

Abstract
The periodontal pathogen Actinobacillus actinomycetemcomitans expresses a cytolethal distending toxin (CDT) that typically arrests the growth of eukaryotic cells at either the G0/G1 or G2/M phase of the cell cycle. It was previously found that CDT failed to arrest the growth of human periodontal ligament fibroblasts (HPLFs) when grown in pure culture. In contrast, proliferation of an oral epithelial cell line was rapidly inhibited by the toxin. In this study, the feasibility of using mixed-cell cultures and cell-specific markers to evaluate the response of oral cells, when in heterogeneous populations, to CDT was established. Proliferation of epithelial cells was rapidly inhibited and the cells were selectively eliminated in co-culture with HPLFs or cementoblasts by 24–48 h post-intoxication. Epithelial cells and HPLFs were detected and counted in co-cultures following cell-specific immunolabelling with antibodies against simian virus 40 large T antigen and the Ab-1 surface antigen, respectively. These results demonstrated that the activities of potential virulence factors, such as CDT, from periodontal pathogens can be successfully examined in mixed-cell cultures. This approach is especially relevant to infectious diseases that affect tissues with a diverse cellular composition, such as the periodontium.
INTRODUCTION
Cytolethal distending toxin (CDT) of Actinobacillus actinomycetemcomitans is a secreted protein toxin that inhibits the proliferation of a wide variety of cell types and cell lines. The holotoxin from this bacterium is assembled from the products of three genes (cdtA, cdtB and cdtC) that have homologues in at least five other pathogenic Gram-negative bacterial genera (Sugai et al., 1998; Mayer et al., 1999; Mao & DiRienzo, 2002). Eukaryotic cells that are sensitive to the toxin are usually arrested at the G0/G1 or G2/M phase of the growth cycle (Comayras et al., 1997; Whitehouse et al., 1998; Cortes-Bratti et al., 1999; Shenker et al., 1999). CDT triggers the block in cell-cycle progression through the action of a DNase I-like nuclease (CdtB) that causes double-strand breaks in the host-cell DNA (Elwell & Dreyfus, 2000; Lara-Tejero & Galán, 2000; Cortes-Bratti et al., 2001; Frisan et al., 2003; Hassane et al., 2003).
A. actinomycetemcomitans is a facultative, Gram-negative bacterium that has long been associated with localized aggressive periodontitis. A. actinomycetemcomitans is one of the few periodontal pathogens capable of active tissue invasion (Blix et al., 1992; Meyer et al., 1996). The bacterium expresses a number of gene products that can be defined as virulence factors based on their in vitro biological activities or similarities to products produced by other pathogens (Henderson et al., 2003). Although CDT is not unique to A. actinomycetemcomitans, this bacterium is the only member of the oral microbial flora identified to date that carries and expresses the toxin locus (Yamano et al., 2003). CDT is prevalent in A. actinomycetemcomitans strains. Ahmed et al. (2001) found that 43 of 50 strains from periodontitis patients contained all three cdt genes and expressed CDT activity. In another study, PCR of subgingival plaque samples revealed that 13 of 106 diseased sites in 146 patients with aggressive and chronic periodontitis contained A. actinomycetemcomitans expressing all three cdt genes (Tan et al., 2002). Fabris et al. (2002) reported that 39 of 40 A. actinomycetemcomitans isolates from a mix of healthy and periodontal diseased subjects expressed activity that caused the distension of CHO cells.
We were interested in characterizing the biological effects of A. actinomycetemcomitans CDT on cells of the human periodontium. If this toxin has a primary role in periodontal disease, it most likely occurs through interactions with the various cellular components of the periodontium including fibroblasts, epithelial cells and cementoblasts. In conjunction with other resident cell types, these cells are responsible for maintaining the structural integrity of the periodontium, as well as facilitating the repair and/or regeneration of these tissues following injury (Lekic et al., 1997). We recently showed that fibroblasts derived from human periodontal ligament (HPLFs) did not display growth inhibition, cell-cycle arrest or dsDNA damage typically associated with CDT (Kanno et al., 2005). In contrast, a human oral epithelial cell line, GMSM-K, was exquisitely sensitive to the toxin. The differential responses of these various cell types to CDT has important implications for the initial colonization of the periodontium by A. actinomycetemcomitans, as well as the health and remodelling of periodontal tissues. The ability of CDT to inhibit the growth of oral epithelial cells may lead to disruption of the protective barrier formed by these cells, facilitating invasion and perturbation of the underlying connective tissue.
In this study, we examined the ability of CDT specifically to inhibit the proliferation of oral epithelial cells when grown in co-culture with HPLFs and cementoblasts. We have shown that cell-specific markers can be used to identify and quantify distinct cell types in CDT-treated co-cultures. The potential application of this novel approach to the characterization of virulence factors of periodontal pathogens is discussed.
METHODS
Cells and tissue-culture conditions. An immortalized human oral epithelial cell line, GMSM-K, was obtained from Valerie Murrah of the Department of Diagnostic Sciences and General Dentistry, University of North Carolina at Chapel Hill, USA. This cell line was used because attempts to isolate and maintain pure primary cultures of human gingival epithelial cells were inconsistent. Cells were routinely grown in T-75 flasks in keratinocyte serum-free medium (Invitrogen) as reported previously (Kanno et al., 2005; Gilchrist et al., 2000). Cells were passaged in modified Eagle's medium (Medium 199; Gibco), containing 10 % fetal calf serum (FCS), antibiotics (10 000 U penicillin G ml−1, 25 μg streptomycin ml−1) and an antimycotic (0·85 % fungizone). Cells were passaged in Medium 199 twice prior to use in co-culture experiments. Cultures were examined by light microscopy to confirm growth based on an increase in cell number. The growth medium was changed every 3 days until the cells were approximately 80 % confluent. At that time, cells were detached from the plates by treatment with trypsin (1 mg ml−1 in PBS) for 10 min followed by treatment with soybean trypsin inhibitor (2 mg ml−1 in PBS; Sigma). Detached cells were collected by centrifugation, washed twice with Ca2+/Mg2+-free PBS and suspended in 10 ml fresh medium for use in co-culture experiments.
HPLFs were derived from periodontal ligament explants using a technique similar to that described by Somerman et al. (1988). Clinically healthy premolars and third molars were obtained from five patients (three males and two females with ages ranging from 23 to 28 years) who presented to the Oral Surgery Clinic of the University of Pennsylvania School of Dental Medicine. Immediately following extraction, the teeth were placed into Medium 199 supplemented with penicillin G (10 000 U ml−1), streptomycin (25 μg ml−1) and fungizone (0·85 %). Within 1 h of extraction, the teeth were washed twice with fresh medium and the attached gingival tissue removed. The teeth were then placed in a Petri dish containing medium and the periodontal ligament dissected away from the mid-third of the root surface by gentle scraping with a disposable surgical scalpel.
The collected tissue was centrifuged at 1000 r.p.m. for 10 min at 4 °C. The tissue was suspended in 5 ml growth medium supplemented with 10 % FCS and incubated at 37 °C in 5 % CO2. Monolayers of HPLFs were typically observed 2–3 weeks after the initial seeding of cultures. Upon reaching confluence, the primary cultures were treated with trypsin (2·5 mg ml−1 in PBS) to detach the cells for passage. Cells were stored in 2 ml 10 % (v/v) DMSO in Medium 199 in liquid nitrogen. For growth experiments, the medium was changed every 3 days until cells were approximately 80 % confluent. Cells were detached from the plates as described for the epithelial cells except that 2·5 mg trypsin ml−1 was used. The detached cells were then placed immediately into 5 ml fresh growth medium containing FCS for use in the co-culture experiments.
Primary human cementoblasts (C11-26M) were maintained in alpha minimal essential medium supplemented with 10 % FCS and antibiotics as described previously (Grzesik et al., 1998, 2000). Cementoblasts were grown in T-75 flasks in Medium 199 and an increase in cell number was confirmed using light microscopy. The growth medium was changed every 3 days until the cells were approximately 80 % confluent. At that time, cells were detached from the plates as described for HPLFs. Detached cells were collected by centrifugation and suspended in 10 ml fresh medium for use in the co-culture experiments.
All protocols for obtaining and handling human oral tissues were reviewed and approved by the Institutional Review Board of the University of Pennsylvania.
Bacterial strains, growth conditions and preparation of CDT. A recombinant clone containing the A. actinomycetemcomitans cdt locus, E. coli BL21(DE3)(pET15bcdt), has been constructed and characterized previously (Mao & DiRienzo, 2002). For preparation of CDT-containing extracts, the recombinant clone was grown in Luria–Bertani medium containing 100 μg ampicillin ml−1 at 37 °C with vigorous shaking. Late exponential-phase cultures (OD600 0·8–1·0) were treated with IPTG (Fisher Scientific) at a final concentration of 1 mM and the cultures were incubated for an additional 4–5 h. Bacteria were harvested by centrifugation, washed twice with PBS and suspended in PBS. Bacteria were disrupted by sonication in an ice bath using three 30 s pulses each separated by a 30 s rest (Braun-Sonic 2000; B. Braun Biotech). The preparations were centrifuged at 12 000 g for 10 min (Spinco model SS-34 rotor) to remove unbroken cells and sterilized by passage through a 0·45 μm pore size filter (Millipore). Total protein was determined with the Micro BCA Protein Assay kit (Pierce Biotechnology). The toxic dose (TD)50 concentration (at which 50 % of the cell population is killed) was determined as described previously (Mayer et al., 1999). Expression of cdt genes was verified by analysis of total cell protein using 10–20 % polyacrylamide Tris/HCl Ready Gels (Bio-Rad Laboratories) as described previously (Kanno et al., 2005).
Light microscopy. For mixed cultures of epithelial cells and either HPLFs or cementoblasts, cells were detached from plates and suspended in fresh Medium 199 supplemented with 10 % FCS. Cell suspensions were adjusted so that 6 × 104 GMSM-K cells were added to each of two T-25 flasks. An equivalent number of HPLFs or cementoblasts was added to each of the epithelial-cell cultures. Both flasks of each set of co-cultures (GMSM-K/HPLFs and GMSM-K/cementoblasts) were incubated for 24 h at 37 °C in an atmosphere containing 5 % CO2. After 24 h, a TD50 equivalent of filter-sterilized CDT-containing extract [4·5 μg total protein (ml medium)−1] from E. coli BL21(DE3)(pET15bcdt) was added to one culture flask of each set. The second flask of each set did not receive toxin and served as a control. The cultures were incubated for up to 72 h post-intoxication. Digital photographs of both the control and the CDT-treated cultures were taken at 0, 24, 48 and 72 h post-intoxication using a Nikon TMS-F inverted microscope and a Fuji Finepix S-2 camera.
Cell-cycle analysis. Cell-cycle arrest was determined by flow cytometry as described previously (Kanno et al., 2005). Cementoblast (C11-26M) cultures (1 × 106 cells) were untreated or treated with total soluble protein from E. coli BL21(DE3)(pET15bcdt) at a concentration of 18·4 μg (ml medium)−1. Cultures were incubated for up to 72 h post-intoxication. Nuclei from washed cells were isolated and stained with propidium iodide (Sigma Chemicals) by a standard procedure (Vindelov, 1977) as described previously (Kanno et al., 2005). Stained nuclei were analysed on a FACSCalibur flow cytometer at the University of Pennsylvania Cancer Center Flow Cytometry and Cell Sorting Shared Resource Facility. Data from 30 000 events were analysed using ModFit 3.0 (Verity Software House).
PFGE. Cementoblast cultures (5 × 106 cells) were incubated overnight. One culture received a TD50 equivalent dose of total soluble protein from E. coli BL21(DE3)(pET15bcdt) at a concentration of 4·5 μg (ml medium)−1. A second culture did not receive any extract. Cells were collected after incubation for 72 h post-intoxication and prepared for analysis by electrophoresis as described previously (Kanno et al., 2005). Electrophoresis was performed at 175 V (starting voltage) for 40 h at 4 °C.
Validation of cell-specific markers. GMSM-K cells (1·5 × 104 cells per well), HPLFs (1·0 × 104 cells per well) and cementoblasts (1·0 × 104 cells per well) were grown separately in Medium 199 in eight-well chamber slides (Nalgene Nunc International). Slides were incubated for 24 h at 37 °C in a moist atmosphere containing 5 % CO2. Cells were washed twice with cold PBS, fixed in 10 % formalin for 10 min at room temperature and made permeable with 0·2 % Triton X-100 in PBS (pH 7·0). Slides were washed three times with PBS and then treated with 200 μl 1 % BSA in PBS per well as a blocking buffer for 30 min at room temperature. Slides were washed twice with PBS and each well of one set of wells containing either HPLFs, GMSM-K cells or cementoblasts was incubated with 200 μl fibroblast antigen (Ab-1; Oncogen Research Products) or anti-cytokeratin mAb (AE1; Zymed Laboratories) at a 1 : 200 dilution in 1 % BSA/PBS for 1 h at room temperature. Unbound antibody was removed by washing the slides three times with PBS. Two hundred microlitres of a 1 : 1000 dilution of Alexa Fluor 488 goat anti-mouse IgG (heavy and light chain) conjugate (Molecular Probes) in 1 % BSA/PBS was then added to each well and the slides incubated for 30 min in the dark. Wells containing GMSM-K cells were also counterlabelled with the DNA-binding fluorochrome 4,6-diamidino-2-phenylindole (DAPI) at a concentration of 1 μg (ml mounting solution)−1. Three other sets of wells containing either HPLFs, GMSM-K cells or cementoblasts were treated with 200 μl per well of a 1 : 200 dilution of mouse anti-simian virus 40 (SV40) T-antigen mAb (Chemicon International) and a 1 : 1000 dilution of Alexa Fluor 488 goat anti-mouse IgG in 1 % BSA/PBS. Slides were stored in the dark for 30 min at room temperature. HPLFs and cementoblasts were also counterlabelled with Texas red-X–phalloidin (Molecular Probes) using a 1 : 1000 dilution.
Slides were washed with PBS, dried at room temperature and treated with one drop of mounting solution (Prolong Anti-Fade; Promega). Cover slips were placed on the slides and viewed under a Nikon Eclipse E600 fluorescence microscope with filters FITC-HYQ for Alexa Fluor 488 conjugate (excitation wavelengths 460–500 nm), Texas red-HYQ (excitation wavelengths 532–587 nm) and UV-2E/C-DAPI (excitation wavelengths 330–380 nm). Images were captured by filtering for individual fluorochromes and then digitally overlaid.
Immunofluorescent detection of CDT-treated cells. GMSM-K cells (1·5 × 104 cells per well) and HPLFs (1·0 × 104 cells per well) were mixed in Medium 199 in eight-well chamber slides. Slides were incubated for 24 h at 37 °C in a moist atmosphere containing 5 % CO2. The growth medium was then removed and cells were incubated with 200 μl protein extract from E. coli BL21(DE3)(pET15bcdt) at a final concentration of 15 μg (ml medium)−1. Cells incubated with diluted extract from E. coli BL21(DE3)(pET15b) were used as controls. In some experiments, slides were incubated for 24, 48 and 72 h post-intoxication without removal of excess toxin (continuous exposure). In other experiments, slides were incubated with toxin-containing extract for 15 min, after which the cells were washed and incubated in fresh medium for 0, 3, 6, 24 and 48 h post-intoxication. At the end of the incubation periods, cells were washed twice with cold PBS, fixed and made permeable as described above. Slides were washed three times with PBS and then treated with 200 μl 1 % BSA/PBS blocking buffer per well for 30 min at room temperature. Slides were washed twice with PBS and incubated with 200 μl Ab-1 mAb per well at a 1 : 200 dilution in 1 % BSA/PBS for 1 h at room temperature. Unbound antibody was removed by washing the slides three times with PBS. Two hundred microlitres of a 1 : 1000 dilution of Alexa Fluor 488 goat anti-mouse IgG conjugate in 1 % BSA/PBS was then added to each well and the slides incubated for 30 min in the dark. Slides were washed with PBS followed by 200 μl of a 1 : 200 dilution of anti-SV40 T-antigen biotin-labelled mAb (BD Biosciences-Pharmingen) per well diluted in 1 % BSA/PBS. Slides were stored in the dark for 1 h at room temperature, washed three times with PBS and incubated with a 1 : 1000 dilution of streptavidin conjugated to Texas red-X (Molecular Probes) in 1 % BSA/PBS at room temperature for 30 min. Finally, slides were washed with PBS, dried at room temperature and mounted with coverslips.
Co-cultures of epithelial cells and cementoblasts were treated similarly. GMSM-K cells (1·5 × 104 cells per well) and C11-26M cells (1·0 × 104 cells per well) were suspended in Medium 199 and added to each well of an eight-well chamber slide. Cultures were incubated for 24 h at 37 °C in a moist atmosphere containing 5 % CO2 and prepared as described for the epithelial cell/HPLF co-cultures. Cells were made permeable and incubated with 200 μl of a 1 : 200 dilution of anti-SV40 T-antigen mAb in 1 % BSA/PBS per well in the dark for 1 h at room temperature. The secondary antibody was a 1 : 1000 dilution of Alexa Fluor 488 goat anti-mouse IgG in 1 % BSA/PBS. Slide cultures were also stained for F-actin at room temperature for 30 min with Texas red-X–phalloidin diluted 1 : 1000.
Slides containing CDT-treated HPLFs (1·0 × 104 cells per well) and cementoblasts (C11-26M; 1·0 × 104 cells per well) were treated with a 1 : 200 dilution of Ab-1 mAb, a 1 : 1000 dilution of Alexa Fluor 488 goat anti-mouse IgG and a 1 : 1000 dilution of Texas red-X–phalloidin.
Images were captured by filtering for individual fluorochromes and then digitally overlaid.
RESULTS
Microscopic evaluation of CDT-treated human oral cells grown in co-culture
Equivalent cell numbers of GMSM-K cells and HPLFs were seeded in duplicate cultures. One set of cultures was left untreated and the other set was treated with recombinant CDT-containing bacterial extract. Growth of both sets of cultures was documented at 0, 24, 48 and 72 h of growth post-intoxication by bright-field microscopy. Both types of cells were present and increased in cell number through 72 h of growth in the untreated cultures (Fig. 1, left column). By 24 h, the HPLFs had begun to overgrow the GMSM-K cells in the CDT-treated cultures (Fig. 1, right column). After 72 h of growth, no epithelial cells were observed, especially after the cultures had been washed to remove non-adherent cells (Fig. 1, inset in lower right panel). Identical results were obtained when GMSM-K cells were grown in co-culture with cementoblasts (data not shown). In this case, cementoblasts predominated the cultures at 72 h post-intoxication.
Fig. 1.

Microscopic evaluation of the effects of CDT on HPLFs and oral epithelial cells (GMSM-K) grown in co-culture. Two sets of cell cultures were grown overnight in Medium 199. One set was left undisturbed (−CDT), while CDT-containing extract from E. coli BL21(DE3) (pET15bcdt) at a concentration of 0.9 mg protein (200 ml medium)−1 was added to the second set (+CDT). The cultures were examined microscopically after incubation for an additional 0, 24, 48 and 72 h. At the end of the experiment, the 72 h cultures were washed to remove dead cells and re-examined (insets). Images are representative of multiple microscopic fields examined. Magnification × 10.
Resistance of cementoblasts to CDT was confirmed by flow cytometry at 48 and 72 h post-intoxication (Fig. 2). Eighty-three per cent of the cells had a DNA content of 2n (G0/G1) and 0 % had a content of 4n (G2/M) after exposure to recombinant CDT-containing bacterial extract for 72 h, indicative of no cell-cycle arrest. Cementoblast cultures exposed to the CDT-containing extract for 72 h also showed no signs of DNA damage. All of the DNA remained in the well after PFGE (Fig. 2, inset).
Fig. 2.

Examination of CDT-treated cementoblasts for cell-cycle arrest and dsDNA damage. Cementoblast cultures (1 × 106 cells) were left untreated (a) or were treated (b, c) with medium containing protein extract from E. coli BL21(DE3)(pET15bcdt) at a concentration of 18.4 μg ml−1. Cell nuclei were collected and stained at 48 h (b) and 72 h (c) post-intoxication. Cell-cycle profiles obtained by flow cytometry are shown. The DNA profile determined by propidium iodide staining is shown as the open tracing marked PI. G0/G1 and G2/M (filled) and S (hatched) peaks were determined by computer analysis. The percentages of cells in the population in the diploid G1, diploid G2 and diploid S states are shown. CV is the coefficient of variance. The insets show PFGE of untreated and CDT-treated (72 h) cultures. Gels were stained with ethidium bromide. The position of the wells in the agarose gel is marked.
Identification and validation of cell-specific markers
The simple microscopic evaluation of CDT-treated cells in co-culture gave a clear indication of the differential effects of the toxin. However, the availability of cell-specific markers provides a more definitive method for tracking changes in mixed-cell populations. Fibroblasts express a cell-surface antigen, Thy-1 (CD90), that is a potential candidate for a specific marker (Saalbach et al., 1996, 1999). Since the GMSM-K cell line is virally transformed, the cells express the SV40 large T antigen (Gilchrist et al., 2000). To determine the specificity of these potential cell markers, HPLFs and GMSM-K cells were grown separately in culture and immunolabelled using commercially available primary antibodies against each of these proteins (Fig. 3). When HPLFs and GMSM-K cells were individually labelled with anti-Ab-1 mAb and Alexa Fluor 488-conjugated secondary antibody, only HPLFs were detected (Fig. 3, left column). Counter-staining the GMSM-K cultures with DAPI showed that cells were present but not recognized by the Ab-1 mAb. A mouse anti-cytokeratin mAb (AE1) was also tested for specific recognition of HPLFs. However, this antibody preparation yielded poor results in immunofluorescence experiments (data not shown). Opposite results were obtained with mouse anti-SV40 T-antigen mAb and Alexa Fluor 488-conjugated secondary antibody (Fig. 3, right column). Counterstaining the HPLFs with Texas red-X–phalloidin demonstrated that the cells were present but not recognized by the SV40 T-antigen antibodies. These results established that specific markers could be used to distinguish epithelial cells from fibroblasts when grown in co-culture. These antibodies failed to recognize cementoblasts; currently no specific markers have been identified for this cell type.
Fig. 3.

Validation of cell-specific markers for HPLFs and GMSM-K cells. HPLFs and GMSM-K cells were grown separately in Medium 199 for 24 h on chamber slides. Slides were treated with either Ab-1 or mouse anti-SV40 T-antigen mAb (diluted 1 : 1000) followed by Alexa Fluor 488 goat anti-mouse IgG (diluted 1 : 1000). GMSM-K cells that were treated with Ab-1 mAb were also labelled with DAPI (GMSM-K inset). HPLFs that were treated with SV40 T-antigen mAb were also labelled with Texas red-X– phalloidin (HPLF inset). All slides were examined using a fluorescence microscope and images recorded with a digital camera. Magnification ×60.
Use of specific immunofluorescence tagging to confirm the differential effects of CDT on the growth of human oral cells in mixed cultures
The specific T-antigen and Thy-1 markers were used to evaluate the effects of CDT on mixed cultures. GMSM-K cells and HPLFs were co-grown and examined at 24 h post-intoxication with CDT. HPLFs were labelled with Ab-1 mAb and Alexa Fluor 488 goat anti-mouse IgG conjugate (green fluorescence). GMSM-K cells were labelled with SV40 T-antigen biotin-labelled mAb and streptavidin–Texas red-X conjugate (red fluorescence). No epithelial cells were detected in co-labelled mixed cultures that were treated with CDT (Fig. 4, upper right panel). These data confirmed the results initially obtained using light microscopy. The same results were obtained with mixed cultures of GMSM-K cells and cementoblasts (Fig. 4, middle row). In this case, the epithelial cells were tagged with mouse anti-SV40 large T-antigen mAb and Alexa Fluor 488 goat anti-mouse IgG conjugate (green fluorescence). The cementoblasts were non-specifically stained with Texas red-X–phalloidin (red fluorescence). As expected, there was no effect of CDT on mixed cultures of HPLFs and cementoblasts (Fig. 4, lower row). The HPLFs were tagged with Ab-1 mAb and Alexa Fluor 488 goat anti-mouse IgG conjugate (green fluorescence) and the cementoblasts were stained with Texas red-X–phalloidin. Images captured by filtering for the individual fluorochromes (insets) are shown for all panels.
Fig. 4.
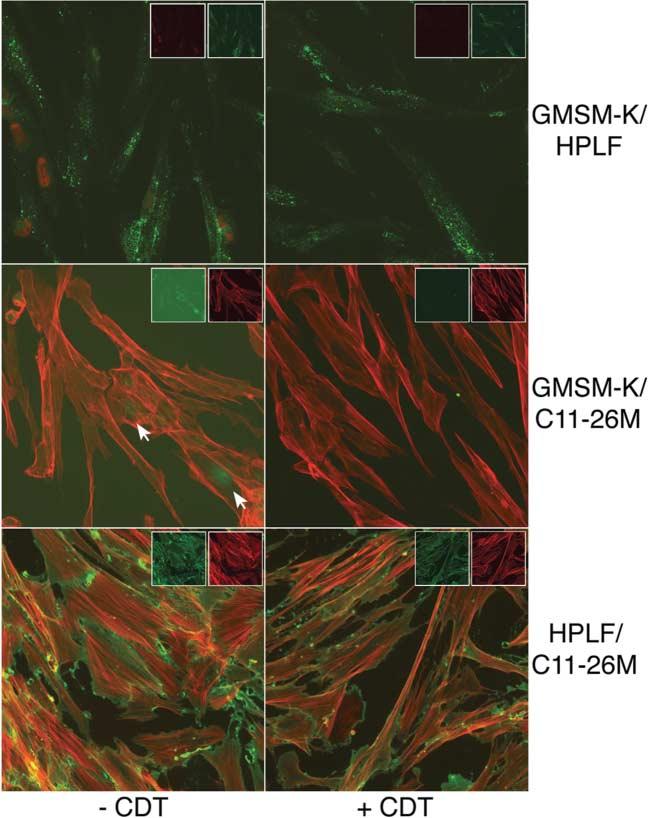
Use of immunospecific staining to determine the effects of CDT on proliferation of oral cells grown in co-culture. GMSM-K cells, HPLFs and cementoblasts (C11-26M) were grown in pairs for 24 h on chamber slides. The paired cultures were either untreated (left column) or treated with CDT (right column). GMSM-K/HPLF co-cultures were labelled with anti-SV40 T-antigen biotin-labelled mAb and streptavidin conjugated to Texas red-X (red fluorescence) and with Ab-1 and Alexa Fluor 488 goat anti-mouse IgG conjugate (green fluorescence). GMSM-K/C11-26M co-cultures were labelled with anti-SV40 T-antigen mAb and Alexa Fluor 488 conjugate (green fluorescence) and Texas red-X–phalloidin (red fluorescence). HPLF/C11-26M co-cultures were labelled with Ab-1 and Alexa Fluor 488 conjugate (green fluorescence) and Texas red-X–phalloidin (red fluorescence). Arrows mark the positions of labelled GMSM-K cells. Insets show fluorescence of the individual fluorochromes. Images are representative of multiple microscopic fields examined. Magnification ×60.
When co-cultures of GMSM-K cells and HPLFs were treated with toxin and observed over time, a clear pattern of epithelial-cell inhibition was observed. An increase in epithelial-cell number was evident in untreated co-cultures up to 72 h post-intoxication (Fig. 5, left column). Inhibition of epithelial cell proliferation was observed in CDT-treated co-cultures (Fig. 5, right column). Epithelial cells were eradicated between 24 and 48 h of growth. Elimination of epithelial cells in co-cultures was relatively rapid, even after a short exposure (15 min) to toxin. No epithelial cells were observed after 24 h of growth post-intoxication, as was the case with long-term exposure (data not shown).
Fig. 5.
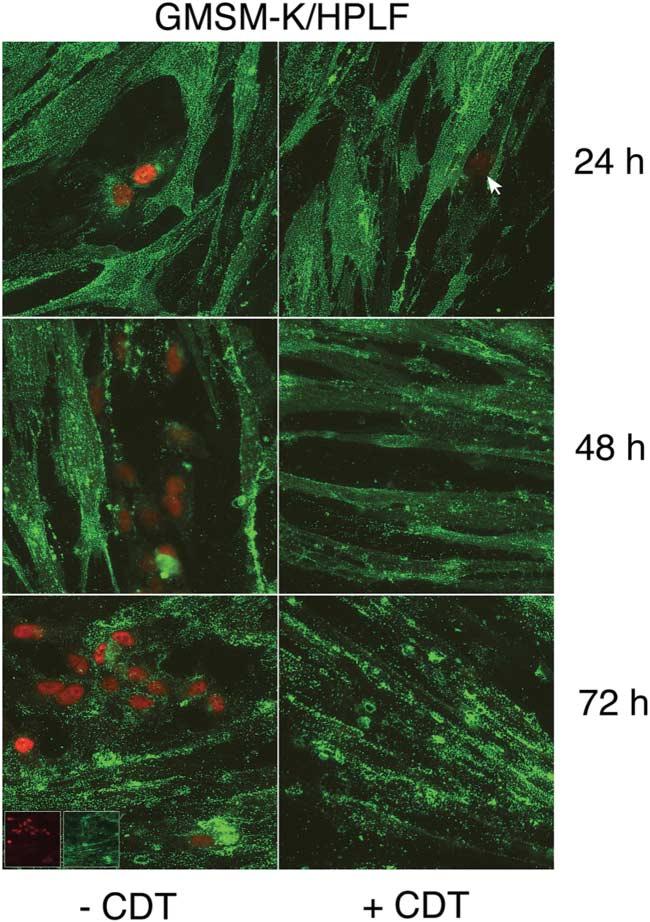
Time course of untreated and CDT-treated co-cultures of HPLFs and oral epithelial cells. HPLFs and GMSM-K cells were grown for 24 h on chamber slides and left untreated (left column) or treated with CDT (right column). Cultures were incubated and examined at 24, 48 and 72 h post-intoxication. Co-cultures were labelled with anti-SV40 T-antigen biotin-labelled mAb and streptavidin–Texas red-X (red fluorescence), and Ab-1 and Alexa Fluor 488 conjugate (green fluorescence). The arrow marks the position of a labelled GMSM-K cell in a CDT-treated co-culture. Immunofluorescence of the individual fluorochromes is shown in the insets in the lower left panel. Images are representative of multiple microscopic fields examined. Magnification ×60.
DISCUSSION
A. actinomycetemcomitans is a well-studied, periodontal pathogen that produces an impressive array of potential virulence factors including toxins such as leukotoxin and CDT (Henderson et al., 2003). It has been a challenge to prove that these toxins play an active role in disease. We were interested in the role of CDT in the virulence potential of A. actinomycetemcomitans and its possible contribution to periodontal disease. Although it has not yet been experimentally established that CDT is involved in periodontal disease, prevalence of the cdt locus in clinical isolates of the bacterium and the unusual cytotoxic properties of CDT indirectly support the argument that the toxin is potentially an important virulence factor. There is also evidence, from DNA sequence analysis of chromosomal regions that flank the cdt locus, that the cdt genes may have been acquired by A. actinomycetemcomitans via horizontal gene transfer (Mayer et al., 1999; Heywood et al., 2005). Taken together, these observations support the concept that there is environmental pressure on the bacterium in the human oral cavity to maintain and express the cdt genes. It is expected that the cdt locus would have been eliminated from the A. actinomycetemcomitans population if it did not impart some advantage to the bacterium in the periodontium.
The periodontium is composed of a number of distinct cell types including epithelial cells, fibroblasts, cementoblasts and osteoblasts, amongst others (Hassel, 1993). From a functional standpoint, the periodontium can be broadly broken down into two tissue compartments referred to as gingiva and the attachment apparatus. Gingiva, which is composed of epithelial cells and the adjacent connective tissue, is attached to a tooth via hemidesmosomes formed by cells of the junctional epithelium. This provides a physical barrier that protects the underlying attachment apparatus from the potentially harmful host as well as microbial and/or environmental factors found within the oral cavity proper. In view of the diverse cellular make-up of the periodontium, we recently examined the response of oral epithelial cells and HPLFs to CDT produced by A. actinomycetemcomitans. We found that CDT selectively inhibited the proliferation of human oral epithelial cells, while HPLFs were resistant to the classical activities characteristic of this family of toxins (Kanno et al., 2005). HPLFs challenged with CDT in culture did not arrest at either the G0/G1 or G2/M phase of the cell cycle, nor did they exhibit dsDNA damage. It was previously reported that CDT of A. actinomycetemcomitans is involved in the non-lethal inhibition of proliferation of HPLFs and human gingival fibroblasts (Belibasakis et al., 2002). More recently it was suggested that inhibition was associated with cell-cycle arrest at both the G0/G1 and G2/M phases of growth (Belibasakis et al., 2004). However, these effects were not observed using either crude extracts or purified recombinant A. actinomycetemcomitans CDT in our studies (Kanno et al., 2005). In contrast, proliferation of human epithelial cell lines was rapidly inhibited by CDT (Akifusa et al., 2001; DiRienzo et al., 2002; Kanno et al., 2005). Growth arrest of the human oral epithelial cells occurred at the S phase of the cell cycle, indicative of a block in DNA replication, and was accompanied by extensive dsDNA damage (Kanno et al., 2005).
Studies of the effects of A. actinomycetemcomitans CDT on human oral cells described above were done using pure cultures. While this approach is a required first step in elucidating the complex biological mechanisms of a microbial toxin, it is a somewhat artificial system, especially when considering an environment as complex as the human periodontium. The interaction of cells and cell products can affect or alter the activities of microbial factors. For example, microbial factors can adhere to cells or cell products, rendering the factor inactive or unavailable to other cells. In an attempt to address some of these issues, we evaluated the feasibility of using mixed cultures for examining the response of oral cells to A. actinomycetemcomitans CDT.
Co-cultures of oral cells have been used to evaluate the irritancy of dental materials (Schmalz et al., 1997), expression of growth factors (Gron et al., 2002) and the effects of cancer cells on normal cells (Sawaki et al., 2003). However, to the best of our knowledge, the cell-specific effects of bacterial toxins have not been examined in co-culture systems. Our earlier finding that there was a differential response of the oral epithelial cells and HPLFs to CDT provided a testable co-culture model.
We first successfully established that there was no cross inhibition of growth or overgrowth of one cell type when the epithelial cells and fibroblasts or epithelial cells and cementoblasts were grown in co-culture. Cementoblasts showed the same response to CDT as HPLFs when grown as pure cultures and this response seems to be typical of oral fibroblast-like cells. Secondly, it was necessary to identify and validate cell-specific markers for the simultaneous detection of each cell type in co-cultures. An SV40 T-antigen mAb was used specifically to detect the GMSM-K cell line. This cell line is immortalized by SV40 transfection and expresses the SV40 large T antigen (Gilchrist et al., 2000). It was found that a mAb against the human Thy-1 (CD90) antigen was specific for the fibroblasts (Saalbach et al., 1996). This antigen is a 35 kDa glycoprotein found on the extracellular membrane of fibroblasts, neurons and some CD34+ blood stem cells (Saalbach et al., 1999). A cementoblast-specific marker was not identified.
Using this approach, we clearly demonstrated that the epithelial cells increased in number during co-growth with HPLFs. However, proliferation of the epithelial cells was selectively and rapidly inhibited by CDT in co-culture with either HPLFs or cementoblasts. This selective inhibition was readily observed by light microscopy of CDT-treated co-cultures and confirmed by immunofluorescence microscopy. No epithelial cells were detected by 24 h post-intoxication in co-cultures exposed to CDT for relatively short times (15 min). These results demonstrated that the response of epithelial cells, fibroblasts and cementoblasts to CDT was the same whether the cells were cultured separately or together.
The ability to measure the response of individual cell types to bacterial toxins in mixed cultures has important applications in the study of the pathogenesis of periodontal disease. In the context of pathogenesis, the differential responses of the various cell types observed in mixed-cell culture suggest that, in vivo, CDT has the potential to compromise the integrity of gingival epithelium, allowing the organism and/or virulence factors to gain access to the underlying connective tissue. Furthermore, there may be indirect effects of CDT that are mediated via the release of molecules derived from dying and/or dead epithelial cells that could have detrimental effects on the other types of cells within the periodontium. This is a testable hypothesis in vitro using mixed cultures. The results of this study suggest that this does not occur with respect to HPLFs and cementoblast proliferation. This does not rule out the possibility of altered cellular function, which will be addressed in future studies. Along these lines, it will be interesting to examine the expression of inflammatory mediators and other factors known to have key roles in advancing the degradation of the various components of the periodontium. It has been reported that CDT of A. actinomycetemcomitans regulates the expression of receptor activator of NF-κB ligand (RANKL) and osteoprotegerin by periodontal ligament cells (Belibasakis et al., 2005). Since expression of these molecules is involved in osteoclast differentiation, they have a central role in the regulation of bone resorption, a key component of periodontitis (Liu et al., 2003; Mogi et al., 2004).
In summary, we have (i) demonstrated that HPLFs, cementoblasts and human oral epithelial cells can be grown together in culture; (ii) established the utility of using specific markers for the simultaneous detection of several of these oral cell types in mixed cultures; and (iii) used these markers to demonstrate that the same cell-specific responses to CDT identified in pure cultures could be observed in heterogeneous cell populations. The approach described in this study will be useful for advancing studies of more complex interactions between microbial virulence factors and those cells targeted in periodontitis.
ACKNOWLEDGEMENTS
We thank Valerie Murrah and Eric Gilchrist (University of North Carolina at Chapel Hill) for the GMSM-K cell line, Pamela Howard (University of Pennsylvania) for providing mAbs and Edward Macarak (University of Pennsylvania) for use of a fluorescence microscope. This study was supported by USPHS grants R01-DE-012593 (J. M. D.), DE-13475 (W. G.) and DE-14600 (W. G.) from the zNational Institutes of Health.
Abbreviations
- CDT
cytolethal distending toxin
- HPLF
human periodontal ligament fibroblast
- PDL
periodontal ligament
- SV40
simian virus 40
- TD50
toxic dose50
REFERENCES
- Ahmed HJ, Svensson LA, Cope LD, Latimer JL, Hansen EJ, Ahlman K, Bayat-Turk J, Klamer D, Lagergard T. Prevalence of cdtABC genes encoding cytolethal distending toxin among Haemophilus ducreyi and Actinobacillus actinomycetemcomitans strains. J Med Microbiol. 2001;50:860–864. doi: 10.1099/0022-1317-50-10-860. [DOI] [PubMed] [Google Scholar]
- Akifusa S, Poole S, Lewthwaite J, Henderson B, Nair SP. Recombinant Actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect Immun. 2001;69:5925–5930. doi: 10.1128/IAI.69.9.5925-5930.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belibasakis G, Johansson A, Wang Y, Claesson R, Chen C, Asikainen S, Kalfas S. Inhibited proliferation of human periodontal ligament cells and gingival fibroblasts by Actinobacillus actinomycetemcomitans: involvement of the cytolethal distending toxin. Eur J Oral Sci. 2002;110:366–373. doi: 10.1034/j.1600-0722.2002.21350.x. [DOI] [PubMed] [Google Scholar]
- Belibasakis GN, Mattsson A, Wang Y, Chen C, Johansson A. Cell cycle arrest of human gingival fibroblasts and periodontal ligament cells by Actinobacillus actinomycetemcomitans: involvement of the cytolethal distending toxin. APMIS. 2004;112:675–685. doi: 10.1111/j.1600-0463.2004.apm1121006.x. [DOI] [PubMed] [Google Scholar]
- Belibasakis GN, Johansson A, Wang Y, Chen C, Kalfas S, Lerner UH. The cytolethal distending toxin induces receptor activator of NF-κB ligand expression in human gingival fibroblasts and periodontal ligament cells. Infect Immun. 2005;73:342–351. doi: 10.1128/IAI.73.1.342-351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blix IJS, Hars R, Preus HR, Helgeland K. Entrance of Actinobacillus actinomycetemcomitans into HEp-2 cells in vitro. J Periodontol. 1992;63:723–728. doi: 10.1902/jop.1992.63.9.723. [DOI] [PubMed] [Google Scholar]
- Comayras C, Tasca C, Pérès SY, Ducommun B, Oswald E, De Rycke J. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun. 1997;65:5088–5095. doi: 10.1128/iai.65.12.5088-5095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Bratti X, Chaves-Olarte E, Lagergard T, Thelestam M. The cytolethal distending toxin from the chancroid bacterium Haemophilus ducreyi induces cell-cycle arrest in the G2 phase. J Clin Invest. 1999;103:107–115. doi: 10.1172/JCI3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Bratti X, Karlsson C, Lagergard T, Thelestam M, Frisan T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J Biol Chem. 2001;276:5296–5302. doi: 10.1074/jbc.M008527200. [DOI] [PubMed] [Google Scholar]
- DiRienzo JM, Song M, Wan LSY, Ellen RP. Kinetics of KB and HEp-2 cell responses to an invasive, cytolethal distending toxinproducing strain of Actinobacillus actinomycetemcomitans. Oral Micro-biol Immunol. 2002;17:245–251. doi: 10.1034/j.1399-302x.2002.170407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Dreyfus LA. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol. 2000;37:952–963. doi: 10.1046/j.1365-2958.2000.02070.x. [DOI] [PubMed] [Google Scholar]
- Fabris AS, DiRienzo JM, Wïkstrom M, Mayer MPA. Detection of cytolethal distending toxin activity and cdt genes in Actinobacillus actinomycetemcomitans isolates from geographically diverse populations. Oral Microbiol Immunol. 2002;17:231–238. doi: 10.1034/j.1399-302x.2002.170405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisan T, Cortes-Bratti X, Chaves-Olarte E, Stenerlow B, Thelestam M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol. 2003;5:695–707. doi: 10.1046/j.1462-5822.2003.00311.x. [DOI] [PubMed] [Google Scholar]
- Gilchrist EP, Moyer MP, Shillitoe EJ, Clare N, Murrah VA. Establishment of a human polyclonal oral epithelial cell line. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:340–347. doi: 10.1067/moe.2000.107360. [DOI] [PubMed] [Google Scholar]
- Gron B, Stoltze K, Andersson A, Dabelsteen E. Oral fibroblasts produce more HGF and KGF than skin fibroblasts in response to co-culture with keratinocytes. APMIS. 2002;110:892–898. doi: 10.1034/j.1600-0463.2002.1101208.x. [DOI] [PubMed] [Google Scholar]
- Grzesik WJ, Kuznetsov SA, Uzawa K, Mankani M, Robey PG, Yamauchi M. Normal human cementum-derived cells: isolation, clonal expansion, and in vitro and in vivo characterization. J Bone Miner Res. 1998;13:1547–1554. doi: 10.1359/jbmr.1998.13.10.1547. [DOI] [PubMed] [Google Scholar]
- Grzesik WJ, Cheng H, Oh JS, Kuznetsov SA, Mankani M, Uzawa K, Robey PG, Yamauchi M. Cementum-forming cells are phenotypically distinct from bone-forming cells. J Bone Miner Res. 2000;15:52–59. doi: 10.1359/jbmr.2000.15.1.52. [DOI] [PubMed] [Google Scholar]
- Hassane DC, Lee RB, Pickett CL. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect Immun. 2003;71:541–545. doi: 10.1128/IAI.71.1.541-545.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel T. Tissues and cells of the periodontium. Periodontol 2000. 1993;3:9–38. doi: 10.1111/j.1600-0757.1993.tb00230.x. [DOI] [PubMed] [Google Scholar]
- Henderson B, Nair SP, Ward JM, Wilson M. Molecular pathogenicity of the oral opportunistic pathogen Actinobacillus actinomycetemcomitans. Annu Rev Microbiol. 2003;57:29–55. doi: 10.1146/annurev.micro.57.030502.090908. [DOI] [PubMed] [Google Scholar]
- Heywood W, Henderson B, Nair SP. Cytolethal distending toxin: creating a gap in the cell cycle. J Med Microbiol. 2005;54:207–216. doi: 10.1099/jmm.0.45694-0. [DOI] [PubMed] [Google Scholar]
- Kanno F, Korostoff J, Volgina A, DiRienzo JM. Resistance of human periodontal ligament fibroblasts to the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. J Periodontol. 2005 doi: 10.1902/jop.2005.76.7.1189. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Tejero M, Galán JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- Lekic PC, Pender N, McCulloch CA. Is fibroblast heterogeneity relevant to the health, diseases, and treatments of periodontal tissues? Crit Rev Oral Biol Med. 1997;8:253–268. doi: 10.1177/10454411970080030201. [DOI] [PubMed] [Google Scholar]
- Liu D, Xu JK, Figliomeni L, Huang L, Pavlos NJ, Rogers M, Tan A, Price P, Zheng MH. Expression of RANKL and OPG mRNA in periodontal disease: possible involvement in bone destruction. Int J Mol Med. 2003;11:17–21. doi: 10.3892/ijmm.11.1.17. [DOI] [PubMed] [Google Scholar]
- Mao X, DiRienzo JM. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell Microbiol. 2002;4:245–255. doi: 10.1046/j.1462-5822.2002.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MPA, Bueno LC, Hansen EJ, DiRienzo JM. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun. 1999;67:1227–1237. doi: 10.1128/iai.67.3.1227-1237.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DH, Lippmann JE, Fives-Taylor PM. Invasion of epithelial cells by Actinobacillus actinomycetemcomitans: a dynamic, multistep process. Infect Immun. 1996;64:2988–2997. doi: 10.1128/iai.64.8.2988-2997.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Otogoto J, Ota N, Togari A. Differential expression of RANKL and osteoprotegerin in gingival crevicular fluid of patients with periodontitis. J Dent Res. 2004;83:166–169. doi: 10.1177/154405910408300216. [DOI] [PubMed] [Google Scholar]
- Saalbach A, Anderegg U, Bruns M, Schnabel E, Herrmann K, Haustein UF. Novel fibroblast-specific monoclonal antibodies: properties and specificities. J Invest Dermatol. 1996;106:1314–1319. doi: 10.1111/1523-1747.ep12349035. [DOI] [PubMed] [Google Scholar]
- Saalbach A, Wetzig T, Haustein UF, Anderegg U. Detection of human soluble Thy-1 in serum by ELISA: fibroblasts and activated endothelial cells are a possible source of soluble Thy-1 in serum. Cell Tissue Res. 1999;298:307–315. doi: 10.1007/s004419900079. [DOI] [PubMed] [Google Scholar]
- Sawaki K, Yamaai T, Mizukawa N, Yoshimoto T. Mortality of human epidermal keratinocytes in co-culture with oral squamous cell carcinoma cells. Anticancer Res. 2003;23:79–84. [PubMed] [Google Scholar]
- Schmalz G, Arenholt-Bindslev D, Hiller KA, Schweikl H. Epithelium-fibroblast co-culture for assessing mucosal irritancy of metals used in dentistry. Eur J Oral Sci. 1997;105:86–91. doi: 10.1111/j.1600-0722.1997.tb00185.x. [DOI] [PubMed] [Google Scholar]
- Shenker BJ, McKay TL, Datar S, Miller M, Chowhan R, Demuth D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J Immunol. 1999;162:4773–4780. [PubMed] [Google Scholar]
- Somerman MJ, Archer SY, Imm GR, Foster RA. A comparative study of human periodontal ligament cells and gingival fibroblasts in vitro. J Dent Res. 1988;67:66–70. doi: 10.1177/00220345880670011301. [DOI] [PubMed] [Google Scholar]
- Sugai M, Kawamoto T, Pérès SY, Ueno Y, Komatsuzawa H, Fujiwara T, Kurihara H, Suginaka H, Oswald E. Cell cycle-specific growth-inhibitory factor produced by Actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect Immun. 1998;66:5008–5019. doi: 10.1128/iai.66.10.5008-5019.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K-S, Song K-P, Ong G. Cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Occurrence and association with periodontal disease. J Periodont Res. 2002;37:268–272. doi: 10.1034/j.1600-0765.2002.01618.x. [DOI] [PubMed] [Google Scholar]
- Vindelov LL. Flow microfluorometric analysis of nuclear DNA in cells from solid tumors and cell suspensions: a new method for rapid isolation and staining of nuclei. Arch B Cell Pathol. 1977;24:227–242. [PubMed] [Google Scholar]
- Whitehouse CA, Balbo PB, Pesci EC, Cottle DL, Mirabito PM, Pickett CL. Camphylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect Immun. 1998;66:1934–1940. doi: 10.1128/iai.66.5.1934-1940.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano R, Ohara M, Nishikubo S. Prevalence of cytolethal distending toxin production in periodontopathogenic bacteria. J Clin Microbiol. 2003;41:1391–1398. doi: 10.1128/JCM.41.4.1391-1398.2003. 10 other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]