

Abstract
Bidirectional mismatch repair directed by a strand break located 3′ or 5′ to the mispair has been reconstituted using seven purified human activities: MutSα, MutLα, EXOI, RPA, PCNA, RFC, and DNA polymerase δ. In addition to DNA polymerase δ, PCNA, RFC, and RPA, 5′-directed repair depends on MutSα and EXOI, whereas 3′-directed mismatch correction also requires MutLα. The repair reaction displays specificity for DNA polymerase δ, an effect that presumably reflects interactions with other repair activities. Since previous studies have suggested potential involvement of the editing function of a replicative polymerase in mismatch-provoked excision, we have evaluated possible participation of DNA polymerase δ in the excision step of repair. RFC and PCNA dramatically activate polymerase δ-mediated hydrolysis of a primer-template. Nevertheless, the contribution of the polymerase to mismatch-provoked excision is very limited, both in the purified system and in HeLa extracts, as judged by in vitro assay using nicked circular heteroplex DNAs. Thus, excision and repair in the purified system containing polymerase δ are reduced 10-fold upon omission of EXOI or by substitution of a catalytically dead form of the exonuclease. Furthermore aphidicolin inhibits both 3′- and 5′-directed excision in HeLa nuclear extracts by only 20-30%. Although this modest inhibition could be due to nonspecific effects, it may indicate limited dependence of bidirectional excision on an aphidicolin-sensitive DNA polymerase.
Mismatch repair stabilizes the genome by correction of DNA biosynthetic errors, by ensuring the fidelity of genetic recombination, and in mammalian cells by participation in the cellular response to some classes of DNA damage. Basic features of the reaction responsible for replication error correction are conserved from bacteria to mammals. Study of partial reactions has demonstrated that repair can be divided into three major steps: mismatch recognition, excision, and repair DNA synthesis (1-4). E. coli mismatch repair has been reconstituted using purified components. In the bacterial reaction MutS is responsible for mismatch recognition and recruits MutL to the heteroduplex in an ATP-dependent manner (5-13). Assembly of the MutL•MutS•heteroduplex complex activates the d(GATC) endonuclease activity of MutH, which incises the unmethylated strand of the heteroduplex (14). This strand break serves as an entry point for the excision system, which is comprised of DNA helicase II and an appropriate single-strand exonuclease (15-18). A 3′ to 5′ exonuclease is required when the nick that directs excision is located 3′ to the mismatch, while a 5′ to 3′ hydrolytic activity is necessary when the strand break is 5′ to the mispair. DNA polymerase III holoenzyme is sufficient to repair the ensuing gap, and covalent integrity is restored to the helix by DNA ligase (19).
Analysis of nuclear extracts of human cells has indicated a similar excision repair mechanism for nick-directed mismatch correction in higher cells (20,21), and several reconstituted systems that rely on purified human proteins have been described that support nick-directed mismatch-provoked excision (22,23). In a simple system comprised of MutSα (MSH2•MSH6 heterodimer), EXOI, and RPA, hydrolysis is mismatch-provoked and terminates upon mismatch removal, but always proceeds 5′ to 3′ from the nick that directs excision (22). Although MutLα (MLH1•PMS2 heterodimer) is not required for EXOI activation in this system, it does enhance the mismatch dependence of the reaction. Supplementation of MutSα, MutLα EXOI, and RPA with PCNA and RFC yields a system that supports bidirectional excision, i.e. hydrolysis that is directed in an appropriate manner by a nick located either 5′ or 3′ to the mismatch (23). In contrast to the simpler 5′ to 3′ reconstituted reaction, 3′-directed excision in this six component system is absolutely dependent on MutLα. RFC is thought to play two roles in 3′-directed excision (23). It functions as a clamp loader, with the loaded form of PCNA necessary to activate 3′-directed excision, but it also acts directly to suppress EXOI mediated 5′ to 3′ hydrolysis from a strand break located 3′ to the mismatch.
While analysis of these purified systems has yielded useful information on mechanism, other activities may play significant roles in mismatch repair in the cell. For example, HMGB1 has been implicated in mismatch repair in nuclear extracts (24), although it is not required in the purified systems (23). Furthermore, analysis of EXOI-depleted extracts and EXOI-deficient cell lines has indicated that other excision activities may function in a manner that is redundant with respect to EXOI (22,25-27).
Depletion of human cell extracts and genetic studies in yeast have also implicated DNA polymerase δ in eukaryotic mismatch repair, with the former studies indicating involvement in the repair synthesis step of the reaction (28,29). This enzyme displays both polymerase and 3′-exonuclease activities (30), with the human enzyme comprised of at least four subunits (p125, p66, p50, and p12) (31-34). Inasmuch as polymerase δ depends on PCNA for processivity (35), it is not surprising that PCNA has been implicated in the repair synthesis step of eukaryotic mismatch correction (36).
Analysis of mutant yeast strains with defects in the polymerase δ editing exonuclease has suggested that this activity may also play a role in mismatch-provoked excision (37,38). Although other studies have suggested that the genetic instability associated with such defects may be due to aberrant checkpoint activation (39), the observation that 3′-directed mismatch-provoked excision in HeLa extracts is partially inhibited by aphidicolin has also been attributed to editing exonuclease involvement in the excision step of mismatch repair (21).
In this study we demonstrate that a seven component system comprised of MutSα, MutLα EXOI, RPA, RFC, PCNA, and DNA polymerase δ supports bidirectional mismatch repair directed by a strand break located either 3′ or 5′ to the mispair. While repair directed by a 5′-strand break can occur in the absence of MutLα, this activity is required for mismatch correction directed by a 3′-strand break. Repair DNA synthesis by polymerase δ depends on presence of RPA, PCNA, and RFC for optimal activity. Importantly, EXOI is required for both 3′-and 5′-directed repair, and no significant involvement of the polymerase δ editing exonuclease is evident in mismatch-provoked excision in the purified system. Analysis of aphidicolin sensitivity of 3′- and 5′-directed excision reactions in HeLa nuclear extracts also indicates that the editing functions of polymerases δ or ε play little if any role in mismatch-provoked excision as scored on nicked circular heteroduplex DNAs.
MATERIALS AND METHODS
Proteins and extracts -- All fractionation was performed at 0 to 4°C in the presence of a set of protease inhibitors at the following final concentrations: 0.5 μg/ml aprotinin, 1 μg/ml E64, 1 μg/ml leupeptin, 5 μg/ml pepstatin, 100 μg/ml pefabloc, and 0.1% phenylmethylsulfonyl fluoride (relative to a saturated solution in isopropanol). With the exception of phenylmethylsulfonyl fluoride (USB), all protease inhibitors were from Roche. Human MutSα, MutLα, EXOIb, hydrolytically defective EXOIb D173A, RPA, PCNA, and RFC were isolated according to Dzantiev et al. (23) and references therein. HeLa nuclear extracts were prepared as described (40). Partially purified human DNA polymerase δ was isolated from HeLa cells as described previously (28). T7 DNA polymerase was from Amersham Biosciences.
The recombinant, four subunit form of DNA polymerase δ was expressed using a set of baculoviruses (kindly provided by Dr. Vladimir Podust, Vanderbilt University) and isolated by a modification of the published procedure (34). Six hundred ml of SF9 cells at 1 × 106/ml in HyQ-SFX medium (HyClone, Inc.) were co-infected with baculoviruses that express p125, His tagged-p66, p50, and p12 subunits of human polymerase δ at multiplicities of infection of 6, 1, 1, and 1 respectively. Cells were harvested 48 hours after infection, washed with cold 2 mM KH2PO4, 10 mM Na2HPO4, 2.7 mM KCl, 137 mM NaCl, and lysed in 20 ml Buffer A (20 mM Tris-HCl, pH 7.5, 0.2% NP40, 100 mM NaCl) by Dounce homogenization (pestle B). Cytosolic extract was cleared by centrifugation at 10,000 g for 10 min and mixed with 1 ml Ni-NTA agarose resin (Invitrogen) for 1.5 hours. After packing into a small column, the resin was washed with Buffer A and bound proteins eluted with Buffer B (20 mM Tris-HCl, pH 7.5, 0.02% vol/vol NP40, 100 mM NaCl, 10% vol/vol glycerol, 200 mM imidazole). Fractions containing polymerase δ were collected, diluted 5-fold with Buffer Q (20 mM Tris-HCl, pH 7.5, 0.02% (vol/vol) NP40, 10% (vol/vol) glycerol, 1 mM dithiothreitol (DTT)), and loaded onto 1 ml MonoQ column (Amersham Pharmacia) equilibrated in Buffer Q containing 20 mM NaCl. The column was eluted with a gradient from 20-400 mM NaCl in Buffer Q. Fractions containing polymerase δ (eluted at 260 mM NaCl) were combined and loaded directly onto a 1 ml HiTrap Heparin column (Amersham Pharmacia) equilibrated in Buffer C (20 mM HEPES, pH 7.5, 5% (vol/vol) glycerol, 0.5 mM DTT, 250 mM NaCl). The column was eluted with a gradient of 250-800 mM NaCl in Buffer C. Polymerase δ eluted at 0.51 M KCl and the final preparation was estimated to be ∼ 70-80% pure by Coomassie staining and contained all four subunits of polymerase δ as judged by Western analysis.
Goat polyclonal antibody against the catalytic subunit of polymerase δ p125 (C-20) was from Santa Cruz Biotechnology. Rabbit polyclonal anti-p50 against full length p50 protein, rabbit polyclonal anti-p68 against peptide GHGPPASKQVSQQPKG, and rabbit polyclonal anti-p12 against peptide GLEPPPEVWQVLKYHPGD (31,33) were generous gifts from Marietta Lee (New York University).
Construction of DNA substrates -- 5′G-T and 3′G-T heteroduplexes (and corresponding G•C and A•T homoduplex control DNAs, respectively) were prepared from f1 bacteriophage derivatives as described (20,23,41). The 5′-heteroduplex contained single strand break in the complementary DNA strand 128 base pairs 5′ to the mismatch (shorter path in the circular DNA). 3′-heteroduplex DNA contained single strand break in the complementary strand 141 base pairs 3′ to the mispair. Gapped homoduplex DNA was prepared by a similar method (22,41). The latter DNA contained 220-nucleotide gap in the complementary strand between the Sau96I and SwaI cleavage sites.
Primed single-stranded DNAs for assay of the polymerase δ editing exonuclease were prepared by hybridization of a 5′-32P-labeled 35 residue oligonucleotide to circular single-stranded phage f1MR3 DNA (6). Oligonucleotide C5650 (d(CCGAATTTCTAGACTCGAGAGCTTGCTAGCAATTC)) is a perfect complement to the viral DNA, while the oligonucleotide C5650-GA (d(CCGAATTTCTAGACTCGAGAGCTTGCTAGCAATGA)) yields a hybrid in which the 3′-terminal dinucleotide is unpaired. Annealing was at 60°C for 30 min in 50 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA, pH 8.0, and the molar ratio of circular viral strand to oligonucleotide was 1.1:1 (110 nM viral DNA and 100 nM oligonucleotide).
DNA synthesis assays -- Synthesis on a poly(dA)•oligo(dT) primer-template was performed in 20 μl reactions containing 20 mM Tris-HCl, pH 7.6, 60 mM KCl, 1.5 mM ATP, 1 mM glutathione, 20 μM dTTP, 0.165 μM [α-33P]dTTP (3000 Ci/mmol), 5 mM MgCl2, 150 μg/ml BSA, 2% (vol/vol) glycerol, 625 ng of poly(dA)•oligo(dT)12-18 (Amersham Biosciences), 80 fmol of recombinant DNA polymerase δ (or a comparable amount of HeLa DNA polymerase δ as judged by quantitative western analysis), and 580 fmol homotrimer PCNA (as indicated).Two-μl samples were quenched in 10 μl of 50 mM NaOH, 10 mM EDTA and spotted on a Hybond N+ membrane (Amersham Bioscience). Unincorporated label was removed by washing with 0.5 M Na2HPO4, the membrane was rinsed with water, dried, and analyzed by PhosphoImaging.
Gap repair DNA synthesis was assayed in 20 μl reactions containing 20 mM Tris-HCl, pH 7.6, 50 mM NaCl, 75 mM KCl, 1.5 mM ATP, 1 mM glutathione, 100 μM each of dGTP, dATP, dTTP, 10 μM dCTP, and 0.165 μM [α-33P]dCTP (3000 Ci/mmol), 5 mM MgCl2, 150 μg/ml BSA, 2% (vol/vol) glycerol, 100 ng (24 fmol) circular f1 DNA containing a 220 nucleotide gap (above), and as indicated 80 fmol recombinant DNA polymerase δ, 290 fmol homotrimer PCNA, 100 fmol of RFC, and 900 fmol RPA. Two-microliter samples were quenched and incorporated label determined as described above.
Assay of mismatch-provoked excision and repair -- Mismatch repair and mismatch-provoked excision in nuclear extracts was performed as described previously (20,25,40). For analysis of aphidicolin inhibition, reactions were supplemented with 90 μM drug. A 27 mM aphidicolin (A. G. Scientific) stock, which was used within 2 months, was prepared by dissolving the compound in DMSO and storing as aliquots at −80°C. The stock solution was diluted to 900 μM with H20 immediately prior to use.
Mismatch repair using purified proteins was performed by minor modifications of previous procedures (23). Repair reactions (20 μl) contained 24 fmol 3′-G-T or 5′-G-T heteroduplex DNA (Fig. 2), 200 μM each of dATP, dCTP, dGTP, dTTP (Amersham), and as indicated 100 ng (390 fmol) MutSα, 70 ng (390 fmol) MutLα, 4 ng (42 fmol) EXOI, 200 ng (1800 fmol) RPA, 66 ng (220 fmol) RFC, 25 ng (290 fmol homotrimer) PCNA, and 20 ng (80 fmol) polymerase δ. After incubation for 5 min (5′-reaction) or 8 min (3′-reaction) at 37°C, reactions were stopped by adding 40 μl of 30 mM EDTA, 180 μg/ml proteinase K, and 0.4 mg/ml glycogen. After incubation at 55°C for 60 min reactions were extracted with phenol:choloroform:isoamylalcohol (25:24:1), followed by chloroform extraction. DNA products recovered by ethanol precipitation were cleaved with HindIII and ClaI, subjected to electrophoresis through a 1% agarose gel, and visualized by ethidium bromide staining. Analysis of mismatch-provoked excision was performed in a similar manner except dNTPs were omitted from the reaction, and excision products were scored by cleavage with NheI and ClaI (23,25; see Fig. 2).
Fig. 2.
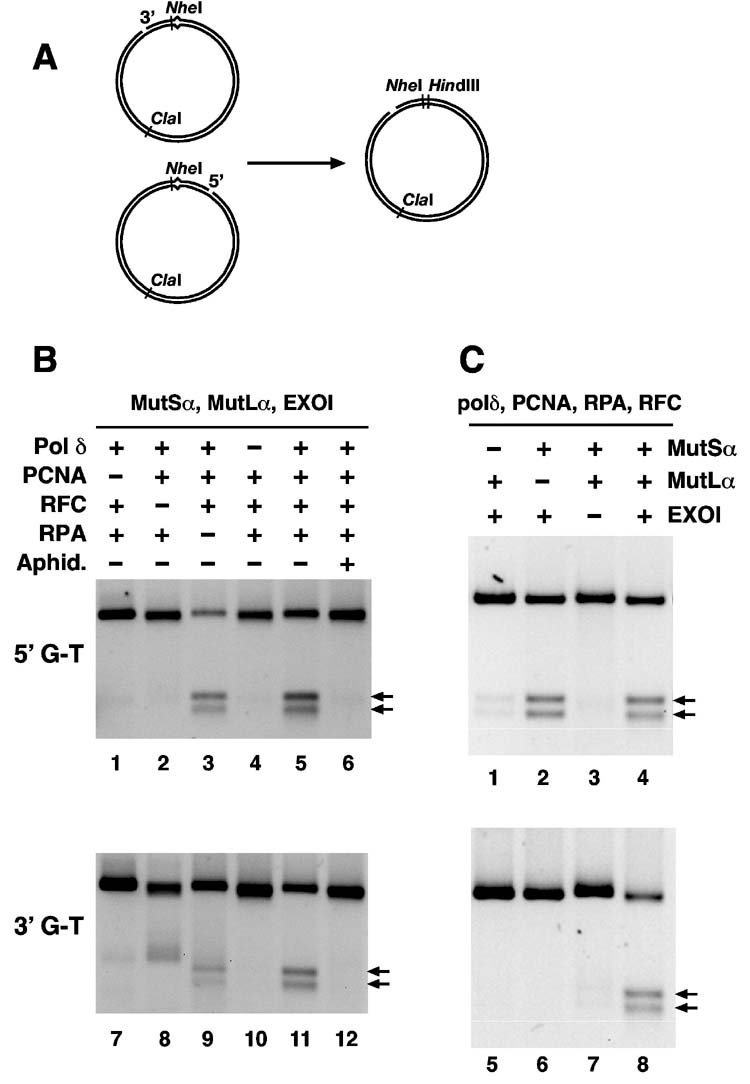
Reconstitution of bidirectional mismatch repair with purified human proteins. (A) The assay for mismatch repair utilizes a 6,440 base pair circular heteroduplexes containing a G-T mismatch within overlapping restriction sites for HindIII and XhoI (6,23) and a site-specific nick in the complementary DNA strand 141 base pairs 3′ to the mismatch (shorter path, 3′-G-T) or 128 base pairs 5′ to the mispair (5′-G-T). Since repair occurring on the incised strand renders the DNAs sensitive to HindIII, cleavage of repaired molecules with HindIII and ClaI yields two rapidly migrating fragments (arrows in panels B and C). The heteroduplexes contain a NheI site 5 base pairs from the mismatch. Because the gaps produced by mismatch-provoked excision span this site, conversion of the heteroduplex to an NheI-resistant form provides a simple assay for mismatch-provoked excision (23,25). (B) Reactions (Materials and Methods) contained a 3′-G-T or 5′-G-T heteroduplex, MutSα, MutLα, and EXOI. DNA polymerase δ, PCNA, RFC, RPA, and aphidicolin were present as indicated. (C) Reactions as in panel B contained DNA polymerase δ, PCNA, RFC, and RPA. MutSα, MutLα, and EXOI were included as indicated. Although not shown, no detectable mismatch rectification was observed on the continuous strand of 3′- or 5′-heteroduplexes when incubated with complete, seven component system.
Excision and repair synthesis tract endpoints were localized by indirect end labeling (23,25,41). Briefly, reaction products were cleaved with an appropriate restriction endonuclease, resolved through alkaline agarose, and transferred to Hybond N+ membranes, which were probed with 32P-labeled oligonucleotides that hybridize to complementary strand sequences on the appropriate side of the restriction enzyme site. 32P-labeled probe V5891 (d(ATTTAACGCGAATTTTAACAA)) was used to localize the 3′-termini on 5′- heteroduplex products following SspI digestion. 32P-labeled probe V194 (d(AACATGTTGAGCTACAGCACC)) was used to map the 3′-termini on 3′-heteroduplex products after BstYI digestion.
Fine mapping of excision and repair DNA synthesis tracts -- Excision and repair reactions in nuclear extracts and with purified proteins were performed as described above. Excision reactions using HeLa nuclear extract were supplemented with 90 μM aphidicolin to inhibit limited DNA synthesis supported by trace dNTP levels present in extract preparations (20). Repair reactions contained 20 μM α-thio-dCTP (TriLink), 180 μM dCTP, and 200 μM each of dATP, dGTP, and dTTP. After incubation for 8 min at 37°C, reactions were processed as described above and DNA digested with SspI. The 10 μl restriction digestion reaction mixture was diluted to 180 μl with TE (10 mM Tris-HCl, pH 7.5, 1 mM EDTA) and divided into two 90 μl aliquots. One aliquot was supplemented with 10 μl of 50 mM iodine in ethanol, which cleaves at sites of α-thio-dCMP incorporation (42), with the other aliquot serving as control. After 5 min at room temperature, DNA was recovered by ethanol precipitation, resuspended in 5 μl of 10 mM Tris-HCl, pH 8.0, 1 mM EDTA and supplemented with 10 μl of 90% formamide (v/v), 89 mM Tris-borate, 1 mM EDTA. Reaction products were separated on a 6% denaturing polyacrylamide gel, electrotransferred to a Hybond N+ membrane, and probed with V5891.
Assay of polymerase δ editing exonuclease -- Editing exonuclease was assayed using primed single-strands which were either perfectly paired or contained a 3′-terminal unpaired dinucleotide. Buffer conditions were identical to those used for assay of reconstituted mismatch-provoked excision (23) except that ATP concentration was 0.1 mM. Reactions (20 μl) contained 24 fmol DNA, 80 fmol polymerase δ, and when indicated 1160 fmol PCNA (trimer), 440 fmol RFC, and 180 μM aphidicolin. Incubation was at 30°C. 3 μl samples were removed as a function of time and quenched by addition of 9 μl of 90% formamide (v/v), 89 mM Tris-borate, 1 mM EDTA. DNA was separated on a 20% polyacrylamide gel containing 8M urea in the Tris-borate buffer. DNA products were visualized by phosphoimaging.
RESULTS
In vitro reconstitution of bidirectional mismatch repair -- A single-strand break located 3′ or 5′ to the mismatch is sufficient to determine the stand specificity of mismatch repair or mismatch-provoked excision in extracts of mammalian cells (20,21). Several reconstituted systems have been recently described that support mismatch-provoked excision using purified human proteins. 5′-directed excision can be mediated by EXOI in a reaction that is controlled by MutSα and RPA, with specificity enhanced by MutLα (22). A six-component system consisting of MutSα, MutLα EXOI, RPA, RFC, and PCNA is sufficient to support mismatch-provoked excision directed by a strand break located either 3′ or 5′ to the mismatch (23). Because previous work has implicated DNA polymerase δ in the repair synthesis step of mismatch correction in HeLa extract fractions (28), we have asked whether supplementation of this six component system with polymerase δ will yield a system that supports mismatch repair.
To address this question, we have used the recombinant, four-subunit form of the polymerase (34). As judged by immunoblotting, the preparation used (Materials and Methods) contained all four subunits (p125, p66, p50, and p12) present in polymerase δ isolated from HeLa cells, with three of these subunits corresponding to major protein bands in the elecrophoretic profile of the isolated recombinant activity (Fig. 1A). The biochemical properties of this preparation are consistent with those described for this enzyme (33,34). As expected (Fig. 1B), the recombinant enzyme was activated by PCNA on a poly(dA)•oligo(dT) primer-template (34,35), with the level of activity similar to that observed with comparable amounts of DNA polymerase δ partially purified from HeLa cells (28). The recombinant enzyme also supported efficient synthesis on a gapped circular DNA that is similar to the excision intermediate produced during the course of in vitro mismatch repair (Fig. 1C). Although gap repair displayed only a modest dependence on RPA, RFC and PCNA were essential for the reaction, which was abolished by aphidicolin, consistent with previous findings (33,35).
Fig. 1.
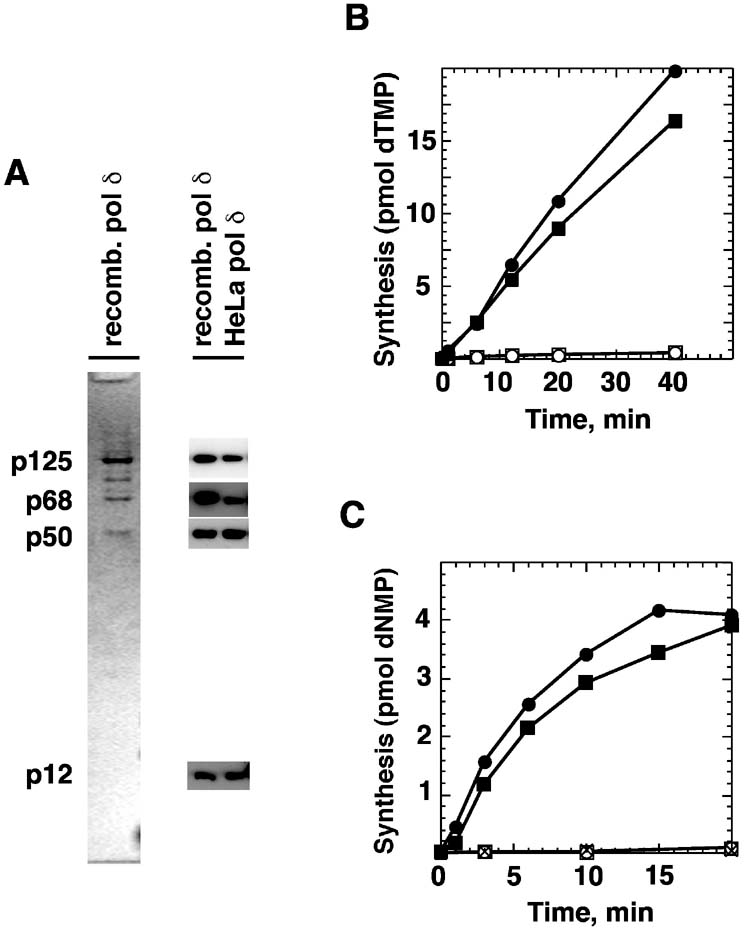
Properties of recombinant human DNA polymerase δ. (A) Purified recombinant DNA polymerase δ was subjected to electrophoresis through a 10% SDS gel and stained with Coomassie blue (left panel). Positions of the four subunits of the protein (p125, p68, p50 and p12) are shown. Presence of each of these subunits in the isolated recombinant and the partially purified HeLa preparation was confirmed by Western analysis with antibodies against the individual subunits (right panel). (B) DNA synthesis on a poly(dA)•oligo(dT) template-primer was determined as described under Materials and Methods using 80 fmol of recombinant polymerase δ (, ○), or a comparable amount (as judged by quantitative Western blot) of HeLa polymerase δ (■, □). Reactions were performed in the absence (○, □) or presence (, ■) of 580 fmol of PCNA. (C) DNA synthesis by recombinant polymerase δ was determined on a circular 6440 base pair DNA containing a 220 nucleotide gap (Materials and Methods). The complete reaction contained DNA polymerase δ, RFC, PCNA, and RPA (); RPA omitted (■); PCNA omitted (□); RFC omitted (○); complete plus 90 μM aphidicolin (X). Complete gap repair would correspond to 5.3 pmol of DNA synthesis.
Supplementation of MutSα, MutLα EXOI, RPA, RFC, and PCNA with recombinant DNA polymerase δ is sufficient to reconstitute nick-directed mismatch rectification of 5′- and 3′-heteroduplexes in vitro. As shown in Fig. 2B, repair of both 5′- and 3′-G-T heteroduplexes was dependent on PCNA (lanes 1 and 7), RFC (lanes 2 and 8) and DNA polymerase δ (lanes 4 and 10), with RPA acting in a stimulatory fashion (compare lane 3 with 5, and lane 9 with 11). Aphidicolin inhibited repair of both heteroduplexes (lanes 6 and 12), an effect that we attribute to inhibition of the DNA synthetic activity of polymerase δ (see below). Because excision on a 5′-heteroduplex can occur in the absence of PCNA and RFC (22,23), the requirements for these two proteins in 5′-heteroduplex repair may solely reflect their involvement in the repair synthesis step. However, since 3′-directed excision requires both PCNA and RFC (23), reconstituted 3′-heteroduplex repair presumably depends on function of the two proteins in both the excision and repair synthesis steps of the reaction. Such a multifunctional role for RFC accounts for the anomalous product produced on the 3′-substrate in the absence of this activity (Fig. 2B lane 8). Absence of RFC not only blocked repair DNA synthesis (Fig. 1), but also prevented activation of 3′-directed excision (23). Under these conditions, mismatch-provoked hydrolysis occurs with 5′ to 3′ polarity (23), yielding the product observed in lane 8.
MutSα, MutLα, and EXOI requirements for reconstituted repair mirror those previously observed for reconstituted excision (22,23). Thus, repair of the 5′-G-T heteroduplex occured efficiently in the absence of MutLα, although MutSα and EXOI were required for the reaction (Fig. 2C lanes 1-3). As can be seen, significant background repair on the 5′-substrate occured in the absence of MutSα (about 15% of that observed in the presence of the mismatch recognition protein; lanes 1 and 4). This is consistent with the previous finding that reconstituted 5′-directed excision displays a mismatch dependence of only 3 to 5-fold, an effect attributed to deficiency of as yet unidentified component(s) that contribute to the specificity of 5′-directed excision (22,23). By contrast, mismatch rectification directed by a 3′-strand break required both MutSα and MutLα (lanes 5 and 6) and was reduced 10-fold upon omission of EXOI (lane 7). It is also important to note that the repair reaction shown in Fig. 2 was strongly directed to the nicked DNA strand; no detectable repair on the continuous strand of the heteroduplex was observed in the presence of the complete seven-component system (not shown).
Dependence of excision and repair on DNA polymerase δ -- As noted above, omission of EXOI from the reconstituted repair system largely abolished both 3′- and 5′-directed mismatch rectification, suggesting that the 3′ to 5′ hydrolytic function of DNA polymerase δ does not contribute significantly to mismatch-provoked excision in this system. A more comprehensive analysis of the dependence of the reconstituted reaction on the polymerase is shown in Fig. 3. In the presence of MutSα, MutLα EXOI, RPA, RFC, and PCNA, mismatch repair increased continuously with polymerase concentration up to 4 nM for both 3′- and 5′-heteroduplexes (Fig. 3A). In the absence of dNTPs, neither the 3′ nor the 5′-directed excision was altered to a significant degree by presence of the polymerase. Identical results were obtained when polymerase δ isolated from HeLa cells was substituted for the recombinant enzyme used in these experiments (not shown). These findings indicate that the contribution of the polymerase δ editing exonuclease to excision in the purified system is limited.
Fig. 3.
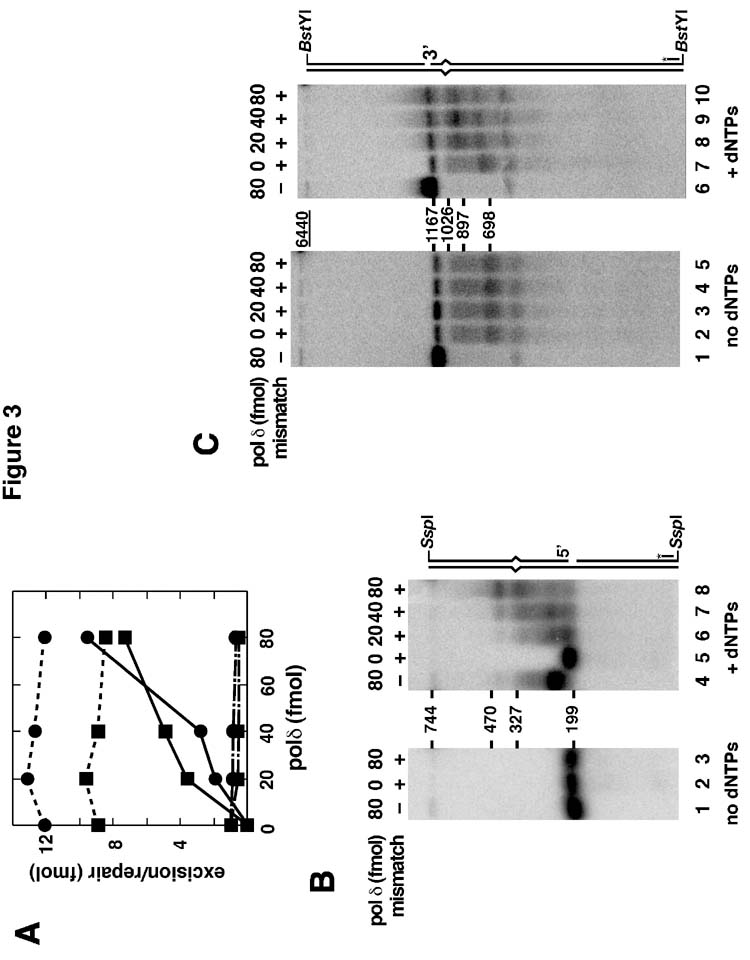
Dependence of excision and repair DNA synthesis on DNA polymerase δ. (A) Reactions containing 5′-G-T () or 3′-G-T (■) heteroduplexes (Fig. 2A) were performed as described under Materials and Methods except that hydrolytically defective EXOI D173A was substituted for EXOI as indicated, DNA polymerase δ concentration was varied as shown, and dNTPs were omitted from those reactions used to score excision. Mismatch repair (solid lines) was scored by cleavage with ClaI and HindIII. Mismatch-provoked excision was determined by cleavage with ClaI and NheI (dashed and hyphenated lines indicate excision occurring in the presence of wild type EXOI or hydrolytically defective EXOI D173A, respectively). (B) Reactions using a 5′-heteroduplex or an otherwise identical G•C homoduplex control (lanes 1 and 4) were performed as in panel A in the absence (lanes 1-3) or presence of dNTPs (lanes 4-8). DNA polymerase δ concentration was varied as indicated. Reaction products were digested with SspI, denatured, resolved by electrophoresis through 1.8% alkaline agarose, and probed with 32P-labeled oligonucleotide V5891 (Materials and Methods). As illustrated schematically in the diagram on the right, this oligonucleotide hybridizes to the 5′-end of the incised strand of the SspI fragment that contains the mismatch. This probe thus permits visualization of the fate of the 3′-terminus at the strand break. (C) Reactions in the absence (lanes 1-5) or presence of dNTPs (lanes 6-10) and indicated amounts of DNA polymerase δ were performed as in panel B except that a 3′-G-T heteroduplex or the corresponding A•T homoduplex control, (lanes 1 and 6) were used. After cleavage with BstYI and denaturation, products were separated and probed with 32P-labeled oligonucleotide V194 (Materials and Methods) to monitor the fate of the 3′-termini in the incised strand of the heteroduplex.
To explore the possibility that EXOI hydrolysis might mask a significant polymerase δ contribution to excision, we performed similar experiments using hydrolytically defective EXOI D173A. This mutant exonuclease is well behaved and purifies like the wild type protein (23). It also inhibits excision in the presence of wild type EXOI, suggesting that it supports assembly of repair complexes. As shown in Fig. 3A, substitution of the mutant protein for wild type EXOI abolished excision, irrespective of presence of the DNA polymerase, indicating that the latter activity plays little if any role in mismatch-provoked hydrolysis in the purified system.
Indirect end-labeling was also used to assess effects of DNA polymerase δ on the fate of heteroduplex 3′-termini during the course of reconstituted excision and repair reactions (Fig. 3, panels B and C). Under excision conditions in the absence of dNTPs, no significant 3′ to 5′ degradation was observed on 5′- homoduplex or heteroduplex substrates in the absence or presence of polymerase δ (Fig. 3B, lanes 1-3). However, mismatch-dependent extension of the 3′-terminus of the strand was readily evident when the 5′ heteroduplex was incubated with increasing amounts of polymerase δ in the presence of dNTPs (Fig. 3B, lanes 5-8). This repair DNA synthesis terminated 100-150 nucleotides beyond the location of the mispair, consistent with the previous localization of 5′-directed excision tracts in HeLa nuclear extracts (20). Extension of the 3′-terminus of virtually all molecules of the homoduplex control was also observed, but repair synthesis tracts in this case were much shorter (extension of the 3′-terminus by about 65 nucleotides, compare lanes 1 and 4). Extension of the homoduplex 3′-terminus in this manner may be due to conversion of the nick to small gaps via limited nonprocessive 5′ to 3′ hydrolysis by EXOI (22).
We have previously shown that 3′-directed hydrolytic tracts produced on heteroduplex DNA in the reconstituted excision system display a broader distribution of sizes than those observed in HeLa nuclear extracts (23). As shown in Fig. 3C, similar results are obtained in the absence of dNTPs when MutSα, MutLα EXOI, RPA, RFC, and PCNA are supplemented with DNA polymerase δ (Fig. 3C, lanes 2-5). As can be seen, hydrolytic tracts in the purified system extend from 100 to 500 nucleotides beyond the mismatch, whereas in Hela extracts these tracts extend to only about 150 nucleotides beyond the mispair (20). Nevertheless, excision in this manner was highly mismatch-dependent (Fig. 3C, compare lane 1 with lanes 2-5), and DNA polymerase δ was without significant effect on either the extent of excision or the distribution of excision endpoints. In the presence of dNTPs, DNA products increased in size with increasing amounts of polymerase (lanes 7-10), presumably reflecting extension of recessed 3′-termini that result from excision. At the higher concentrations of polymerase δ, these DNA synthesis tracts extend to and just beyond the mismatch, within the error of the gel method used (lanes 8-10). As observed with the 5′-homoduplex control (Fig. 3B, lane 4), significant extension of the 3′-terminus of the strand break also occurred with 3′-homoduplex control (Fig, 3C, compare lane 1 and lane 6). As noted above, this may be a consequence of limited nonspecific hydrolysis by EXOI.
Fine structure analysis of repair tracts -- Excision repair tracts produced in HeLa nuclear extracts and in the reconstituted repair system were compared using high resolution sequencing gels. In these experiments excision tracts were determined under conditions of DNA synthesis block (20), while repair synthesis tracts were analyzed by partial substitution of dCTP[α]S for dCTP. The latter method exploits the fact that sites of dCMP[α]S incorporation can be visualized by virtue of the sensitivity of phosphothiodiester bonds to cleavage with iodoethanol (42). After digestion with SspI, which yields a 744 bp fragment spanning the mismatch and strand break, DNA products were treated with ethanol or iodoethanol, resolved on a urea polyacrylamide gel, and products visualized by indirect end-labeling using a 32P-oligonucleotide complementary to the 5′-end of the incised strand of the SspI fragment. As shown diagrammatically in Fig. 4, this method monitors the fate of 3′-termini on the incised DNA strand.
Fig. 4.
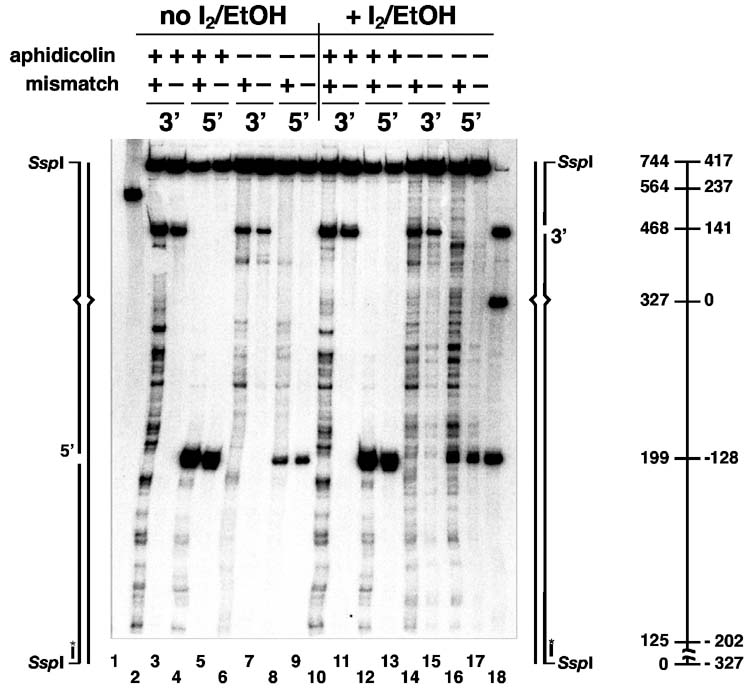
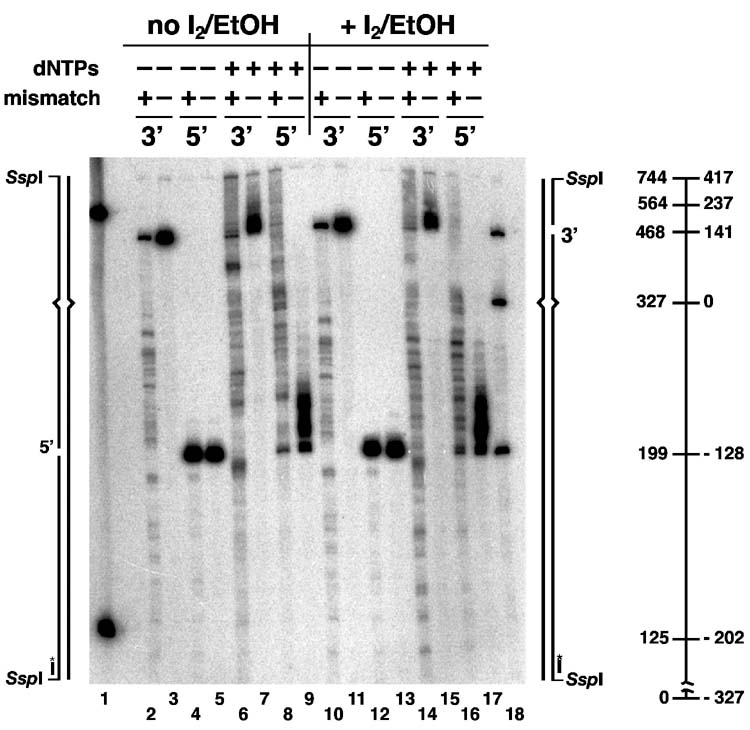
Fine mapping of excision and re-synthesis tracts. (A) Reactions containing 200 μg HeLa nuclear extract and 96 fmol (400 ng) 3′-G-T heteroduplex, 3′-A•T homoduplex, 5′-G-T heteroduplex, or 5′-G•C homoduplex DNA as indicated, were carried out in 40 μl volume under excision or repair conditions (Materials and Methods) for 10 min at 37°C. Repair reactions (lanes 6-9 and 14-17) contained 100 μM each of dATP, dGTP, TTP, 90 μM dCTP, and 10 μM dCTP[αS]. Excision reactions (lanes 2-5 and 10-13) were performed in a similar manner except that dNTPs were omitted and reactions were supplemented with 90 μM aphidicolin to suppress DNA synthesis supported by endogenous nucleotides present in extracts (20). After digestion with SspI, DNA samples were untreated (lanes 2-9) or treated with 5 mM iodine in ethanol (lanes 10-17), products resolved on a denaturing gel and probed with 5′-32P-labeled oligonucleotide V5891 (Materials and Methods). As illustrated schematically in the diagrams to each side of the gel, this probe hybridizes to the incised complementary strand adjacent to the SspI site and can be used to locate 3′-termini on both 3′- and 5′-DNA substrates. Numerical coordinates shown in the diagram on the right indicate fragment size in nucleotides (left axis) or distance from the mismatch (right axis). The 744 nucleotide species is the product of ligation. Lanes 1 and 18 are markers. (B) Reactions (40 μl) containing 200 ng (780 fmol) MutSα, 140 ng (780 fmol) MutLα, 8 ng (84 fmol) EXOI, 400 ng (3600 fmol) RPA, 50 ng (580 fmol homotrimer) PCNA, 130 ng (440 fmol) RFC, 40 ng (160 fmol) DNA polymerase δ and 200 ng (48 fmol) 3′-G-T heteroduplex, 3′-A•T homoduplex, 5′-G-T heteroduplex, or 5′-G•C homoduplex DNA (Materials and Methods) were incubated for 8 min at 37°C. Repair reactions (lanes 6-9 and 14-17) contained 100 μM each of dATP, dGTP, TTP, 90 μM dCTP, and 10 μM dCTP[αS]. Excision reactions (lanes 2-5 and 10-13) were performed in a similar manner except that dNTPs were omitted. Samples were processed and analyzed as in (A). Lanes 1 and 18 are markers.
Fig. 4A shows such an analysis of excision and repair in HeLa nuclear extracts. Results obtained with 5′-heteroduplex and homoduplex controls are shown in pairs in lanes 4-5, 8-9, 12-13, and 16-17. With the exception of ligation of a subset of molecules to produce a 744 nucleotide product, 3′-termini in the majority of the 5′-DNAs were stable in HeLa extract under conditions of DNA synthesis block, although very limited hydrolysis occurred on the 3′-side of the strand break with heteroduplex, but not homoduplex DNA (compare lanes 4 and 5). As expected, the distribution of these products was unaffected by iodoethanol treatment (lanes 12 and 13). Under conditions permissive for repair DNA synthesis, unligated intermediates resulting from extension of 3′-termini were produced on heteroduplex, but to a much lesser extent on homoduplex DNA (lanes 8 and 9). Iodoethanol treatment resulted in extensive fragmentation of the heteroduplex product within a region extending from the strand break to about 150 nucleotides beyond the original location of the mismatch (compare lane 16 with lane 8), implying that repair DNA synthesis occurred throughout this region. Iodoethanol treatment also resulted in modest fragmentation of the homoduplex product (compare lane 17 with lanes 9 and 16). This observation is in agreement with the previous finding that some background repair DNA synthesis occurs on homoduplex DNA in HeLa nuclear extracts (40).
Similar results were obtained with 5′-substrates in the reconstituted reaction (Fig. 4B), although differences from the HeLa extract reaction are evident in electrophoretic profiles because ligation cannot occur in the purified system. As in HeLa extracts, limited hydrolysis to the 3′-side of the strand break is evident on heteroduplex DNA when repair synthesis was blocked by omission of dNTPs (lanes 4-5, and lanes 12-13). In the presence of dNTPs, about 90% of the 3′-termini in the 5′-G-T heteroduplex were extended, with repair synthesis intermediates apparent in the region from the strand break to well beyond the mispair (lane 8). The fact that some repair synthesis intermediates fail to reach the mismatch suggests that DNA polymerase δ may be limiting in this system, consistent with the results shown in Fig. 3A. As can be seen in lane 9, a large fraction of 3′-termini in the homoduplex control were also extended in this system, but extension in this case was only about 50 nucleotides as compared to the much more extensive elongation that occured on the heteroduplex. As mentioned above, this probably reflects nonprocessive 5′ to 3′ hydrolysis by EXOI from the homoduplex strand break, an effect that has been observed earlier (22). Treatment with iodoethanol revealed the sites of dCMP[α]S incorporation due to these DNA biosynthetic events (lanes 16 and 17).
Similar experiments were performed with 3′-heteroduplex and homoduplex DNAs, but results in this case are more difficult to interpret because excision products that fail to undergo elongation by repair synthesis cannot always be distinguished from intermediates that undergo partial elongation. As observed previously (20) incubation of 3′-substrates with HeLa nuclear extract under conditions of DNA synthesis block resulted in mismatch-dependent production of excision intermediates that extend to at least 200 nucleotides beyond the mispair (Fig. 4A, lanes 2 and 3). Bands corresponding to these excision products are generally attenuated under conditions permissive for DNA synthesis (lane 6), suggesting that the excision products shown in lane 2 serve as precursors for repair DNA synthesis. This idea is consistent with the fact that treatment of the repair products (lane 6) with iodoethanol resulted in extensive fragmentation throughout the region where excision endpoints are observed (compare lanes 2, 6, and 14). The homoduplex control also displays some iodoethanol sensitivity, but to a much lower degree than that observed with the GT heteroudplex (compare lanes 14 and 15). As mentioned above, significant background repair synthesis is known to occur on homoduplex DNA in HeLa nuclear extracts (40).
Comparable experiments with the 3′-heteroduplex in the purified system are shown in Fig. 4B. While the homoduplex control was not subject to significant hydrolysis in the reconstituted reaction in the absence of dNTPs, extensive excision occurred on the heteroduplex under these conditions (lanes 2 and 3). Supplementation of reconstituted reactions with dNTPs resulted in appearance of a distinct product spectrum (compare lanes 2 and 6), with the major product located about 100 nucleotides 3′ to the original location of the mismatch. This suggests that excision intermediates like those in lane 2 are subject to polymerase δ elongation to yield the repair synthesis products of lane 6. In fact, iodoethanol treatment of the material shown in lane 6 resulted in a new fragmentation pattern indicative of dCMP[α]S incorporation throughout the region defined by the excision tract endpoints (compare lane 14 with lanes 2 and 6). As mentioned above, significant dCMP[α]S incorporation occurred on the 3′-homoduplex in HeLa extract (Fig. 4A, lane 15). By contrast, incorporation of the thionucleotide into the homoduplex control in the defined system was almost nonexistent in the region 3′ to the strand break (Fig. 4B, compare lanes 14 and 15). However, as observed with the 5′-homoduplex, the 3′-terminus at the homoduplex strand break was also subject to modest elongation in the purified system (compare lanes 3 and 7).
Specific requirement for polymerase δ in repair DNA synthesis -- A specific requirement for polymerase δ in mismatch repair has been suggested based on extract fractionation studies (28) and genetic studies have implicated the POL32 subunit of yeast polymerase δ in mismatch repair in this organism (29). To assess the specificity of the requirement for DNA polymerase δ in the purified system, we tested the ability of the DNA polymerase encoded by bacteriophage T7 to substitute for polymerase δ in the reconstituted mismatch repair system. The amount of T7 polymerase used in this analysis was determined based on its ability to support repair of the 220 nucleotide gap in the circular DNA used in Fig. 1C. As judged by this method, 4 fmol of the T7 enzyme is comparable in activity to the 80 fmol of DNA polymerase δ (in the presence of RFC and PCNA) that is used in the reconstituted mismatch repair system (not shown).
As demonstrated above, repair does not occur when DNA polymerase δ is omitted from the reconstituted system (Fig. 2B). However, if DNA products produced in the absence of the polymerase were deproteinized and then incubated with the enzyme in the presence of RFC, PCNA, and RPA, repaired molecules were produced with an efficiency similar to that of the complete system for both 3′- and 5′-heteroduplexes (Fig. 5A). Substitution of T7 DNA polymerase for polymerase δ in the reconstituted repair system yielded different results (Fig. 5B). As mentioned above, MutSα, MutLα, EXOI and RPA are sufficient to support 5′-directed excision (RFC and PCNA are not required) (22,23). Supplementation of these four proteins with T7 polymerase resulted in a modest level of repair, although repair products were aberrant in the sense that the smaller restriction fragment scored by the repair assay (see Fig. 2A) was recovered in low yield (lane 1). The addition of RFC and PCNA suppressed 5′-directed repair in the presence of the T7 enzyme to about 20% of that observed in the presence of polymerase δ (compare lane 3 in panel B with lane 1 in panel A). However, when 5′-excision products produced in the absence of T7 polymerase (in the presence or absence of RFC and PCNA) were deproteinized and then incubated with T7 DNA polymerase, repair products were produced in high yield (panel B lanes 2 and 4). Similar results were obtained with the 3′-heteroduplex (lanes 5-8). No detectable repair occurred when the 3′-substrate was incubated with MutSα, MutLα, EXOI, RPA, RFC and PCNA in the presence of T7 DNA polymerase (lane 7), although incubation of the 3′-heteroduplex with the excision system, followed by deproteinization and then incubation with the polymerase did yield repair products (lane 8). As shown in lane 6, repair in this manner was dependent on the presence of RFC and PCNA during the initial incubation; these two proteins are required for 3′-directed excision (23).
Fig. 5.
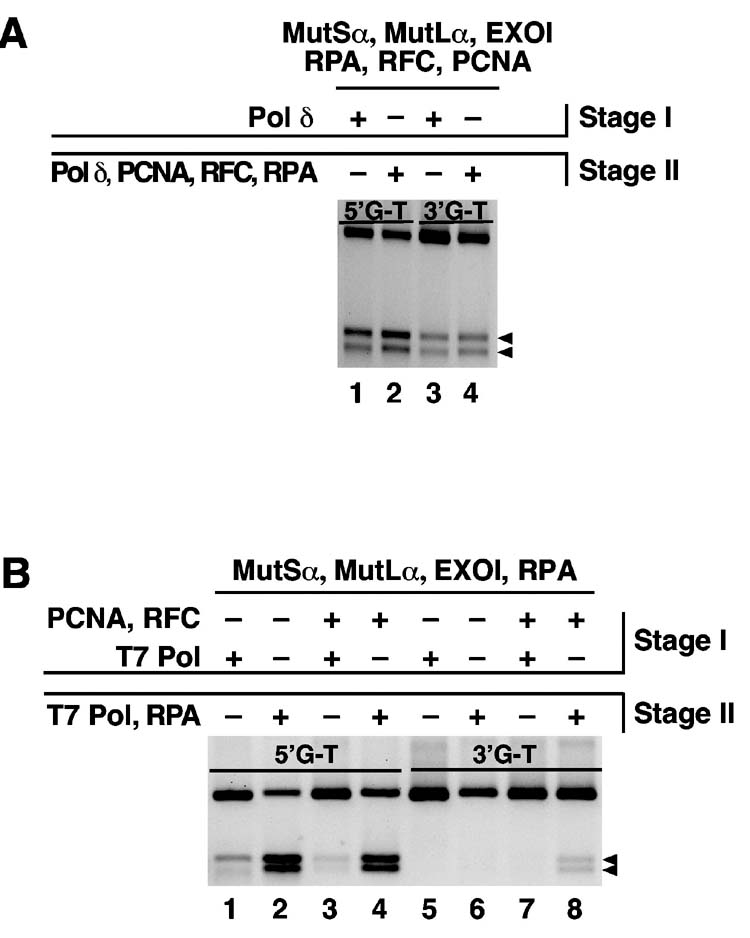
T7 DNA polymerase cannot substitute for DNA polymerase δ in mismatch repair. (A) Reactions (20 μl) were carried out in two stages. Stage I reactions containing 24 fmol 5′-G-T or 3′-G-T heteroduplex DNA were performed as described (Materials and Methods) in the presence or absence of 80 fmol DNA polymerase δ as indicated. Incubation was for 8 min at 37°C. For stage II incubations, reactions corresponding to lanes 2 and 4 were deproteinized by treatment with proteinase K, phenol, and chloroform, collected by ethanol precipitation, and then incubated with RFC, PCNA, DNA polymerase δ, and RPA as in stage I except that MutSα, MutLα, and EXOI were omitted. To score repair, DNA products from all reactions were digested with HindIII and ClaI, subjected to electrophoresis through 1% agarose, and visualized with ethidium bromide. Arrows indicate repair products. (B) Stage I reactions were performed as in panel A except that T7 DNA polymerase (4 fmol) was substituted for polymerase δ, and RFC and PCNA were present only as indicated. After deproteinization, products corresponding to lanes 2, 4, 6, and 8 were incubated in stage II reactions with T7 DNA polymerase and RPA in the absence of other proteins. Repair was scored as in panel A.
We have ruled out two trivial explanations for these findings. The inability of the T7 enzyme to support repair in the presence of the other activities is not due to an inability to copy a RPA-bound template strand because the stage II T7 polymerase reactions shown in Fig. 5B were performed in the presence of the single-stranded DNA binding protein. The possibility that T7 DNA polymerase might inhibit mismatch-provoked excision was also excluded by scoring the extent of excision in the stage I incubations of Fig. 5B; in the reactions shown in lanes 3 and 7, excision occurred on 71% and 47% of the heteroduplex molecules, respectively (not shown). These findings indicate that primer-template access in repair intermediates is restricted, presumably via interaction with one or more repair activities.
Aphidicolin effects on mismatch repair and the hydrolytic function of DNA polymerase δ -- DNA polymerases δ and ε possess a 3′ to 5′ hydrolytic activity that provides a proofreading function during DNA synthesis (35). The polymerase activity of both enzymes is inhibited by aphidicolin (43-46), and on certain DNA substrates, the 3′ exonuclease function is also subject to inhibition by the drug. Studies with polymerase δ have shown that aphidicolin inhibits the hydrolysis of oligo(dT) in a poly(dA)•oligo(dT) substrate, but fails to block hydrolysis of single-stranded oligo(dT) or removal of 3′-terminal mispaired nucleotides in a substrate of the form poly(dA)•oligo(dT)-(dGMP)4 (43-45). Similar studies have been done with polymerase ε. As observed with polymerase δ, hydrolysis of single-stranded DNA is not significantly inhibited by aphidicolin (46). However, in contrast to the results obtained with polymerase δ using poly(dA)•oligo(dT), polymerase ε hydrolysis of a defined oligonucleotide primer-template was inhibited by only 20% (46).
These aphidicolin effects on editing exonuclease function have been exploited to ask whether this activity contributes to the excision step of mismatch repair. Hays and colleagues have reported that aphidicolin inhibits 3′-directed excision in HeLa nuclear extracts by about 70% but is without effect on 5′-directed hydrolysis (21). Because the aphidicolin effects on editing exonuclease function summarized above were determined in the absence of RFC and PCNA and since the studies described here have failed to indicate a significant role for polymerase δ in mismatch-provoked excision, we have reevaluated effects of the drug on editing exonuclease function and on the excision step of mismatch repair as it occurs in nuclear extracts.
The polymerase δ 3′ to 5′ hydrolytic activity was determined on 35 residue oligonucleotides hybridized to circular single-stranded f1MR3 DNA, such that the hybrid was either perfectly paired or contained an unpaired 3′-terminal dinucleotide. In the absence of RFC and PCNA, mispaired 3′-terminal residues were subject to removal by an aphidicolin resistant reaction, but the perfectly paired hybrid was much more resistant to hydrolysis (Fig. 6, left panels), results in agreement with previous studies (45,46). However, different results were obtained in the presence of RFC and PCNA. The clamp loader and replication clamp promoted extensive hydrolysis of the hybridized oligonucleotide without regard to the paired or unpaired nature of 3′-terminal nucleotides (right panels). We attribute this hydrolysis to polymerase δ because RFC and PCNA failed to support detectable hydrolysis in the absence of the polymerase (not shown). Furthermore, hydrolysis of the perfectly paired hybrid was abolished by aphidicolin, and in the presence of the drug, hydrolysis of the mispaired hybrid was restricted to the 3′-terminal unpaired nucleotides. These findings indicate that in the absence of dNTPs, RFC and PCNA modulate activity of the polymerase δ editing exonuclease, presumably via interaction of the clamp with the enzyme.
Fig. 6.
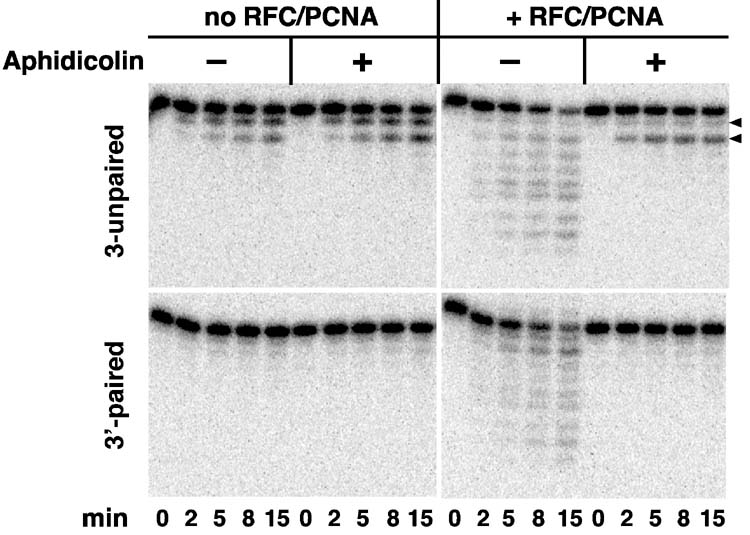
Effects of aphidicolin on the DNA polymerase δ exonuclease. Reactions (Materials and Methods) contained DNA polymerase δ and a 5′-32P-labeled oligonucleotide hybridized single-stranded circular f1 MR3 DNA such that the 3′-terminus was either perfectly paired or contained an unpaired dinucleotide (Materials and Methods). RFC, PCNA, and aphidicolin were present as indicated, and reactions were sampled as a function of time. Arrows in the upper panel indicate products produced by removal of one or both unpaired nucleotides.
Despite the inhibitory effects of aphidicolin on duplex DNA hydrolysis by polymerase δ in the presence of RFC and PCNA, effects of the drug on both 5′- and 3′-directed excision in HeLa nuclear extracts are modest. As shown in Table I, aphidicolin inhibited the initial rate of 3′-directed excision by 30% and the extent of the reaction by 22%. However, these inhibitory effects were not restricted to the 3′-reaction. 5′-directed excision was inhibited to a similar degree (22% inhibition of initial rate and 20% inhibition of extent). The results shown in Table I were obtained with nuclear extracts for which we were able to observe significant inhibition of the excision reaction. With some extracts, we have been unable to detect aphidicolin inhibition of either 3′- or 5′-directed excision (not shown). Our failure to observe substantial inhibition of the 3′-excision reaction is not due to a problem with the drug preparation used, because it was effective in abolishing DNA synthesis by polymerase δ (Fig. 1C), inhibiting of 3′ to 5′ hydrolysis of duplex DNA by polymerase δ (Fig. 6), as well as overall mismatch repair (Table I legend). These findings differ from those of Wang and Hays (21), but the basis of this discrepancy is unclear. Our results suggest that an aphidicolin-sensitive DNA polymerase (α, δ, or ε) might contribute in a modest but undefined manner to both 3′- and 5′-directed, mismatch-provoked excision in nuclear extracts. However, it is also possible that the inhibitory effects described here, as well as that described by Hays and Wang (21) for 3′-excision, could be due to some sort of nonspecific effect on the extract system.
TABLE 1.
Aphidicolin effects on rates and extents of excision in HeLa nuclear extracts
| Excision Extent (fmol) | Excision Rate (fmol/min) | |||
|---|---|---|---|---|
| Heteroduplex | − Aphidicolin | + Aphidicolin | − Aphidicolin | + Aphidicolin |
| 5′-G-T | 11.1 ± 0.6 | 8.9 ± 0.8 | 3.3 ± 0.3 | 2.6 ± 0.2 |
| 3′-G-T | 6.0 ± 0.5 | 4.7 ± 0.4 | 0.98 ± 0.02 | 0.69 ± 0.01 |
Extents of excision were determined in the absence of exogenous dNTPs in reactions (Materials and Methods) containing 24 fmol 5′-G-T or 3′-G-T heteroduplex DNA and 100 μg HeLa nuclear extract. Extents of excision, which were determined after a 5 min incubation at 37°C, are limited primarily by ligation of unprocessed substrate. Values shown are the average of 4 determinations ± one standard deviation. Corresponding values for extents of repair determined in the presence of dNTPs (100 μM each) but in the absence of aphidicolin were: 11.5 ± 1 and 5.2 ± 0.4 fmol for 5′- and 3′-substrates, respectively. Repair in the presence of aphidicolin was undetectable (< 1 fmol). Initial rate values were determined by sampling scaled up reactions and are based on the linear portion of the progress curve. Values shown are the average of two independent determinations, with errors shown corresponding to the range of values observed.
DISCUSSION
The studies described here are based on two previous observations; the demonstration that MutSα, MutLα, EXOI, RPA, RFC, and PCNA are sufficient to support bidirectional mismatch-provoked excision (23), and the finding that DNA polymerase δ is required for human mismatch repair in nuclear extracts (28). The latter study implicated polymerase δ in the reaction, but involvement of polymerases α and ε was not excluded because the depleted extracts used contained a normal complement of polymerase α and trace levels of ε. While the experiments described here do not exclude significant participation of DNA polymerases α and ε in mismatch repair, they do show that the δ enzyme is sufficient to meet the polymerase requirement in a reconstituted system. As mentioned above, genetic studies in S. cerevisiae have also indicated involvement of POL32, a subunit of polymerase δ, in mismatch repair (29).
Analysis of the repair synthesis on gapped circular DNA and the use of staged reactions (Figs. 1 and 5) implicate polymerase δ in the repair synthesis step of mismatch repair in the reconstituted system. This reaction also requires PCNA and RFC, and is stimulated by RPA, proteins that also play key roles in mismatch-provoked excision (22,23,47). These conclusions are consistent with previous findings based on use of crude nuclear fractions. Thus, the depleted extracts used to demonstrate polymerase δ involvement in mismatch repair were shown to be defective in a step subsequent to mismatch-provoked excision (28), and PCNA has been implicated in both excision and repair synthesis steps of the crude extract reaction (36).
As further evidence for the specificity of the polymerase δ requirement in mismatch repair, we have found that T7 DNA polymerase substitutes poorly for the human polymerase in reconstituted mismatch repair. However, the T7 enzyme is capable of efficient gap repair synthesis on deproteinized excision intermediates in the presence of RPA. Based on this finding, we presume that access to the excision intermediate is restricted in some way by one or more repair activities and that this nucleoprotein complex serves to recruit polymerase δ to the primer terminus.
MutSα, MutLα, EXOI, RPA, RFC and PCNA are sufficient to support bidirectional mismatch-provoked excision in a defined system (23). Inasmuch as EXOI hydrolyzes DNA with 5′ to 3′ polarity (48-50), it can account for 5′-directed excision mediated by these proteins. The activity responsible for 3′-directed hydrolysis in this reconstituted system has not been identified. Because the other components used were free of significant exonuclease activity, it was suggested that a cryptic 3′ to 5′ hydrolytic activity of EXOI might be responsible for 3′-directed excision in this system (23). However, the seven component system described in this study does include a 3′ to 5′ hydrolytic activity, the editing exonuclease of DNA polymerase δ. In fact, the strong synergistic increase in mutation rates observed with S. cerevisiae exoI pol3-01 and exoI pol2-4 double mutants, as compared to those observed with exoI,pol3-01, or pol2-4 single mutants, has led to the suggestion that in addition to their editing function, the 3′ to 5′ exonucleases of polymerases δ and ε participate in the excision step of mismatch repair (37). Given the multiple functions of EXOI (51), it is also possible that these genetic results indicate co-participation of these exonucleases in a mutation avoidance pathway distinct from mismatch repair (38). Indeed, the hypermutability associated with the pol3-01 editing exonuclease defect is dependent on S phase checkpoint activation (39). Nevertheless, it has also been reported that 3′-, but not 5′-directed, mismatch-provoked excision in HeLa nuclear extracts is inhibited to a substantial degree by aphidicolin, an effect attributed to participation of polymerase δ and/or ε editing exonuclease(s) in the excision step of mismatch repair (21).
In an attempt to further clarify possible function of the polymerase δ editing exonuclease in mismatch repair, we have asked whether this activity contributes to excision in the purified system. However, we were unable to detect significant excision in reactions that contained the DNA polymerase but lacked EXOI or in reactions that included both the polymerase and a catalytically dead form of EXOI. These findings strongly suggest that the polymerase 3′ to 5′ hydrolytic activity does not contribute substantially to excision in the defined system. We have also repeated previous experiments on aphidicolin effects on mismatch-provoked excision (20,21). In our hands, the effects of the drug on mismatch-provoked excision in HeLa nuclear extracts are limited and extract dependent. With some extracts, we have been unable to detect any inhibition, and in the case of those extracts where significant inhibition was observed, the effect was modest and evident with both 3′- and 5′-heteroduplexes. One explanation for these results is that the aphidicolin effects on excision are nonspecific in nature, although these findings could also indicate a limited dependence of both 3′- and 5′-directed excision on an aphidicolin-sensitive DNA polymerase. It is important to note in this regard that the substrates used to study in vitro mismatch repair are non-replicating DNAs. If editing exonuclease involvement in mismatch repair was dependent on a replication fork context, the nicked circular heteroduplexes used for in vitro assay may preclude detection of such an effect.
ACKNOWLEDGEMENTS
We thank Bruce Stillman and Mark Wold for providing expression plasmids for PCNA and RPA, Vladimir Podust and Ellen Fanning for polymerase δ baculoviruses, Marietta Lee for antiserum against the subunits of polymerase δ, and Elisabeth Penland for tissue culture support. We also thank Sally York and Vickers Burdett for useful suggestions, and Jochen Genschel for provision of EXOI and EXOI D173A.
Footnotes
This study was supported in part by grant GM45190 from the National Institutes of Health. P. M. is an Investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.Modrich P, Lahue R. Ann. Rev. Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 2.Jiricny J. Mutat. Res. 1998;409(3):107–121. doi: 10.1016/s0921-8777(98)00056-1. [DOI] [PubMed] [Google Scholar]
- 3.Buermeyer AB, Deschenes SM, Baker SM, Liskay RM. Annu. Rev. Genet. 1999;33:533–564. doi: 10.1146/annurev.genet.33.1.533. [DOI] [PubMed] [Google Scholar]
- 4.Kunkel TA, Erie DA. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 5.Su S-S, Modrich P. Proc. Natl. Acad. Sci. U. S. A. 1986;83(14):5057–5061. doi: 10.1073/pnas.83.14.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su S-S, Lahue RS, Au KG, Modrich P. J. Biol. Chem. 1988;263(14):6829–6835. [PubMed] [Google Scholar]
- 7.Parker BO, Marinus MG. Proc. Natl. Acad. Sci. U. S. A. 1992;89:1730–1734. doi: 10.1073/pnas.89.5.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grilley M, Welsh KM, Su S-S, Modrich P. J. Biol. Chem. 1989;264:1000–1004. [PubMed] [Google Scholar]
- 9.Galio L, Bouquet C, Brooks P. Nucleic Acids Res. 1999;27(11):2325–2331. doi: 10.1093/nar/27.11.2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spampinato C, Modrich P. J. Biol. Chem. 2000;275(13):9863–9869. doi: 10.1074/jbc.275.13.9863. [DOI] [PubMed] [Google Scholar]
- 11.Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P. J. Biol. Chem. 2001;22:28291–28299. doi: 10.1074/jbc.M103148200. [DOI] [PubMed] [Google Scholar]
- 12.Acharya S, Foster PL, Brooks P, Fishel R. Mol. Cell. 2003;12(1):233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 13.Selmane T, Schofield MJ, Nayak S, Du C, Hsieh P. J. Mol. Biol. 2003;334(5):949–965. doi: 10.1016/j.jmb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Au KG, Welsh K, Modrich P. J. Biol. Chem. 1992;267(17):12142–12148. [PubMed] [Google Scholar]
- 15.Cooper DL, Lahue RS, Modrich P. J. Biol. Chem. 1993;268(16):11823–11829. [PubMed] [Google Scholar]
- 16.Dao V, Modrich P. J. Biol. Chem. 1998;273:9202–9207. doi: 10.1074/jbc.273.15.9202. [DOI] [PubMed] [Google Scholar]
- 17.Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P. Proc. Natl. Acad. Sci. U. S. A. 2001;98(12):6765–6770. doi: 10.1073/pnas.121183298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viswanathan M, Burdett V, Baitinger C, Modrich P, Lovett ST. J. Biol. Chem. 2001;276(33):31053–31058. doi: 10.1074/jbc.M105481200. [DOI] [PubMed] [Google Scholar]
- 19.Lahue RS, Au KG, Modrich P. Science. 1989;245(4914):160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 20.Fang W.-h., Modrich P. J. Biol. Chem. 1993;268(16):11838–11844. [PubMed] [Google Scholar]
- 21.Wang H, Hays JB. J. Biol. Chem. 2002;277(29):26143–26148. doi: 10.1074/jbc.M200358200. [DOI] [PubMed] [Google Scholar]
- 22.Genschel J, Modrich P. Mol. Cell. 2003;12(5):1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- 23.Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P. Mol. Cell. 2004;15(1):31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 24.Yuan F, Gu L, Guo S, Wang C, Li GM. J. Biol. Chem. 2004;279:20935–20940. doi: 10.1074/jbc.M401931200. [DOI] [PubMed] [Google Scholar]
- 25.Genschel J, Bazemore LR, Modrich P. J. Biol. Chem. 2002;277:13302–13311. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- 26.Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou H, Jr., Kneitz B, Yang G, Kunkel TA, Kolodner RD, Cohen PE, Edelmann W. Genes Dev. 2003;17(5):603–614. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo S, Presnell SR, Yuan F, Zhang Y, Gu L, Li GM. J. Biol. Chem. 2004;279:16912–16917. doi: 10.1074/jbc.M313213200. [DOI] [PubMed] [Google Scholar]
- 28.Longley MJ, Pierce AJ, Modrich P. J. Biol. Chem. 1997;272(16):10917–10921. doi: 10.1074/jbc.272.16.10917. [DOI] [PubMed] [Google Scholar]
- 29.Amin NS, Nguyen MN, Oh S, Kolodner RD. Mol. Cell. Biol. 2001;21(15):5142–5155. doi: 10.1128/MCB.21.15.5142-5155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hubscher U, Maga G, Spadari S. Annu. Rev. Biochem. 2002;71:133–163. doi: 10.1146/annurev.biochem.71.090501.150041. [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Mo J, Rodriguez-Belmonte EM, Lee MY. J. Biol. Chem. 2000;275(25):18739–18744. doi: 10.1074/jbc.M001217200. [DOI] [PubMed] [Google Scholar]
- 32.Mo J, Liu L, Leon A, Mazloum N, Lee MY. Biochemistry. 2000;39(24):7245–7254. doi: 10.1021/bi0000871. [DOI] [PubMed] [Google Scholar]
- 33.Xie B, Mazloum N, Liu L, Rahmeh A, Li H, Lee MY. Biochemistry. 2002;41(44):13133–13142. doi: 10.1021/bi0262707. [DOI] [PubMed] [Google Scholar]
- 34.Podust VN, Chang LS, Ott R, Dianov GL, Fanning E. J. Biol. Chem. 2002;277:3894–3901. doi: 10.1074/jbc.M109684200. [DOI] [PubMed] [Google Scholar]
- 35.Waga S, Stillman B. Annu. Rev. Biochem. 1998;67:721–751. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- 36.Gu L, Hong Y, McCulloch S, Watanabe H, Li GM. Nucleic Acids Res. 1998;26(5):1173–1178. doi: 10.1093/nar/26.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran HT, Gordenin DA, Resnick MA. Mol. Cell. Biol. 1999;19(3):2000–2007. doi: 10.1128/mcb.19.3.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin YH, Garg P, Stith CM, Al-Refai H, Sterling JF, Murray LJ, Kunkel TA, Resnick MA, Burgers PM, Gordenin DA. Mol. Cell. Biol. 2005;25(1):461–471. doi: 10.1128/MCB.25.1.461-471.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datta A, Schmeits JL, Amin NS, Lau PJ, Myung K, Kolodner RD. Mol. Cell. 2000;6(3):593–603. doi: 10.1016/s1097-2765(00)00058-7. [DOI] [PubMed] [Google Scholar]
- 40.Holmes J, Clark S, Modrich P. Proc. Natl. Acad. Sci. U. S. A. 1990;87(15):5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang W.-h. Strand-specific mismatch repair in human cells. Duke University; 1993. [Google Scholar]
- 42.Huang J-C, Svoboda DL, Reardon JT, Sancar A. Proc. Natl. Acad. Sci. U. S. A. 1992;89:3664–3668. doi: 10.1073/pnas.89.8.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goscin LP, Byrnes JJ. Biochemistry. 1982;21(10):2513–2518. doi: 10.1021/bi00539a034. [DOI] [PubMed] [Google Scholar]
- 44.Lee MY, Tan CK, Downey KM, So AG. Biochemistry. 1984;23(9):1906–1913. doi: 10.1021/bi00304a003. [DOI] [PubMed] [Google Scholar]
- 45.Sabatino RD, Bambara RA. Biochemistry. 1988;27(7):2266–2271. doi: 10.1021/bi00407a003. [DOI] [PubMed] [Google Scholar]
- 46.Cheng CH, Kuchta RD. Biochemistry. 1993;32(33):8568–8574. doi: 10.1021/bi00084a025. [DOI] [PubMed] [Google Scholar]
- 47.Umar A, Buermeyer AB, Simon JA, Thomas DC, Clark AB, Liskay RM, Kunkel TA. Cell. 1996;87(1):65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- 48.Wilson DM, 3rd, Carney JP, Coleman MA, Adamson AW, Christensen M, Lamerdin JE. Nucleic Acids Res. 1998;26(16):3762–3768. doi: 10.1093/nar/26.16.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee BI, Wilson DM., 3rd J. Biol. Chem. 1999;274(53):37763–37769. doi: 10.1074/jbc.274.53.37763. [DOI] [PubMed] [Google Scholar]
- 50.Lee B, Nguyen LH, Barsky D, Fernandes M, Wilson DM., 3rd Nucleic Acids Res. 2002;30(4):942–949. doi: 10.1093/nar/30.4.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tran PT, Erdeniz N, Symington LS, Liskay RM. DNA Repair (Amst) 2004;3(12):1549–1559. doi: 10.1016/j.dnarep.2004.05.015. [DOI] [PubMed] [Google Scholar]