

Summary
The Cdt is a family of gram-negative bacterial toxins that typically arrest eukaryotic cells in the G0/G1 or G2/M phase of the cell cycle. The toxin is a heterotrimer composed of the cdtA, cdtB and cdtC gene products. Although it has been shown that the CdtA protein subunit binds to cells in culture and in an enzyme-linked immunosorbent assay (CELISA) the precise mechanisms by which CdtA interacts with CdtB and CdtC has not yet been clarified. In this study we employed a random mutagenesis strategy to construct a library of point mutations in cdtA to assess the contribution of individual amino acids to binding activity and to the ability of the subunit to form biologically active holotoxin. Single unique amino acid substitutions in seven CdtA mutants resulted in reduced binding of the purified recombinant protein to Chinese hamster ovary cells and loss of binding to the fucose-containing glycoprotein, thyroglobulin. These mutations clustered at the 5′- and 3′-ends of the cdtA gene resulting in amino acid substitutions that resided outside of the aromatic patch region and a conserved region in CdtA homologues. Three of the amino acid substitutions, at positions S165N (mutA81), T41A (mutA121) and C178W (mutA221) resulted in gene products that formed holotoxin complexes that exhibited a 60% reduction (mutA81) or loss (mutA121, mutA221) of proliferation inhibition. A similar pattern was observed when these mutant holotoxins were tested for their ability to induce cell cycle arrest and to convert supercoiled DNA to relaxed and linear forms in vitro. The mutations in mutA81 and mutA221 disrupted holotoxin formation. The positions of the amino acid substitutions were mapped in the Haemophilus ducreyi Cdt crystal structure providing some insight into structure and function.
Introduction
The cytolethal distending toxin (Cdt) of the periodontal pathogen Actinobacillus actinomycetemcomitans is a typical member of a family of secreted gram-negative bacterial protein toxins that classically arrest the growth of many types of eukaryotic cells or cell lines at either the G0/G1 or G2/M phase of the cell cycle (reviewed in references Pickett and Whitehouse, 1999; Young and Schauer, 2000; Cortes-Bratti et al., 2001a; De Rycke and Oswald, 2001; Lara-Tejero and Galán, 2002; De Rycke and Ducommun, 2003; Ohara et al., 2004; Heywood et al., 2005; Oswald et al., 2005). Biologically active toxin, from many of the diverse bacterial genera that express the Cdt, is a heterotrimer composed of approximately 18–25 (CdtA), 31 (CdtB) and 21 (CdtC) kDa protein subunits expressed from a polycistronic operon (Akifusa et al., 2001; Lara-Tejero and Galán, 2001; Saiki et al., 2001; Mao and DiRienzo, 2002; Lee et al., 2003; Shenker et al., 2004). One of the many intriguing characteristics of the cdt genes is that they appear to have a eukaryotic rather than prokaryotic heritage. The cdt gene products exhibit deduced amino acid sequence and structure/function similarities (albeit weak) to those of eukaryotic proteins. Furthermore, introns have been identified in the cdt operon of A. actinomycetemcomitans (Tan et al., 2005). The presence of introns, typical of eukaryotic genes, is a rare occurrence in the eubacteria.
The toxic component of the holotoxin appears to be the CdtB protein subunit which has similarity, based on position-specific iterated (PSI) BLAST (Altschul et al., 1997) comparisons, to mammalian type I deoxyribonucleases (Elwell and Dreyfus, 2000; Lara-Tejero and Galán, 2000; Lara-Tejero and Galán, 2002). Purified recombinant CdtB exhibits nicking or relaxation activities, in vitro, when incubated with supercoiled DNA (Elwell and Dreyfus, 2000; Elwell et al., 2001). DNA damage in the form of double-stranded breaks, as assessed by pulsed-field gel electrophoresis (PFGE), was detected in sensitive cell types exposed to the holotoxin (Cortes-Bratti et al., 2001b; Frisan et al., 2003; Kanno et al., 2005). Mutating conserved DNase I active site residues in CdtB resulted in the loss of both in vitro nuclease activity and in vivo cell cycle arrest (Elwell and Dreyfus, 2000; Lara-Tejero and Galán, 2000).
The CdtA protein subunit also appears to be related to eukaryotic proteins. Based on the initial observations reported by Hofmann et al. (2000) and Hassane et al. (2001), amino acid sequence threading analysis (Lara-Tejero and Galán, 2001) and a reverse position-specific (RPS)-BLAST search of the conserved domain (CD) database (Mao and DiRienzo, 2002) functional similarity was noted between a 60–88 sequence of amino acids in CdtA and the B chain of members of the ricin/abrin A–B toxin family. The B chain of ricin acts in a lectin-like interaction attaching to carbohydrate receptors on the cell surface to promote uptake of the A chain of the toxin (Rutenber et al., 1987). Data from immunofluorescence experiments (Mao and DiRienzo, 2002; Akifusa et al., 2005; McSweeney and Dreyfus, 2005; Kanno et al., 2005) and enzyme-linked immunosorbent assays on live cells (CELISA) (Lee et al., 2003) experiments showed that purified recombinant CdtA binds to cells in culture.
A specific eukaryotic connection for the CdtC subunit protein has not been found. However, CdtC has significant amino acid sequence similarity with CdtA (40% for the Campylobacter jejuni deduced amino acid sequences; Pickett and Whitehouse, 1999; Lee et al., 2003) suggesting a common function. Purified recombinant CdtC also binds to HeLa (McSweeney and Dreyfus, 2005) and HEp-2 (Akifusa et al., 2005) cells in culture and to HeLa cells in the CELISA (Lee et al., 2003). It has recently been shown that both CdtA and CdtC from Escherichia coli are carbohydrate-binding proteins that recognize N-linked fucose moieties on the surface of HeLa cells (McSweeney and Dreyfus, 2005).
The Cdt of Haemophilus ducreyi has been crystalized (NeŠić et al., 2004). The crystal structure supports a ternary complex having three interdependent molecular interfaces. The CdtA and CdtC subunits interact to form two ricin-like lectin domains separated by a deep groove. There is a highly aromatic surface on the CdtA face of the groove. Mutating four aromatic residues (quadruple mutant) in the aromatic patch domain of the CdtA subunit produced a non-toxic reconstituted holotoxin.
Mutagenesis approaches are invaluable for studying the relationship between structure and function. Insertional inactivation of the CdtA gene using antibiotic resistance gene cassette or transposon (TnphoA, Tn5) insertions has been used to demonstrate that this subunit is an important component of the holotoxin (Scott and Kaper, 1994; Lewis et al., 2001; Mao and DiRienzo, 2002; Young et al., 2004). In-frame deletions and site-directed mutagenesis of cdtA have provided more detailed information about the role of putative functional regions in the protein subunit (Lee et al., 2003; NeŠić et al., 2004; Saiki et al., 2004). However, there is still a great deal to be learned about the molecular details of the interactions between CdtA and the CdtB and CdtC subunits and between CdtA and cell surface receptors.
To advance studies of the function and interactions of the CdtA subunit and to identify single amino acid residues critical for CdtA activity, we made a library of randomly generated point mutations. Mutants with unique single amino acid substitutions were examined for changes in binding of isolated recombinant CdtA to Chinese hamster ovary (CHO) cells. Holotoxin was reconstituted with each mutated cdtA gene product and tested for the ability to inhibit CHO cell proliferation, to promote cell cycle arrest and to exhibit DNase I-like nicking activity in vitro. Each mutated cdtA gene product was also tested for the ability to bind to fucose-containing glycoproteins, to compete with wild-type CdtA for binding to CdtB and CdtC and to form a holotoxin complex. The greater than 90% similarity between the deduced amino acid sequences of the CdtA proteins of A. actinomycetemcomitans and H. ducreyi (Cope et al., 1997; Sugai et al., 1998; Mayer et al., 1999) allowed us to map the positions of the amino acid substitutions in the H. ducreyi Cdt crystal structure. The implications of specific amino acid substitutions on predicted relationships between structure and function are discussed.
Results
Selection of point mutations in cdtA and isolation of the mutants
A library of randomly generated point mutations was constructed in the plasmid vector pET15b. The His-tag DNA sequence was removed from the plasmid vector and six codons for histidine were placed in the reverse polymerase chain reaction (PCR) primer used to generate the mutant sequences (Table 1). These steps ensured that full length cdtA gene sequences could be easily identified in the mutant library by screening for the presence of the His-tag on the gene products. The pJDA9 construct (Table 2), encoding wild-type CdtA, was used as the template for mutagenesis and served as a positive control for all experiments. Mutations were introduced at a very low frequency to enhance chances of recovering mutants having a single nucleotide change. Total cell lysates of transformants carrying potential mutated cdtA gene sequences were first screened in a CELISA for reduction in binding to CHO cells relative to an E. coli lysate containing wild-type CdtA. This initial screening assay facilitated the analysis of a large number of potential mutants and provided a positive selection method for loss of biological activity. Those transformants that demonstrated binding at levels below that of the wild-type recombinant strain where examined for the correct size DNA insert by either restriction endonuclease mapping or by PCR. Those transformants that had the expected size plasmid DNA insert (668 bp) were then checked for expression of the cloned gene by Western blotting employing a monoclonal antibody that recognized the carboxy-terminal His-tag. Plasmid DNA from those transformants that contained an immuno-positive polypeptide, having an apparent molecular weight of approximately 27 kDa, were then subjected to DNA sequencing to confirm and identify the mutation.
Table 1.
Oligonucleotide primers used for cdtA-His, cdtB-His and cdtC-His cloning.
| Name | Sequencea |
|---|---|
| cdtA-F | 5′-CCCACCCATGGCTCCGAGGAGAGGTACAATGAAAAAG-3′ |
| cdtA-R | 5′-TTAGGATCCGTGGTGGTGGTGGTGGTGATTAACCGCTGT-3′ |
| cdtB-F | 5′-GCCGCCGCGGTTCCATGGAATGGGTAAAGC-3′ |
| cdtB-R | 5′-TTAGGATCCGTGGTGGTGGTGGTGGTGGCGATCATGAA-3′ |
| cdtC-F | 5′-TCCCGCCGGTTTTGTCCATGGTCGCTAAGGAGAATAC-3′ |
| cdtC-R | 5′-AATGGATCCGTGGTGGTGGTGGTGGTGGCTACCCTGATT-3′ |
Underlined bases mark the NcoI and BamHI restriction endonuclease recognition sites. Bases in bold type mark the codons for the His-tag.
Table 2.
Plasmids used in this study.
| Plasmid | Features | Source or reference |
|---|---|---|
| pCDT1 | cdt locus from A. actinomycetemcomitans cloned in E. coli | Mayer et al. (1999) |
| pJDA9 | cdtA gene containing 6 His codons at 3′-end | This study |
| pJDB7 | cdtB gene containing 6 His codons at 3′-end | This study |
| pJDC2 | cdtC gene containing 6 His codons at 3′-end | This study |
| pMUT65cdtA | CdtAC196S | This study |
| pMUT81cdtA | CdtAS165N | This study |
| pMUT99cdtA | CdtAT88T | This study |
| pMUT112cdtA | CdtAF43S | This study |
| pMUT121cdtA | CdtAT41A | This study |
| pMUT221cdtA | CdtAC178W | This study |
| pMUT239cdtA | CdtAP56Q | This study |
| pMUT241cdtA | CdtAT219A | This study |
| pMUT41cdtC | CdtC | This study |
| pET15b | Cloning vector | Novagen |
Identification and validation of the point mutations
Of 192 transformants, selected at random following mutagenesis, five failed to grow. Sixty-six transformants exhibited binding in the CELISA comparable to that of the negative control. Twenty-six of these 66 transformants had no insert DNA in the plasmid. Of the 40 remaining transformants 21 had very low level expression of CdtA after IPTG induction resulting in a binding-deficient phenotype. As it would not be feasible to recover significant quantities of gene product from these transformants for testing in the bioassays they were not studied further. The plasmid DNA inserts from the remaining 19 transformants were sequenced. Reliable DNA sequence data could not be retrieved, from two to three sequencing runs, from six of the 19 transformants. The remaining 13 transformants expressed a full length cdtA open reading frame (ORF). Two of these transformants (mutA65 and mutA112) each had two nucleotide changes. Only one of these nucleotide changes in both mutA65 and mutA112 resulted in an amino acid substitution. Eleven transformants had a single nucleotide change. Six of these transformants had a corresponding amino acid change. Two transformants (mutA241 and mutA249) had the same nucleotide change (A655G) and corresponding amino acid substitution (T219A). The mutants are listed in Table 3. The cdtA ORFs from seven of the transformants that exhibited binding in the CELISA equivalent to that of the wild type (pJDA9) were also sequenced. No nucleotide changes were found.
Table 3.
Summary of point mutations.
| Mutant designation | Nucleotide change | Amino acid change | Position in H. ducreyi CdtA | Comments |
|---|---|---|---|---|
| mutA62 | A54C | None | – | |
| mutA65 | A198T/T589A | None/C197S | C198 | |
| mutA79 | G348A | None | – | |
| mutA81 | T452A | S165N | S166 | |
| mutA99 | T264A | None | – | |
| mutA112 | T128C/A246G | F43S/none | P43 | |
| mutA121 | A121G | T41A | I41 | |
| mutA183 | G177A | None | – | |
| mutA221 | T534G | C178W | C179 | |
| mutA238 | C142T | None | – | |
| mutA239 | C166A | P56Q | S56 | |
| mutA241 | A655G | T219A | T220 | |
| mutA249 | A655G | T219A | T220 | Same as mutA241 |
| mutC41 | A146T/A261G/ | Q49L/none | NA | |
| T430C/A517T/ | None/S173C |
NA, not applicable.
The hydropathies of the mutant CdtA-His6 proteins were compared with that of the wild-type protein. Hydropathy was increased in the region surrounding the substituted residues in mutA121T41A and mutA241T219A Hydropathy was decreased in the regions surrounding the substituted residues in mutA65C197S, mutA81S165N, mutA112F43S, mutA221C178W and mutA239P56Q The calculated pI of wild-type CdtA-His6 was 8.2164. The pI of mutA65C197S and mutA221C178W was 8.3192. All the other mutant proteins had a pI identical to that of the wild type.
Effect of cdtA point mutations on subunit binding
Wild-type CdtA-His6 demonstrated saturation binding kinetics with CHO cells in the CELISA (Fig. 1A). No difference was observed between the binding of CdtA labelled at either the amino- or carboxy-terminal end of the protein (data not shown). CdtA-His6 was isolated by affinity chromatography from the seven mutants (Table 3) that had confirmed unique single amino acid substitutions. Binding of the isolated mutant proteins to CHO cells was measured in the CELISA and compared with that of the wild-type CdtA-His6 (Fig. 1B). All seven mutant proteins exhibited a statistically significant reduction in binding (P < 0.005 for six mutants, P < 0.01 for one mutant). CdtA-His6 was also isolated from one of the mutants (mutA99) that had a single nucleotide change (T264A) but no corresponding amino acid substitution (Table 3). No statistically significant reduction in binding, relative to the wild-type CdtA-His6, was observed with the gene product from this mutant (Fig. 1B). The analysis of mutA99 demonstrated that using crude bacterial extracts in the CELISA can give false positive results and explains why CdtA from five of 11 binding-deficient clones had no amino acid substitutions.
Fig. 1.

Effect of point mutations on CdtA binding to CHO cells. Affinity-purified CdtA mutant proteins were incubated with CHO cells in a CELISA. A. Saturation curves of wild-type recombinant CdtA-His6, CdtB-His6, CdtC-His6 and total soluble protein from E. coli BL-21(DE3) (pET15b) in a CELISA. Bound Cdt protein subunit was detected with anti-HisTag monoclonal antibody (1:3000 dilution) and anti-mouse IgG horseradish peroxidase conjugate (1:3000 dilution). B. Recombinant CdtA-His6 mutant proteins (1 μg well−1) were incubated with CHO cells in a CELISA. Bound protein was detected as in the experiment in A and compared with wells containing wild-type CdtA-His6, CdtB-His6 and CdtC-His6. Amino acid substitutions are shown for each mutant. All samples were run in triplicate. Statistically significant differences between the mutants and wild-type CdtA-His6 are marked with asterisks (P < 0.005*, P < 0.01**). Results are representative of three trials.
A second binding assay based on immobilized fucose-containing glycoproteins in an ELISA was used to support the CELISA results. Cdt subunit protein binding to thyroglobulin was similar to that observed with CHO cells. Wild-type CdtA-His6 demonstrated saturation kinetics (Fig. 2A). Wild-type CdtB-His6 and CdtC-His6 did not bind and bound poorly respectively. All seven CdtA mutant proteins failed to bind to thyroglobulin (P < 0.005; Fig. 2B). Binding of CdtA-His6 from mutA99 again was not statistically different than that of the wild-type protein.
Fig. 2.
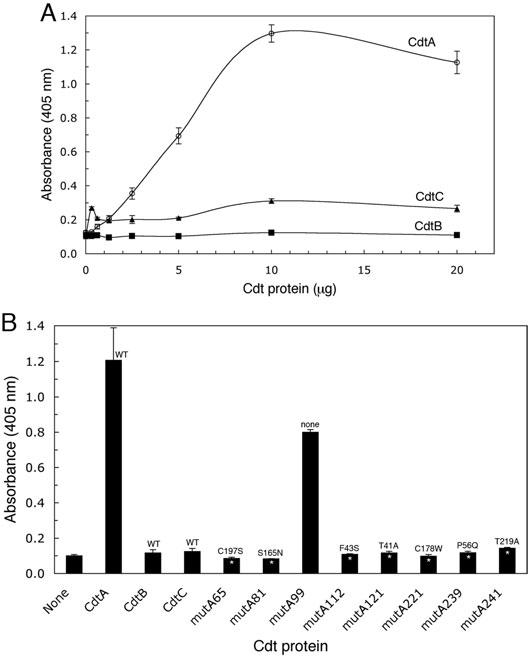
Effect of point mutations on CdtA binding to thyroglobulin. A. Saturation curves of the binding of wild-type recombinant CdtA-His6, CdtB-His6, CdtC-His6 to thyroglobulin-coated wells in ELISA. Bound protein was detected as described in the legend to Fig. 1. B. Affinity-purified recombinant CdtA-His6 mutant proteins (10 μg well−1) were added to wells coated with thyroglobulin. Bound protein was detected as in the experiment in A and compared with wells containing 10 μg well−1 of wild-type CdtA-His6, CdtB-His6 and CdtC-His6. Amino acid substitutions are designated for each mutant. All samples were run in triplicate. Statistically significant differences between the absorbance values for mutant and wild-type CdtA-His6 are marked with an asterisk (P < 0.005). Results are typical of three trials.
Effect of cdtA point mutations on the activities of reconstituted holotoxin
Reconstituted wild-type holotoxin exhibited a dose dependent effect on the proliferation of CHO cells (Fig. 3A). A TD50 was obtained with 1 μg ml−1 of reconstituted holotoxin. Reconstituted holotoxins were prepared in a 1:1:1 ratio using the mutant CdtA-His6 tagged protein subunits and wild-type subunits CdtB-His6 and CdtC-His6. Cultures of CHO cells were treated with the various mutant CdtA-containing holotoxins for 36 h and cell survival was compared with that of cultures treated with holotoxin reconstituted with the wild-type CdtA-His6 (Fig. 3B). Holotoxins made with protein from four of the mutants (mutA65, mutA112, mutA239 and mutA241), and wild-type holotoxin (CdtA-H), inhibited proliferation of CHO cells by 95–100%. Holotoxin prepared with CdtA-His6 from mutA81 reduced CHO cell proliferation by 60%. Holotoxins reconstituted with protein from mutA121 and mutA221 had no detectable effect on proliferation (100% of the input cells survived). CHO cells were also treated with double the concentration (5.0 μg ml−1) of holotoxins reconstituted with protein from mutA81, mutA121 and mutA221 (Fig. 3B, bars labelled 5). The difference in the results were negligible. Holotoxin made with protein from mutA99 (no amino acid substitution) inhibited CHO cell proliferation by 100%. To demonstrate that these results were not an artifact or anomaly associated with CdtA, holotoxins were reconstituted with protein from both mutA81 and mutC41 as well as with wild-type CdtA-His6 and the CdtC-His6 protein from mutC41. The cdtC gene in mutC41 contains four nucleotide changes, two of which have corresponding amino acid substitutions (Q49L and S173C) in the gene product. Both of these hybrid holotoxins had no effect on CHO cell proliferation (Fig. 3B).
Fig. 3.
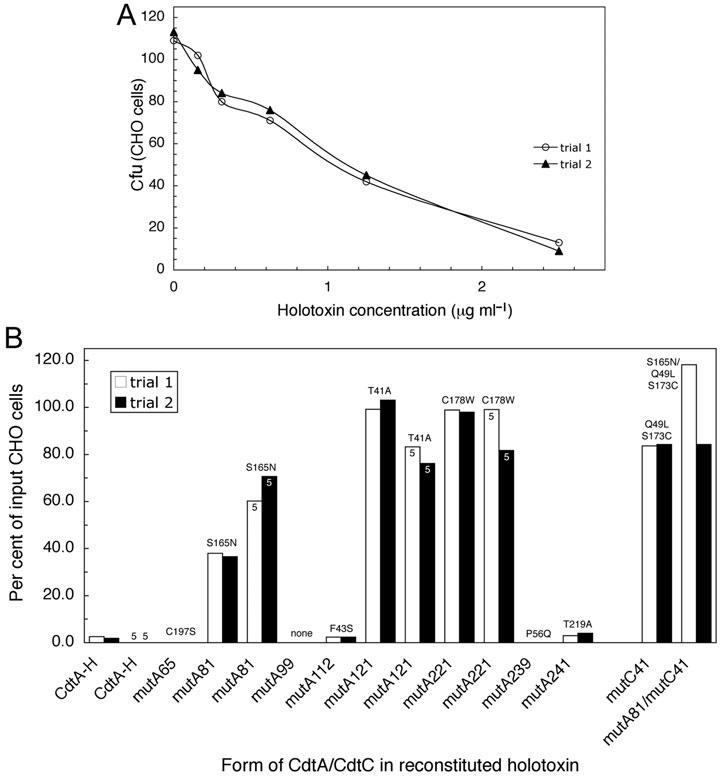
Effect of point mutations in CdtA on the proliferation of CHO cells treated with reconstituted holotoxin. A. Wild-type recombinant CdtA-His6, CdtB-His6 and CdtC-His6 (1:1:1 by weight) were preincubated in folding buffer and increasing concentrations of the mixture was added to CHO cell cultures. Cell colonies were stained and counted after 6 days and the data expressed as cfu. B. Holotoxin was reconstituted as in the experiment in A except that recombinant CdtA-His6 mutant proteins were substituted for wild-type CdtA-His6. In some samples a CdtC-His6 mutant (mutC41) was substituted for wild-type CdtC-His6. Amino acid substitutions are shown for each mutant. MutC41 contained two amino acid substitutions. CdtA-H designates holotoxin reconstituted with wild-type CdtA-His6. All samples were run twice in single wells. Twice the amount of CdtA-His6 protein (5 μg ml−1) was used to treat some cultures. Cfu were obtained as in A and the data is expressed as the percent of input CHO cells.
To further support the observation that several of the CdtA-His6 mutant proteins yielded holotoxins that failed to exhibit biological activity, CHO cell cultures were treated with wild-type and mutant CdtA-containing holotoxin and examined by flow cytometry. CHO cell cultures treated for 36 h with increasing concentrations of wild-type holotoxin showed a dose dependent increase in the percent of cells having a 4n DNA content indicative of arrest at the G2/M phase (Fig. 4A panels a–e). Greater than 90% of the cells (1 × 106 cells treated) had a 4n DNA content when exposed to 10 μg ml−1 of wild-type holotoxin (Fig. 4B). A TD50 was obtained with approximately 4 μg ml−1 of reconstituted holotoxin. FACS analysis of CHO cell cultures treated with holotoxin reconstituted with each of the mutant CdtA-His6 protein subunits gave results that paralleled exactly those of the proliferation assay (Fig. 4C). The percentage of CHO cells having a 4n DNA content following exposure to holotoxins made with mutant CdtA-His6 protein from mutA81 (panel b), mutA121 (panel e) and mutA221 (panel f) was 44, 10 and 15, respectively, compared with 66% for cells treated with wild-type toxin (Table 4). Exposure of CHO cells to equivalent concentrations of holotoxins made with mutA65, mutA112, mutA239 and mutA241 CdtA-His6 proteins yielded 70–79% of the cell population with a 4n DNA content. Only 15% of the cell population had a 4n DNA content following treatment with holotoxin made with wild-type CdtA-His6 and CdtB-His6 and cdtC gene product from mutC41 (panel i).
Fig. 4.

Effect of point mutations in the cdtA gene on the cell cycle of CHO cells treated with reconstituted holotoxin. A. CHO cell cultures (1 × 106) were treated with 0, 2.5, 5.0, 10.0 and 25.0 μg of total holotoxin protein ml−1. Holotoxin was reconstituted from wild-type CdtA-His6, CdtB-His6 and CdtC-His6 (1:1:1) for 36 h post intoxication. DNA profiles obtained by FACS analysis of propidium iodide stained nuclei are shown (solid line). G0/G1 (2n) and G2/M (4n) (solid filled) and S (diagonal line filled) peaks where determined by computer analysis. B. Dose–response curve of the percentage of cells having a 4n DNA content after treatment with each holotoxin concentration. C. CHO cell cultures were treated as in A except that 10 μg of total mutant CdtA or CdtC holotoxin protein ml−1 was used. Panels a, mutA65; b, mutA81; c, mutA99; d, mutA112; e, mutA121; f, mutA221; g, mutA239; h, mutA241; i, mutC41. Results are representative of two experiments.
Table 4.
Cell cycle analysis of CHO cells treated with reconstituted mutant holotoxin.
| Form of CdtA | Diploid G1 (2n) (%) | Diploid G2 (4n) (%) | Diploid S (%) | Coefficient of variance (%) |
|---|---|---|---|---|
| none | 75.35 | 5.93 | 18.72 | 2.71 |
| CdtA-H | 15.42 | 66.00 | 18.59 | 4.18 |
| mutA65 | 4.79 | 70.85 | 24.37 | 9.87 |
| mutA81 | 36.44 | 43.91 | 19.65 | 3.34 |
| mutA99 | 8.71 | 78.14 | 13.16 | 5.04 |
| mutA112 | 9.81 | 79.11 | 11.08 | 4.22 |
| mutA121 | 73.74 | 9.92 | 16.34 | 3.32 |
| mutA221 | 68.04 | 14.86 | 17.10 | 3.73 |
| mutA239 | 4.90 | 79.28 | 15.81 | 5.99 |
| mutA241 | 7.97 | 79.25 | 12.79 | 4.32 |
| mutC41 | 66.03 | 15.43 | 18.54 | 3.78 |
It has been shown that wild-type or properly assembled holotoxin has diminished in vitro DNA nicking activity presumably due to inhibition of CdtB by the N-terminal tail of correctly assembled CdtC (NeŠić et al., 2004). Taking advantage of this semi-quantitative approach, holotoxins reconstituted with recombinant wild-type CdtA-His6 and the CdtA-His6 mutant proteins were examined for the ability to convert supercoiled plasmid DNA to linear and relaxed forms. Holotoxin composed of wild-type subunit proteins had no supercoiled DNA nicking activity relative to CdtB-His6 alone (Fig. 5A). The nicking activity of CdtB-His6 was not affected when this subunit was mixed with either CdtA-His6 or CdtC-His6. CdtA-His6 and CdtC-His6 proteins, alone and in combination, did not display DNA nicking activity. Holotoxins prepared with mutant CdtA-His6 proteins from mutA121 and mutA221 converted all the supercoiled DNA to relaxed and linear forms (Fig. 5B). This was comparable to the nicking activity observed with the free CdtB-His6 subunit and was suggestive of altered holotoxin reconstitution. Holotoxins made with mutant CdtA-His6 proteins from mutA65, mutA81, mutA112, mutA239 and mutA241 showed varying degrees of inhibition of DNA nicking activity. These data indicated that holotoxin reconstitution was altered but not to the extent that CdtB DNA nicking activity was completely inhibited. As expected, the nicking activity of holotoxin made with protein from mutA99 (no amino acid substitution) was inhibited indicative of proper (wild-type) holotoxin assembly. Holotoxin made with mutC41 protein did not exhibit DNA nicking activity (Fig. 5B). However, when both mutA81 and mutC41 were used to make reconstituted holotoxin all of the supercoiled DNA was converted to relaxed form. These results with the CdtA-His6 mutant proteins followed the pattern of inhibition or loss of inhibition of activity observed in the other functional assays.
Fig. 5.

Effect of point mutations in the cdtA gene on the in vitro DNA nicking activity of reconstituted holotoxin. A. Wild-type Cdt subunit proteins alone and in various combinations were incubated with pBluescript II SK(+) DNA under conditions optimized for CdtB-His6 nuclease activity (Experimental procedures). B. Holotoxin prepared with protein from each of the CdtA-His6 mutants and from mutC41 was incubated with plasmid DNA as in A. Samples were examined on 0.8% agarose gels stained with ethidium bromide. Supercoiled (S), linear (L) and relaxed (R) forms of DNA are labelled. Results are representative of three experiments.
Two additional methods were used to determine if the mutant CdtA-His6 proteins formed reconstituted holotoxin. In the first method, the thyroglobulin ELISA, shown in Fig. 2 that is based on the binding of CdtA to fucose-containing glycoproteins (McSweeney and Dreyfus, 2005), was used to evaluate the ability of the mutant proteins to compete with wild-type CdtA-His6 for binding to CdtB-His6 and CdtC-His6. Results from this assay using combinations of wild-type subunit proteins showed that the binding of the subunits was specific and additive (Fig. 6A). An increase in the absorbance following binding of CdtB-His6 to CdtA-His6 in the absence of CdtC-His6 was not statistically significant (absorbance ratio of 1.2). However, the increase in absorbance following binding of CdtC-His6 to CdtA-His6 in the absence of CdtB-His6 was significant (P < 0.01) and had an absorbance ratio of 1.5. When all three subunit proteins were combined the increase in absorbance was statistically significant (P < 0.005) and the absorbance ratio was 3.0. These results demonstrated that all three subunits were bound and that CdtA and CdtC binding was required for binding of CdtB. In competition experiments CdtA-His6 from mutA99, mutA112, mutA121 and mutA239 showed no increase in absorbance (absorbance ratios 1.0–1.4) relative to wild-type CdtA alone suggesting that these mutant proteins formed holotoxin (Fig. 6B). CdtA-His6 from mutA65, mutA81, mutA221 and mutA241 had absorbance ratios below 3.0 (1.8–2.6) suggesting that they may form aberrant or partial holotoxin complexes.
Fig. 6.
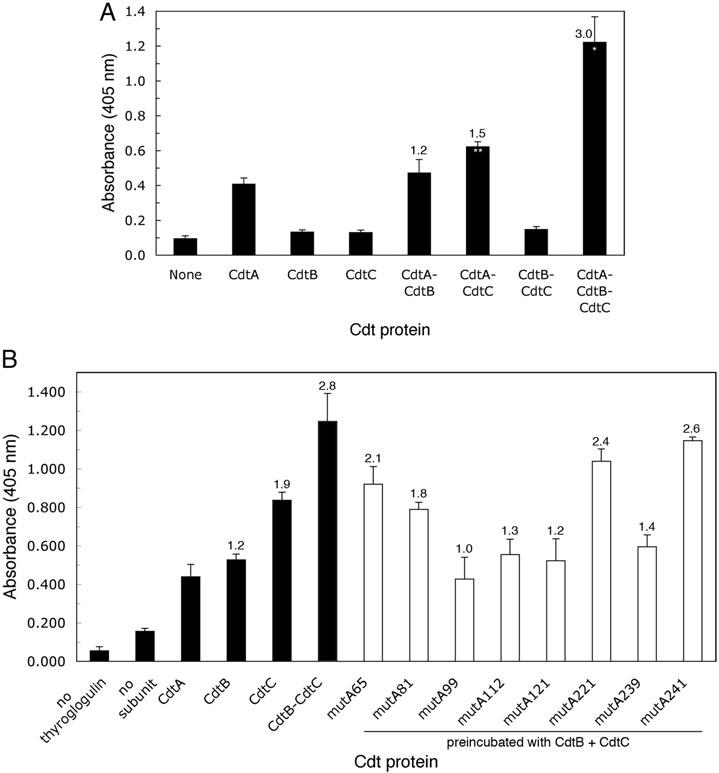
Competitive binding of mutant CdtA-His6 proteins to CdtB-His6 and CdtC-His6. A. Subunit, heterodimer and heterotrimer binding to thyroglobulin. ELISA plates were coated with thyroglobulin followed by the addition of individual or a combination of wild-type subunit proteins. B. Competition assay. ELISA plates were coated with thyroglobulin followed by 4 μg well−1 of wild-type CdtA-His6. Individual wild-type CdtB-His6 and CdtC-His6 subunit proteins, a preincubated mixture of CdtB-His6 with CdtC-His6 and preincubated mixtures of the mutant CdtA-His6 proteins with wild-type CdtB-His6 and CdtC-His6 were added to the plates. The plates were developed and the absorbance determined as described in the legend to Fig. 1. All samples were run in triplicate. Absorbance ratios based on the binding of wild-type CdtA-His6 to thyroglobulin were calculated as described in Experimental procedures. A statistically significant increase in the absorbance values for the homodimers and heterotrimer relative to that of wild-type CdtA-His6 are marked with an asterisks (P < 0.005*, P < 0.01**). Results are representative of two experiments.
A second assay for reconstitution was based on the size-dependent retention of reconstituted holotoxin (estimated molecular weight of 77 kDa) in dialysis tubing that has a 100 kDa molecular weight cut-off. Holotoxin was reconstituted with wild-type and mutant CdtA-His6 proteins and then dialysed for 48 h at 4°C. The material retained in the dialysis bag was then examined on Western blots. Figure 7A shows that the individual Cdt subunit proteins (estimated molecular weights of 21–31 kDa) and preincubated mixtures of wild-type CdtA-His6 and CdtC-His6 (heterodimer) were not retained following dialysis. In contrast, all three wild-type subunit proteins were detected in the dialysis bag, on Western blots, when reconstituted holotoxin (heterotrimer) was used. All three subunit proteins were retained following dialysis (approximately 100% of each protein retained) when CdtA-His6 from mutA121 was substituted for the wild-type protein (Fig. 7B). This result supported heterotrimer formation by this mutant CdtA protein. However, when CdtA-His6 from mutA221 was used to form holotoxin greater than 75% of CdtB and CdtC and 46% of CdtA were lost from the bag following dialysis (Fig. 7B). Approximately 38%, 31% and 64% of CdtA, CdtB and CdtC, respectively, were not retained when CdtA-His6 from mutA81 was used to make holotoxin. Reconstituted mixtures containing the other CdtA-His6 mutant proteins, which were all active in the bioassays, were trapped in the dialysis bag (data not shown). These results were in good agreement with those from the competition assay. A mixture of CdtA-His6 from mutA121, CdtC-His6 from mutC41 and wild-type CdtB-His6 was not trapped following dialysis indicating that a holotoxin complex was not formed by this combination of mutant subunits.
Fig. 7.

Effect of point mutations in the cdtA gene on the reconstitution of holotoxin. Individual wild-type subunit proteins and heterotrimers composed of preincubated mixtures of either wild-type or mutant CdtA-His6 proteins were dialysed for 48 h. The protein composition of the material in the dialysis bag, before and after dialysis, was then examined by Western blotting as described in the Experimental procedures. BD, before dialysis; AD, after dialysis.
Mapping amino acid substitutions that alter CDT activity in the holotoxin structure
The amino acid sequences of CdtA from A. actinomycetemcomitans and H. ducreyi are greater than 91% similar (Cope et al., 1997; Sugai et al., 1998; Mao and DiRienzo, 2002). Figure 8 shows an alignment of the A. actinomycetemcomitans and H. ducreyi CdtA (GenBank Accession number U53215) deduced amino acid sequences. A single gap was introduced in the A. actinomycetemcomitans CdtA sequence at the position of amino acid residue 64 to obtain the alignment. There are 18 conserved amino acid differences between the A. actinomycetemcomitans and H. ducreyi native CdtA. Three of the A. actinomycetemcomitans CdtA mutations (mutA121, mutA112 and mutA239) were at positions that had conserved amino acid differences relative to the H. ducreyi CdtA. The other four CdtA mutations were at positions that were identical in the two protein sequences. However, due to the gap in the A. actinomycetemcomitans sequence at position 64 the H. ducreyi sequence is out of register by one amino acid after this position. Two of the mutants (mutA65 and mutA221) contained a substitution for a cysteine residue at positions C179 and C198, respectively, in the A. actinomycetemcomitans sequence.
Fig. 8.

Alignment of A. actinomycetemcomitans recombinant CdtA-His6 (pJDA9), H. ducreyi CdtA (GenBank Accession number U53215) and CdtA-His6 mutant deduced amino acid sequences (Table 3). Identical residues are boxed. The positions of the four amino acid substitutions in the aromatic patch (ap) mutant described in reference 24 are marked. Amino acid resides 18–56 were disordered in the crystal structure (NeŠić et al., 2004). The arrow marks the amino-terminal end of the 17–18 kDa form of CdtA identified in several studies (Frisk et al., 2001; Saiki et al., 2004; Shenker et al., 2004). The solid line between the two sequences marks the region conserved among the various CdtA proteins (Lee et al., 2003).
Recent publication of the crystal structure of the H. ducreyi Cdt (NeŠić et al., 2004) provided a model for mapping the positions of the substituted residues that alter CdtA binding and holotoxin activity. The crystal structure of the H. ducreyi Cdt is shown in Fig. 9. Panels A–C illustrate the relationship of the CdtA, CdtC and CdtB subunits respectively. The seven unique mutations in the A. actinomycetemcomitans CdtA are mapped on the structure (Fig. 9D). The three mutants mutA121, mutA112 and mutA239, with amino acid substitutions corresponding to the H. ducreyi CdtA positions I41, P43 and S56, respectively, contained substituted residues at the amino terminal end of the protein that was not resolved in the crystal structure. All three of these mutants had a binding-deficient phenotype and were the strongest competitors (absorbance ratio ≈ 1) of wild-type CdtA binding to CdtB and CdtC. Only one of these amino terminal point mutations (mutA121T41A), corresponding to position I41 (underlined) in the H. ducreyi CdtA protein, also abolished proliferation inhibition activity and the ability to send cells into G2/M arrest when incorporated into reconstituted holotoxin. The other four mutations (mutA65C197S, mutA81S165N, mutA221C178W and mutA241/249T219A) were closer to the carboxy-terminal end of the CdtA protein. All of these mutants had a binding-deficient phenotype and competed (absorbance ratios 1.8–2.6) against wild-type CdtA for binding to CdtB and CdtC. Two of the carboxy-terminal point mutations (mutA81S165N and mutA221C178W), corresponding to positions S166 and C179 (underlined), respectively, in the H. ducreyi CdtA protein, resulted in a reduction or loss of proliferation inhibitory activity and the ability to send cells into G2/M arrest. All seven unique mutations reside outside of the aromatic patch region (Fig. 9E).
Fig. 9.

Location of the amino acid substitutions of the CdtA-His6 mutant proteins in the crystal structure of the H. ducreyi Cdt. The coordinates (Protein Data Bank Accession number 1SR4) for the crystal structure (NeŠić et al., 2004) were computer modelled. A–C. Orientation of the CdtA, CdtC and CdtB subunits respectively. The amino-(N) and carboxy-(C) terminal residues in CdtA and CdtC and the theoretical active site in CdtB are labelled. D. Locations of the seven unique amino acid substitutions (black text) in the A. actinomycetemcomitans recombinant CdtA-His6 mutants that affected binding are shown at the positions of the corresponding residues in the H. ducreyi sequence. The positions of the three substitutions that resulted in a non-cytotoxic phenotype are underlined. The CdtA used to crystalize the H. ducreyi Cdt contained residues 18–223 to eliminate a putative secretory signal sequence (NeŠić et al., 2004). Residues 18–56 were disordered in the crystal structure. The two amino acid substitutions in the CdtC-His6 mutant (mutC41) are shown in red. E. Location of the four amino acid substitutions in the aromatic patch (AP) region mutant of NeŠić et al. (2004). F. Locations of the four cysteine residues in CdtA. These cysteines are conserved in the A. actinomycetemcomitans and H. ducreyi CdtA.
Discussion
A number of investigations have provided evidence implicating the CdtA and CdtC subunits in the binding of Cdt to cells. Frisk et al. (2001) found all three recombinant Cdt proteins absorbed to the surface of HEp-2 cells. However, a 17 kDa form of CdtA was the predominant bound species. Direct evidence of CdtA binding to cells was obtained from immunofluorescence experiments (Mao and DiRienzo, 2002; Akifusa et al., 2005; McSweeney and Dreyfus, 2005; Kanno et al., 2005) and CELISA (Lee et al., 2003). These studies examined the ability of purified recombinant CdtA subunit protein and reconstituted holotoxin to bind to CHO and HeLa cells respectively. Deng and Hansen (2003) detected CdtC attached to HeLa cells in cultures treated with a CdtA–CdtC complex. However, they could not detect bound CdtC when the same cells were treated with this subunit alone. McSweeney and Dreyfus (2005) showed that CdtA and CdtC bound to agarose bead-immobilized fucose-containing glycoproteins, such as thyroglobulin. We observed that CdtA-His6 bound with saturation kinetics to thyroglobulin-coated ELISA plates (this study). However, CdtC-His6 bound very poorly under the same conditions unless added to the plates together with CdtA-His6.
Although it is now well established that all three Cdt subunits are required for the toxin to manifest its cell inhibitory effects in vivo (Pickett et al., 1994; Sugai et al., 1998; Deng et al., 2001; Frisk et al., 2001; Lara-Tejero and Galán, 2001; Saiki et al., 2001; Mao and DiRienzo, 2002), several studies have shown that CdtB and CdtC reconstituted in vitro manifests biological activity. HEp-2 cells exposed to a recombinant A. actinomycetemcomitans Cdt reconstituted with CdtB and CdtC were arrested at the G2/M phase of the cell cycle (Akifusa et al., 2001). This result was confirmed with purified recombinant C. jejuni Cdt subunit proteins and HeLa cells (Lee et al., 2003). However, cell cycle arrest was reduced by 75% when reconstituted holotoxin lacked CdtA. It seems clear that there is an interaction between CdtA and either or both CdtC and CdtB subunits that is required to produce a holotoxin that has optimal biological activity (Deng and Hansen, 2003; Lee et al., 2003; NeŠić et al., 2004; Shenker et al., 2005). CdtA and CdtC may compete for binding to the same fucose-containing cell surface receptor (McSweeney and Dreyfus, 2005) supporting the possibility that these two subunits act in concert in vivo to bind toxin to the cell surface. In addition, CdtC appears to be required for internalization of CdtB (Akifusa et al., 2005). However, the precise mechanisms by which CdtA and CdtC each contribute to toxin binding have not yet been clarified.
Deletion mutants and site-directed mutagenesis methods have been employed to provide more detailed information about the binding specificity of CdtA. An in-frame deletion in the C. jejuni CdtA, removing amino acids 126–168, did not affect the ability of the reconstituted toxin to arrest HeLa cells, did not reduce binding of the purified subunit protein to these cells in CELISA and competed for binding to these cells with holotoxin containing wild-type CdtA (Lee et al., 2003). This 43 amino acid region is conserved among CdtA proteins in various bacterial species. Apparently, deletion of this conserved region does not affect the biological activity of the toxin. In another study, a series of in-frame deletion mutants lacking amino acids 19–38, 19–47, 19–57, 19–77 and 19–88 was constructed (Saiki et al., 2004). Deletion mutants CdtAΔ19–38 and CdtAΔ19–47 did not affect the ability of the CdtA mutant proteins to make fully active holotoxin. NeŠić et al. (2004) used site-directed mutagenesis to make substitutions for four aromatic amino acids (W91G, W98G, W100G and W102A), in a single mutant, in the aromatic patch region of the H. ducreyi CdtA. Recombinant Cdt made with this aromatic patch mutant failed to arrest HeLa cells at the G2/M phase of the cell cycle but did not disrupt stability of the holotoxin as measured by gel filtration chromatography.
In the current study we employed a random mutagenesis strategy to construct a library of point mutations in cdtA to assess the contribution of individual amino acids to the binding activity of the subunit and to the ability of the subunit to form active holotoxin. A random mutagenesis strategy rather than a targeted strategy was used initially because, except for the aromatic patch region, no other binding or functional domains have been identified in CdtA. We obtained seven CdtA mutants, with unique point mutations, that produced subunit proteins that had dramatically reduced binding to CHO cells and thyroglobulin. Three of these mutations (mutA121T41A, mutA112F43S and mutA239P56Q) resulted in amino acid substitutions in the amino-terminal region of the protein (corresponding to H. ducreyi CdtA positions I41, P43 and S56, respectively, see Fig. 8). This region (residues 18–56) is disordered in the H. ducreyi Cdt crystal structure so it is difficult to make predictions about the relationship between the locations of the amino acid substitutions and binding activity (NeŠić et al., 2004 and Fig. 9D. It has been proposed that the amino-terminus of CdtA inserts deep in the groove formed between CdtA and CdtC in the crystal structure and may mimic a receptor peptide (NeŠić et al., 2004). However, it is possible that the amino terminus of CdtA is important for formation of the groove and therefore may be essential for recognition of the cell surface receptor. If that is the case the amino acid substitutions in mutA121T41A, mutA112F43S and mutA239P56Q may alter the ability of the amino-terminus to fold properly thus changing or masking the CdtA face of the groove. We did not obtain binding-deficient mutants with amino acid substitutions at positions prior to residue 41. This is in good agreement with the previous observation that in-frame deletion of residues 19–47 did not alter toxin activity (Saiki et al., 2004). In that study a dramatic loss of Cdt activity was observed with the next largest deletion in the series (residues 19–57). It is not surprising that deletion of the amino-terminus up to amino acid 47 does not affect Cdt activity because there are indications that CdtA is post-translationally processed by both A. actinomycetemcomitans and E. coli (Sugai et al., 1998).
The other four binding-deficient mutants (mutA81S165N, mutA221C178W, mutA65C197S and mutA241/249T219A) contained amino acid substitutions toward the carboxy-terminal region of the protein (corresponding to H. ducreyi CdtA positions S166, C179, C198 and T220, respectively, see Fig. 8). No mutants containing amino acid substitutions in the aromatic patch region were obtained (see Fig. 9D and E). The reason for this result is most likely due to our screening method which selected mutants that had a CdtA binding-deficient phenotype. The aromatic patch mutant reported by NeŠić et al. (2004) lost the ability to arrest HeLa cells but not to affect the ability of the mutant protein to form holotoxin. Our data show that single amino acid substitutions outside of the aromatic patch region can affect the binding of free CdtA to cells and to fucose-containing glycoprotein. Several of these mutations affect but do not abolish the ability of CdtA to bind to the other subunits. Two of these mutations (mutA65C197S and mutA221C178W) replaced cysteine residues (corresponding to H. ducreyi CdtA positions C179 and C198, respectively, see Figs 8 and 9F).
Three (mutA81S165N, mutA121T41A and mutA221C178W) of the seven CdtA binding-deficient mutants exhibited significantly altered biological activities yet they apparently formed a holotoxin complex with CdtB and CdtC. These mutations contained substituted amino acids at positions I41 (mutA121T41A) in the amino-terminal region and S166 (mutA81S165N) and C179 (mutA221C178W) in the carboxy-terminal region of the H. ducreyi CdtA (see Figs 8 and 9D). The mutation in mutA121 is particularly interesting because the CdtA-His6 gene product was a strong competitive inhibitor of wild-type CdtA-His6 binding to CdtB-His6 and CdtC-His6 yet holotoxin made with this mutant protein lacked biological activity. The amino acid switch alanine for threonine in mutA121 was relatively conservative in terms of potential physical changes in the protein. There was a slight increase in the relative hydropathy around residue 41 of the altered gene product. However, the calculated pI of the mutant protein was identical to that of the wild-type protein. It is possible that the threonine in this position is important for the recognition of the N-linked fucose-containing cell surface receptor. In contrast, a switch from phenylalanine to serine at neighbouring position 43 (mutA112) did not affect the biological activity of the subunit. This was a more radical mutation in which a non-reactive amino acid replaced an aromatic residue.
MutA81 is also an interesting mutant because there were partial effects on the cell proliferation inhibition, cell cycle arrest and CdtB DNA nicking activities of holotoxin made with this CdtA-His6 protein. This partial loss of activity correlated with a reduced ability to form heterotrimer. There was a switch from serine to asparagine at position 165 in mutA81. The change from a neutral to acidic amino acid could be significant. However, there was no change in the calculated pI of the mutant protein compared with that of the wild-type protein. The mutation resulted in a slight decrease in the relative hydropathy surrounding this residue. Position 165 in CdtA in the H. ducreyi Cdt crystal structure is close to the end of the conserved region in CdtA proteins (Lee et al., 2003). Other than mutA81S165N, we did not find binding-deficient mutations with amino acid substitutions in this conserved region. These findings support the results obtained by Lee et al. (2003) with their 43 amino acid deletion mutant.
An amino acid substitution at position 178 in CdtA (mutA221) also resulted in the loss of holotoxin activity. This residue (C179) in the H. ducreyi crystal structure is not near the putative receptor groove between CdtA and CdtC nor does it appear to be in contact with the CdtB subunit. However, it is likely that an intrachain disulphide bridge between C179 and C198 was disrupted (see Fig. 9F). The substitution in mutA221 was a switch from cysteine to tryptophan. Not only was a reactive cysteine residue removed but it was replaced with a large heterocyclic amino acid. The amino acid switch increased the pI of the protein and decreased the hydropathy of the region around residue 178. However, it is interesting to note that a more reserved cysteine substitution in mutA65C197S did not affect the biological activity of CdtA. This mutant protein had a increased calculated pI (same as that of mutA221C178W) and reduced hydropathy relative to the wild-type protein. Based on the difference between the activities of holotoxins reconstituted with protein from mutA221C178W and mutA65C197S, it appears that a disulphide bridge in this region of CdtA may not be required for optimum Cdt activity. The loss of activity in holotoxin made with protein from mutA221C178W is more likely due to the introduction of a large reactive amino acid that changes the folding of CdtA so that it no longer forms a holotoxin complex. This is supported by the results of the competition experiments in which this mutant protein was a relatively weak competitive inhibitor of wild-type CdtA-His6 binding to the other subunits and the dialysis experiments.
In summary, we have identified point mutations in the cdtA gene of the A. actinomycetemcomitans Cdt that reduce or abolish binding of the subunit protein to CHO cells and fucose-containing glycoproteins such as thyroglobulin. These mutations resulted in amino acid substitutions that clustered at the amino- and carboxy-terminal regions of the protein. Three of these point mutations resulted in a statistically significant reduction or loss of biological activity of reconstituted holotoxin. Therefore, mutations that affect subunit binding to cells and receptor-like glycoproteins can reside outside of the aromatic patch region. Several of the point mutations resulted in gene products that formed holotoxin complexes that lost the ability to arrest cell growth. In the case of some, but not all of these mutations, the loss of activity could be due to the inability of the mutant protein to properly assemble. The next step is to use a site-directed mutagenesis approach to determine if these mutations reside in more extensive domains in CdtA critical for proper holotoxin assembly and binding to the cell surface.
Experimental procedures
Plasmids and oligonucleotide primers
Based on the sequence of the cdt locus of A. actinomycetemcomitans Y4 (Mayer et al., 1999; GenBank Accession number AF006830), synthetic oligonucleotide primer pairs (Table 1) were designed and used to amplify the three cdt gene sequences. Plasmid pCDT1 (Mayer et al., 1999), containing the cdtABC ORFs cloned in pBluescript II SK(+) (Stratagene, La Jolla, CA) was used as template DNA. Oligonucleotide primers were purchased from Integrated DNA Technologies (IDT, Coralville, IA). Codons for six histidine residues (His-tag) were added to the 3′-end of each CdtA, CdtB and CdtC gene sequence using the reverse primers (bases in bold type in Table 1). Products amplified by PCR were cloned into the NcoI and BamHI sites (underlined bases in Table 1) of pET15b (Novagen, EMD Biosciences, San Diego, CA) so that the His-tag in the vector was removed. The amplified DNA was used to transform competent E. coliDH5α [supE44 ΔlacU169 (ϕ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1] (Invitrogen, Carlsbad, CA). Transformants were selected on Luria–Bertani (LB) agar plates containing 75 μg ml−1 of ampicillin. Positive clones were identified by the restriction endonuclease digestion pattern. Plasmid DNA was then isolated from one clone containing each Cdt gene and transformed into E. coli BL21(DE3) [F− ompT hsdSB (rB− mB−) gal dcm (DE3)] competent cells (Novagen). The DNA sequence of the insert in the selected plasmids pJDA9, pJDB7 and pJDC2 (Table 2) was obtained to confirm that the cdt gene-containing DNA fragments were cloned in the proper orientation and that no mutations were introduced during PCR. Automated cycle sequencing reactions were conducted by the Genetics Core Facility at the University of Pennsylvania using an Applied Biosystems 377 sequencer with Dye Primer chemistry.
Construction and screening of a cdtA mutant library
The oligonucleotide primer pair cdtA-F/cdtA-R (Table 1) and pJDA9 DNA (Table 2) were used with the GeneMorph Random Mutagenesis Kit (Stratagene), according to the manufacturer's instructions, to generate a set of mutations, at low frequency, in the cdtA gene. Mutated cdtA DNA fragments were then cloned into the NcoI and BamHI sites of pET15b and transformed into Library Efficiency E. coli DH5α Competent Cells (Invitrogen). The transformants were pooled, the plasmid DNA extracted and used to transform E. coli BL21(DE3) competent cells. Individual colonies were transferred to master plates. Each transformant was grown in 10 ml of LB broth medium containing 75 μg ml−1 of ampicillin at 37°C with vigorous shaking. Isopropyl-β-D-thiogalactopyranoside (IPTG) (Sigma Chemicals, St. Louis, MO) was added to a final concentration of 1 mM when the cultures reached late logarithmic phase (OD600 = 0.8–1.0). The bacteria were grown an additional 4–5 h, collected by centrifugation (6000 g for 10 min), washed with PBS and lysed at 4°C by sonication (three bursts of 1 min duration at the low power setting; Braun-Sonic 2000, B. Braun Biotech, Allentown, PA). The sonicates were centrifuged at 6000 g for 15 min to collect inclusion bodies. Inclusion body pellets were suspended in 200 μl of PBS containing 6 M urea.
A CELISA, modified from that used by Lee et al. (2003), was used to evaluate binding of the cdtA mutants. Briefly, CHO-K1 cells (1.5 × 104 cells well−11) were used to inoculate 96-well plates (Mao and DiRienzo, 2002). The plates were incubated for 48 h at 37°C in an atmosphere containing 5% CO2. The plates were washed with PBS containing 1 mM each MgCl2 and CaCl2 and the cells fixed with 10% formalin for 15 min at room temperature. The inclusion body fractions from the individual cdtA mutant library transformants described above were mixed with 4 volumes of PBS containing 3% BSA (PBS-BSA). These preparations (100 μl) were added to wells in triplicate. The plates were incubated at room temperature for 1 h, washed three times with PBS and then fixed for 15 min at room temperature with 2% formaldehyde and 0.2% glutaraldehyde. The plates were then washed three times with PBS containing 0.1% Tween-20 (PBST) and incubated with PBS-BSA at room temperature for 1 h. A 1:3000 dilution of anti-HisTag monoclonal antibody (Novagen) in PBS-BSA was added to the wells and the plates incubated for 1 h at room temperature. The plates were washed three times with PBS-BSA and incubation was continued with a 1:3000 dilution of anti-mouse IgG horseradish peroxidase conjugate (Amersham Biosciences, Piscataway, NJ) for 1 h at room temperature. The cells were washed three times with PBST followed by the addition of 100 μl of horseradish peroxidase substrate ABTS-100 (Rockland, Gilbertsville, PA). The plates were read in a Perkin Elmer HTS 7000 Bio Assay Reader (Boston, MA) at a wavelength of 405 nm. Absorbance readings were compared with those obtained from triplicate wells containing inclusion body fractions from E. coli BL-21(DE3) (pJDA9) and E. coli BL-21(DE3) (pET15b). There was no detectable difference in CdtA-His6 binding to formalin fixed and non-fixed CHO cells (data not shown).
Expression of the cdtA gene in the individual transformants was assessed by Western blotting. Bacteria collected from 1 ml of IPTG-induced culture were mixed with 100 μl of gel loading buffer (2% SDS, 0.05 M Tris-HCl (pH 6.8), 10% glycerol, 0.01% β-mercaptoethanol), and heated in a boiling water bath for 5 min. Samples (20 μl per lane) were applied to a 10–20% polyacrylamide Tris-HCl Ready Gel (Bio-Rad Laboratories, Hercules, CA). Following electrophoresis, protein bands were transferred to a nitrocellulose membrane. The membrane was blocked with 5% skim milk in TBS buffer [20 mM Tris-HCl (pH 7.6), 0.8% NaCl] for 1 h at room temperature with shaking. The membrane was washed and then incubated with anti-HisTag monoclonal antibody (Novagen), diluted 1:3000 in TBS buffer, for 1 h at room temperature with shaking. The filter was washed and incubated for 1 h with horseradish peroxidase-conjugated anti-mouse IgG (Novagen) diluted 1:3000 in TBS buffer. Immunopositive protein bands were detected with an Enhanced Chemiluminescence Western Blotting Detection Kit (Amersham Pharmacia Biotech, Piscataway, NJ) according to the manufacturer's instructions. Prestained molecular weight standards were obtained from New England Biolabs (Beverly, MA). Lysates from E. coli BL-21(DE3) (pJDA9) and E. coli BL-21(DE3) (pET15b) were used as positive and negative controls respectively.
Plasmid DNA was obtained from the mutant library cdtA clones using a QIAprep Miniprep kit (QIAGEN, Valencia, CA). The plasmid DNA from each clone was digested with restriction endonucleases NcoI and BamHI according to the manufacturer's instructions. The digested samples were examined on 8% agarose gels to confirm the presence and size of the plasmid insert. DNA sequence analysis was performed by the Genetics Core Facility at the University of Pennsylvania as described above.
Isolation of recombinant wild-type His6-tagged CdtA, CdtB and CdtC and mutant CdtA subunit proteins
Recombinant clones E. coli BL-21(DE3) (pJDA9), E. coli BL-21(DE3) (pJDB7), E. coli BL-21(DE3) (pJDC2) and individual clones from the cdtA mutant library were used to prepare the three wild-type His-tagged Cdt protein subunits and mutated His-tagged CdtA proteins as described previously (Mao and DiRienzo, 2002). Yield was approximately 2–4 mg protein per 100 ml of culture. The final dialysed protein preparations were passed through 45 micron filters and quantified with the Micro BCA protein assay kit (Pierce, Rockville, IL). Purity was assessed by analysis on 10–20% polyacrylamide gels. Aliquots of the quantified protein samples were stored at −70°C in a buffer containing 10 mM Tris-HCl (pH 7), 100 mM NaCl, 5 mM MgCl2 and 5 mM imidazole.
Binding of recombinant wild-type His6-tagged CdtA, CdtB and CdtC and mutant CdtA subunit proteins to CHO cells
Saturation binding kinetics were determined for wild-type CdtA-His6, CdtB-His6 and CdtC-His6 using 0–1.25 mg of each protein per millilitre in triplicate in the CELISA. To assess binding activity purified His6-tagged wild-type and mutant proteins (1 μg well−1) were added to triplicate wells in the CELISA. The 96-well plates were prepared and processed as described above for screening of the transformants. These experiments were run a minimum of three times.
Reconstitution and activity of wild-type and mutant CdtA-containing holotoxin
Wild-type holotoxin and holotoxin containing mutant CdtA-His6 proteins were reconstituted as described previously (Mao and DiRienzo, 2002). The recombinant His6-tagged proteins were mixed in equivalent proportions of wild-type CdtA-His6 or mutated CdtA-His6 with wild-type CdtB-His6 and CdtC-His6 and incubated for 1 h at 4°C in reconstitution buffer (10 mM Tris-HCl (pH 7), 100 mM NaCl, 5 mM MgCl2). In some experiments holotoxin was reconstituted with a CdtC-His6 mutant protein. This mutant, mutC41, was obtained by the same methods used for creation of the CdtA mutant library (data not shown). This mutant had the fewest nucleotide changes identified in the CdtC library [four nucleotide changes and two amino acid substitutions (Q49L and S173C)].
Proliferation inhibition
To quantify the effects of reconstituted holotoxin on cell proliferation, CHO cells were seeded in six-well tissue culture plates (300 cells in 3 ml of medium per well in triplicate) as described previously (Mao and DiRienzo, 2002). Cell cultures were incubated for 4 h prior to addition of the toxin to allow the cells to attach to the plates. Holotoxin preparations made with wild-type and mutant CdtA-His6 proteins (2.5 or 5 μg of total protein ml−1) were added to the CHO cell cultures and the plates were incubated for 5–6 days to allow colonies to form. The medium was then removed and the cells were fixed with 10% formalin. After incubation for 5 min, the colonies were stained with crystal violet for 10 min, dried and counted. The number of colonies per well was expressed as colonyforming units (cfu). A dose–response curve for wild-type reconstituted holotoxin was also prepared using 0–2.5 μg of total protein ml−1. Due to limiting amounts of purified proteins samples were run in duplicate wells. These experiments were run twice.
Cell cycle analysis
Cell cycle arrest was determined by flow cytometry as described previously (Kanno et al., 2005). A dose–response curve was obtained with reconstituted wild-type holotoxin using 2.5, 5, 10 and 25 μg ml−1 of reconstituted wild-type holotoxin. CHO cells were treated with 10 μg ml−1 total protein of holotoxin made with mutant CdtA-His6 proteins. Cells were exposed to the toxin in culture for 36 h. Propidium iodide stained nuclei were analysed on an FACSCalibur flow cytometer at the University of Pennsylvania Cancer Center Flow Cytometry and Cell Sorting Shared Resource facility. The data from 30 000 events were analysed with ModFit 3.0 (Verity Software House, NH).
In vitro DNase activity
Supercoiled DNA nicking activity was determined, with minor modifications, as described previously (Elwell and Dreyfus, 2000; Mao and DiRienzo, 2002). Briefly, 1 μg of supercoiled pBluescript II SK(+) DNA was incubated with reconstituted holotoxin (500 ng of each subunit protein per reaction) in 25 mM HEPES (pH 7.0) containing 50 mM MgCl2 at 37°C for 1 h. Holotoxin was reconstituted with wild-type CdtB-His6 and CdtC-His6 and either wild-type CdtA-His6 or one of the CdtA-His6 mutant proteins as described above except that incubation was at room temperature for 1 h. In some reactions CdtC-His6 from mutC41 was used in place of the wild-type subunit.
Glycoprotein ELISA
A glycoprotein ELISA assay was designed based on the immobilized glycoprotein binding assay of McSweeney and Dreyfus (2005). Microtitre plates (96-well) were coated overnight with 75 μg well−1 of thyroglobulin (Sigma). The plates were washed three times with PBST followed by the addition of wild-type or mutant Cdt subunit proteins. All proteins were added to triplicate wells at a concentration predetermined for each experiment (described below). Plates were washed, treated with anti-His Tag monoclonal antibody and horseradish peroxidase conjugate and were processed as described above for the CELISA. Saturation binding kinetics were determined for wild-type CdtA-His6, CdtB-His6 and CdtC-His6 using 0–20 μg of each protein per well. In other experiments, various combinations of subunit proteins using 4 μg of CdtA-His6, 4.5 μg of CdtB-His6 and 3.5 μg of CdtC-His6 were added per well to assess holotoxin assembly. In a third set of experiments, 10 μg of each subunit protein and each CdtA-His6 mutant protein was added per well to examine the ability of the mutant proteins to bind to the glycoprotein. Competition experiments were performed by preincubating 4.5 μg of CdtB-His6 and 3.5 μg of CdtC-His6 with 4 μg of each mutant CdtA-His6 protein. Thyroglobulin-coated plates were incubated with 4 μg of wild-type CdtA-His6 protein well−1 and the plates were washed three times with PBST. The subunit protein mixtures were then added to the plates and the plates processed as described above. An absorbance ratio was calculated by dividing the average (triplicate wells) absorbance value of the CdtB-His6/CdtC-His6 protein or mutant CdtA-His6 protein wells by the average absorbance value of wells containing only bound wild-type CdtA-His6. The binding experiments were repeated two to three times and, except for the competition assay, were repeated with fetuin-coated plates.
Reconstitution (dialysis) assay
Dialysis membrane with a molecular weight exclusion limit of 100 kDa (Spectrum Laboratories, Rancho Dominguez, CA) was soaked and rinsed in distilled water. Each of the subunit proteins alone (100 μg of protein or mutant protein) or reconstituted heterotrimer (300 μg of total protein) were suspended in a total volume of 1 ml of refolding buffer containing 0.3 M urea. Each sample was dialysed against 800 ml of the same buffer for 48 h at 4°C with two buffer changes. An aliquot (35 μl) of each protein or reconstituted protein sample was then examined, before and after dialysis, on Western blots as described above. Bands on Western blots were quantified with ImageJ version 1.34 (http://rsb.info.nih.gov/ij/).
Computer analysis
The European Molecular Biology Open Software Suite (EMBOSS 2.9.0) (http://emboss.sourceforge.net) was used to obtain amino acid sequence alignments and to calculate isoelectric points (pI) and hydropathy (Kyte and Doolittle, 1982; Rice et al., 2000). Deduced amino acid sequences of wild-type and mutant A. actinomycetemcomitans recombinant CdtA-His6 and CdtC-His6 were from nucleic acid sequences obtained during this study. The H. ducreyi CdtA sequence was from GenBank (Accession number U53215).
The crystal structure of the H. ducreyi Cdt (NeŠić et al., 2004) was modelled in the software program iMol 0.30 (http://www.pirx.com/iMol/). Coordinates were obtained from the Protein Data Bank (Accession number 1SR4).
Statistical methods
Mean values and standard deviations were plotted where appropriate. The paired t-test was used to evaluate the data in some experiments.
Acknowledgements
The authors gratefully acknowledge the able technical assistance of Suqing Fan. This work was supported by USPHS Grant DE12593 from the National Institute of Dental and Craniofacial Research.
References
- Akifusa S, Poole S, Lewthwaite J, Henderson B, Nair SP. Recombinant Actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect Immun. 2001;69:5925–5930. doi: 10.1128/IAI.69.9.5925-5930.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akifusa S, Heywood W, Nair SP, Stenbeck G, Henderson B. Mechanism of internalization of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiology. 2005;151:1395–1402. doi: 10.1099/mic.0.27671-0. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope LD, Lumbley S, Latimer JL, Klesney-Tait J, Stevens MK, Johnson LS, et al. A diffusible cytotoxin of Haemophilus ducreyi. Proc Natl Acad Sci USA. 1997;94:4056–4061. doi: 10.1073/pnas.94.8.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Bratti X, Frisan T, Thelestam M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon. 2001a;39:1729–1736. doi: 10.1016/s0041-0101(01)00159-3. [DOI] [PubMed] [Google Scholar]
- Cortes-Bratti X, Karlsson C, Lagergard T, Thelestam M, Frisan T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J Biol Chem. 2001b;276:5269–5302. doi: 10.1074/jbc.M008527200. [DOI] [PubMed] [Google Scholar]
- Deng K, Hansen EJ. A CdtA–CdtC complex can block killing of HeLa cells by Haemophilus ducreyi cytolethal distending toxin. Infect Immun. 2003;71:6633–6640. doi: 10.1128/IAI.71.11.6633-6640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng K, Latimer JL, Lewis DA, Hansen EJ. Investigation of the interaction among the components of the cytolethal distending toxin of Haemophilus ducreyi. Biochem Biophys Res Commun. 2001;285:609–615. doi: 10.1006/bbrc.2001.5223. [DOI] [PubMed] [Google Scholar]
- De Rycke J, Ducommun B. Bacterial cyclostatin, or how do bacteria manipulate the eukaryotic cell cycle. Med Sci (Paris) 2003;19:1128–1136. doi: 10.1051/medsci/200319111128. [DOI] [PubMed] [Google Scholar]
- De Rycke J, Oswald E. Cytolethal distending toxin (CDT): a bacterial weapon to control host cell proliferation? FEMS Microbiol Lett. 2001;203:141–148. doi: 10.1111/j.1574-6968.2001.tb10832.x. [DOI] [PubMed] [Google Scholar]
- Elwell CA, Dreyfus LA. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol. 2000;37:952–963. doi: 10.1046/j.1365-2958.2000.02070.x. [DOI] [PubMed] [Google Scholar]
- Elwell C, Chao K, Patel K, Dreyfus L. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect Immun. 2001;69:3418–3422. doi: 10.1128/IAI.69.5.3418-3422.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisan T, Cortes-Bratti X, Chaves-Olarte E, Stenerlöw B, Thelestam M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol. 2003;5:695–707. doi: 10.1046/j.1462-5822.2003.00311.x. [DOI] [PubMed] [Google Scholar]
- Frisk A, Lebens M, Johansson C, Ahmed H, Svensson L, Ahlman K, Lagergard T. The role of different protein components from the Haemophilus ducreyi cytolethal distending toxin in the generation of cell toxicity. Microb Pathog. 2001;30:313–324. doi: 10.1006/mpat.2000.0436. [DOI] [PubMed] [Google Scholar]
- Hassane DC, Lee RB, Mendenhall MD, Pickett CL. Cytolethal distending toxin demonstrates genotoxic activity in a yeast model. Infect Immun. 2001;69:5752–5759. doi: 10.1128/IAI.69.9.5752-5759.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heywood W, Henderson B, Nair SP. Cytolethal distending toxin: creating a gap in the cell cycle. J Med Microbiol. 2005;54:207–216. doi: 10.1099/jmm.0.45694-0. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Tomiuk S, Wolff G, Stoffel W. Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc Natl Acad Sci USA. 2000;97:5895–5900. doi: 10.1073/pnas.97.11.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno F, Korostoff J, Volgina A, DiRienzo JM. Resistance of human periodontal ligament fibroblasts to the cytolethal distending toxin of Actino-bacillus actinomycetemcomitans. J Periodontol. 2005;76:1189–1201. doi: 10.1902/jop.2005.76.7.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Galán JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Galán JE. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun. 2001;69:4358–4365. doi: 10.1128/IAI.69.7.4358-4365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Tejero M, Galán JE. Cytolethal distending toxin: limited damage as a strategy to modulate cellular functions. Trends Microbiol. 2002;10:147–152. doi: 10.1016/s0966-842x(02)02316-8. [DOI] [PubMed] [Google Scholar]
- Lee RB, Hassane DC, Cottle DL, Pickett C. Interactions of Campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect Immun. 2003;71:4883–4890. doi: 10.1128/IAI.71.9.4883-4890.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Stevens MK, Latimer JO, Ward CK, Deng K, Blick R, et al. Characterization of Haemophilus ducreyi cdtA, cdtB, and cdtC mutants in in vitro and in vivo systems. Infect Immun. 2001;69:5626–5634. doi: 10.1128/IAI.69.9.5626-5634.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSweeney LA, Dreyfus LA. Carbohydrate-binding specificity of the Escherichia coli cytolethal dis-tending toxin CdtA-II and CdtC-II subunits. Infect Immun. 2005;73:2051–2060. doi: 10.1128/IAI.73.4.2051-2060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, DiRienzo JM. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell Microbiol. 2002;4:245–255. doi: 10.1046/j.1462-5822.2002.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MPA, Bueno LC, Hansen EJ, DiRienzo JM. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun. 1999;67:1227–1237. doi: 10.1128/iai.67.3.1227-1237.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NeŠić D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- Ohara M, Oswald E, Sugai M. Cytolethal distending toxin: a bacterial bullet targeted to nucleus. J Biochem (Tokyo) 2004;136:409–413. doi: 10.1093/jb/mvh154. [DOI] [PubMed] [Google Scholar]
- Oswald E, Nougayrede JP, Taieb F, Sugai M. Bacterial toxins that modulate host cell-cycle progression. Curr Opin Microbiol. 2005;8:83–91. doi: 10.1016/j.mib.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Pickett CL, Whitehouse CA. The cytolethal distending toxin family. Trends Microbiol. 1999;7:292–297. doi: 10.1016/s0966-842x(99)01537-1. [DOI] [PubMed] [Google Scholar]
- Pickett CL, Cottle DL, Pesci EC, Bikah G. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect Immun. 1994;62:1046–1051. doi: 10.1128/iai.62.3.1046-1051.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rutenber E, Ready M, Robertus JD. Structure and evolution of ricin B chain. Nature. 1987;326:624–626. doi: 10.1038/326624a0. [DOI] [PubMed] [Google Scholar]
- Saiki K, Konishi K, Gomi T, Nishihara T, Yoshikawa M. Reconstitution and purification of cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiol Immunol. 2001;45:497–506. doi: 10.1111/j.1348-0421.2001.tb02650.x. [DOI] [PubMed] [Google Scholar]
- Saiki K, Gomi T, Konishi K. Deletion and purification studies to elucidate the structure of the Actinobacillus actinomycetemcomitans cytolethal distending toxin. J Biochem (Tokyo) 2004;136:335–342. doi: 10.1093/jb/mvh121. [DOI] [PubMed] [Google Scholar]
- Scott DA, Kaper JB. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect Immun. 1994;62:244–251. doi: 10.1128/iai.62.1.244-251.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker BJ, Besack D, McKay T, Pankoski L, Zekavat A, Demuth DR. Actinobacillus actinomycetemcomitans cytolethal distending toxin (Cdt): evidence that the holotoxin is composed of three subunits: CdtA, CdtB, and CdtC. J Immunol. 2004;172:410–417. doi: 10.4049/jimmunol.172.1.410. [DOI] [PubMed] [Google Scholar]
- Shenker BJ, Besack D, McKay T, Pankoski L, Zekavat A, Demuth DR. Induction of cell cycle arrest in lymphocytes by Actinobacillus actinomycetemcomitans cytolethal distending toxin requires three subunits for maximum activity. J Immunol. 2005;174:2228–2234. doi: 10.4049/jimmunol.174.4.2228. [DOI] [PubMed] [Google Scholar]
- Sugai M, Kawamoto T, Pérès SY, Ueno Y, Komatsuzawa H, Fujiwara T, et al. The cell cycle-specific growth-inhibitory factor produced by Actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect Immun. 1998;66:5008–5019. doi: 10.1128/iai.66.10.5008-5019.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KS, Ong G, Song KP. Introns in the cytolethal distending toxin gene of Actinobacillus actinomycetemcomitans. J Bacteriol. 2005;187:567–575. doi: 10.1128/JB.187.2.567-575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young VB, Schauer DB. Cytolethal distending toxin: a bacterial toxin which disrupts the eukaryotic cell cycle. Chem Res Toxicol. 2000;13:936–939. doi: 10.1021/tx000091d. [DOI] [PubMed] [Google Scholar]
- Young VB, Knox KA, Pratt JS, Cortez JS, MansfieldL S, Rogers AB, et al. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect Immun. 2004;72:2521–2527. doi: 10.1128/IAI.72.5.2521-2527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]