nephrogenic fibrosing dermopathy] Ned Tijdschr Geneeskd. 2003;147:2435–38. (Dutch). [PubMed] [Google Scholar]
- 22.Jan F, Segal JM, Dyer J, LeBoit P, Siegfried E, Frieden IJ. Nephrogenic fibrosing dermopathy: two pediatric cases. J Pediatr. 2003;143:678–81. doi: 10.1067/S0022-3476(03)00538-9. [DOI] [PubMed] [Google Scholar]
- 23.Baron PW, Cantos K, Hillebrand DJ, Hu KQ, Ojogho ON, Nehlsen-Cannarella S, et al. Nephrogenic fibrosing dermopathy after liver transplantation successfully treated with plasmapheresis. Am J Dermatopathol. 2003;25:204–09. doi: 10.1097/00000372-200306000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Perazella MA, Ishibe S, Perazella MA, Reilly RF. Nephrogenic fibrosing dermopathy: an unusual skin condition associated with kidney disease. Semin Dial. 2003;16:276–80. doi: 10.1046/j.1525-139x.2003.16053.x. [DOI] [PubMed] [Google Scholar]
- 25.Mackay-Wiggan JM, Cohen DJ, Hardy MA, Knobler EH, Grossman ME. Nephrogenic fibrosing dermopathy (scleromyxedema-like illness of renal disease) J Am Acad Dermatol. 2003;48:55–60. doi: 10.1067/mjd.2003.78. [DOI] [PubMed] [Google Scholar]
- 26.Streams BN, Liu V, Liegeois N, Moschella SM. Clinical and pathologic features of nephrogenic fibrosing dermopathy: a report of two cases. J Am Acad Dermatol. 2003;48:42–47. doi: 10.1067/mjd.2003.77. [DOI] [PubMed] [Google Scholar]
- 27.Jain SM, Wesson S, Hassanein A, Canova E, Hoy M, Fennell RS, et al. Nephrogenic fibrosing dermopathy in pediatric patients. Pediatr Nephrol. 2004;19:467–70. doi: 10.1007/s00467-003-1380-1. [DOI] [PubMed] [Google Scholar]
- 28.Strobl H, Knapp W. TGFβ regulation of dendritic cells. Microbes Infect. 1999;1:1283–90. doi: 10.1016/s1286-4579(99)00256-7. [DOI] [PubMed] [Google Scholar]
- 29.Gruschwitz MS, Hornstein OP. Expression of transforming growth factor type β on human epidermal dendritic cells. J Invest Dermatol. 1992;99:114–16. doi: 10.1111/1523-1747.ep12611890. [DOI] [PubMed] [Google Scholar]
- 30.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]