

Abstract
The C family G-protein-coupled receptors contain members that sense amino acid and extracellular cations, of which calcium-sensing receptor (CASR) is the prototypic extracellular calcium-sensing receptor. Some cells, such as osteoblasts in bone, retain responsiveness to extracellular calcium in CASR-deficient mice, consistent with the existence of another calcium-sensing receptor. We examined the calcium-sensing properties of GPRC6A, a newly identified member of this family. Alignment of GPRC6A with CASR revealed conservation of both calcium and calcimimetic binding sites. In addition, calcium, magnesium, strontium, aluminum, gadolinium, and the calcimimetic NPS 568 resulted in a dose-dependent stimulation of GPRC6A overexpressed in human embryonic kidney cells 293 cells. Also, osteocalcin, a calcium-binding protein highly expressed in bone, dose-dependently stimulated GPRC6A activity in the presence of calcium but inhibited the calcium-dependent activation of CASR. Coexpression of β-arrestins 1 and 2, regulators of G-protein signaling RGS2 or RGS4, the RhoA inhibitor C3 toxin, the dominant negative Gαq-(305–359) minigene, and pretreatment with pertussis toxin inhibited activation of GPRC6A by extracellular cations. Reverse transcription-PCR analyses showed that mouse GPRC6A is widely expressed in mouse tissues, including bone, calvaria, and the osteoblastic cell line MC3T3-E1. These data suggest that in addition to sensing amino acids, GPRC6A is a cation-, calcimimetic-, and osteocalcin-sensing receptor and a candidate for mediating extracellular calcium-sensing responses in osteoblasts and possibly other tissues.
The extracellular calcium-sensing receptor, which belongs to the C family (also called family 3) of G-protein-coupled receptors (GPCRs),2 plays an important role in the regulation of calcium homeostasis. Inactivating and activating mutations of the receptor in humans have been shown to respectively induce hyper- and hypoparathyrodism (1). CASR is widely expressed in many tissues (2), where it has been shown to play an important role in regulating parathyroid hormone secretion by the parathyroid gland, calcitonin secretion by C-cells in the thyroid, calcium and water transport by kidney epithelia, parathyroid hormone-related protein production and calcium transport in the mammary gland (3), and fluid fluxes in the gastrointestinal tract (4). Although calcium is the physiological ligand for CASR, other divalent and trivalent cations can also activate this receptor. In addition, CASR activity (in the presence of Ca2+) is allosterically modulated by amino acids (5) and small peptides (6, 7) as well as a family of structurally related phenylal-kylamines called calcimimetics and calcilytics (8, 9).
Extracellular calcium-sensing responses are also observed in tissues where the physiological role of CASR is not certain, such as bone. For example, cultured osteoblasts and osteoclasts from bone respond to increased extracellular calcium through a cell surface calcium-sensing mechanism that may be distinct from CASR (10). Although CASR has been detected in these cells by some investigators (11–13), others have been unable to identify CASR in either osteoblasts or osteoclasts (14–17). In addition, CASR null mice exhibit no abnormalities of osteoblastic and osteoclastic function after correction of hyperparathyroidism that results from the deletion of CASR in parathyroid glands (18). More importantly, osteoblasts derived from CASR null mice display unaltered responses to extracellular calcium (19), suggesting the presence of another extracellular calcium-sensing mechanism. Because the bone microenvironment has a higher calcium concentration than the systemic circulation and different calcium binding proteins due to the presence of extracellular matrix proteins, a unique calcium-sensing receptor may be required in osteoblasts. A putative osteoblastic calcium-sensing receptor has been proposed to exist in osteoblasts (Ob. CASR) that is distinguished from CASR by differences in ligand specificity and coupling to signal transduction pathways (10). Recent functional characterization of Ob. CASR indicates its ability to sense cations and amino acids and function like a G-protein-coupled receptor, suggesting that this putative receptor may also belong to the family C of GPCRs, which are characterized by an evolutionarily conserved amino acid-sensing motif linked to an intramembranous 7-transmembrane loop region (10).
The C family of GPCRs consists of eight metabotropic glutamate receptors (mGluR1–8), two γ-aminobutyric acid receptors (GABABR1/2), three taste receptors (T1R1, T1R2, and T1R3) and six orphan receptors (RAIG1, GPRC6A, GPRC5B-5D, and GABABL) in addition to CASR (20–27). Structural homologies and conservation of specific domains in some members of this family of receptors suggest an evolutionary link between extracellular calcium and amino acid-sensing (28). In addition to CASR (29, 30), other members of this receptor family, such as various mGluRs (31) and GABAB receptor (32), are capable of sensing extracellular calcium due to homology within a long amino-terminal domain that contains binding sites for both calcium and amino acids (5, 28). Three serine residues (Ser-170 > Ser-147 > Ser-169) in the extracellular domain and a proline residue (Pro-823) in the 7-transmembrane loop region domain of CASR are necessary for full responsiveness to extracellular calcium (33, 34). The serine 166, corresponding to Ser-147 in CASR, and the proline residue, corresponding to Pro-823, are conserved in mGluR1 and mGluR5, whereas mGluR3 has 2 conserved sites, corresponding to Ser-147, Ser-169, and Pro-823 in CASR (31, 32, 34). A distinct site, Ser-269, mediates the allosteric modulating effects of calcium on the GABAB receptor (35). CASR also senses l-amino acids in vitro (5). Ser-170 in CASR, which corresponds to amino acid binding residue Thr-188 in mGluR, is necessary for amino acid sensing in CASR and mGluRs (22). Calcimimetics, which are believed to be specific allosteric modulators of CASR, require Glu-837 as well as other sites in the transmembrane loop regions (36–39). These calcimimetic binding sites are not conserved in mGluRs, which do not respond to calcimimetics (8).
GPRC6A, an orphan receptor with a long amino-terminal domain, has recently been shown to sense amino acids (40–42), but the physiological ligands and function of this receptor have not been clearly established. In addition, calcium has been shown to augment the amino acid responsiveness of this receptor (5, 43), but the extracellular cation-sensing properties of the full-length native GPRC6A have not been extensively evaluated (40–42). To determine whether GPRC6A is a candidate for an alternative cation-sensing receptor, we examined the response of GPRC6A to a panel of extracellular cations, assessed whether this receptor is expressed in bone and osteoblasts, and determined whether it is responsive to calcimimetics as well as osteocalcin, the most abundant calcium-binding extracellular matrix protein in bone.
EXPERIMENTAL PROCEDURES
Materials
Aluminum chloride (AlCl3·6H2O) was obtained from Fisher. Calcium chloride, gadolinium chloride hexahydrate, magnesium chloride, and strontium chloride were purchased from Sigma. Bovine serum albumin (faction V) was obtained from Roche Applied Science. Osteocalcin (purified from bovine bone) was purchased from Biodesign International (Saco, ME).
Plasmids
GPRC6A cDNA was RT-PCR-amplified from human kidney total RNA and cloned into the expression vector pcDNA3.1(+)-Puro, which is the same as pcDNA3.1(+) (Invitrogen) except that the neomycin resistance gene is replaced by the gene encoding puromycin resistance. RNA was reverse-transcribed using random hexamers and Moloney murine leukemia virus (Promega, Madison, WI) according to the manufacturer’s protocol. A portion of the RT reaction mix, GPRC6A-specific primers (forward, 5′-cgaaacatggcattcttaattatact; reverse, 5′-atcctggaatgtggcatctctcctaag) and Pfu DNA polymerase (Stratagene, La Jolla, CA) were combined in a PCR reaction mix, and the GPRC6A cDNA was amplified using conventional cycling conditions. The 2816-bp cDNA amplimer was purified using Promega Wizard PCR cleanup reagents, then ligated into pcDNA3.1(+)-Puro vector previously digested with BamHI/EcoRV and made blunt-ended by treatment with Klenow. The ligation mix was electroporated into Escherichia coli DH10B, and several ampicillin-resistant colonies were picked and expanded in LB (amp) medium. Plasmid DNAs were purified from individual clones using Qiagen Qiaprep reagents, then digested with BamHI and EcoRV and size-separated by electrophoresis through a 0.8% agarose gel to identify those clones containing the full-length cDNA in the correct orientation relative to the CMV promoter. The integrity of the GPRC6A open reading frame was confirmed by comprehensive DNA sequencing. The Myc-tagged mouse GPRC6A cDNA was kindly provided by Drs. Yao and Hampson (40). Serum response element (SRE)-luciferase construct was a generous gift from Dr. Jeffrey E. Pessin (44). The rat CASR cDNA was obtained from Drs. A. M. Snowman and S. H. Snyder and subcloned in the mammalian expression vector pcDNA3 (Invitrogen) as previously described (45, 46). The Gαq-(305–359) minigene construct that corresponds to the COOH-terminal peptide sequence of Gαq residues 305–359 (47) and constructs of pcDNA3-Flag-rBarr1 and pcDNA3-FLAG-rBarr2 (48) were kindly provided by Drs. Louis M. Luttrell and Robert J. Lefkowitz from Duke University. All of the G protein α-subunits and C3 toxin (generously provided by Dr. J. Silvio Gutkind) were in pcDNA I expression vectors (49). RGS2, RGS4, and RGS12 in the p-cytomegalovirus (pCMV) expression vector as previously described (50) were generous gifts of Dr. Dianqing Wu (Department of Pharmacology and Physiology, University of Rochester).
PCR and RT-PCR Analysis
Mouse cDNA tissues panels were purchased from Clontech (Palo Alto, CA). The PCR primers for GPRC6A application are GPRC6A.F737 (5′-tgtgcattgccttcaaagag) and GPRC6A.R1812 (5′-gagagccaaggagtcatccc). Mouse glyceraldehyde-3-phosphate dehydrogenase was amplified as a control. Amplification products were resolved by electrophoresis on a 1.0% agarose gel and visualized by ethidium bromide staining.
RNA was isolated from mouse kidney, testis, epididymis, bone, and fat by grinding snap-frozen tissues in liquid nitrogen and then extracting total RNA using Trizol reagent (Molecular Research Center, Inc., Cincinnati, OH). RNA samples pretreated with DNase were further cleaned using an RNeasy spin column (Qiagen, Inc., Valencia CA), and the yield was quantified using a Ribogreen RNA quantitation kit (Molecular Probes, Eugene, OR). RT-PCR was performed using a two-step RNA PCR procedure by modification of previously described methods (19). Briefly, in separate reactions 2.0 μg of DNase-treated total RNA was reverse-transcribed into cDNA with the respective reverse primers specified below, and Moloney murine leukemia virus reverse transcriptase (Invitrogen). Reactions were carried out at 42 °C for 60 min followed by 94 °C for 5 min and 5 °C for 5 min. The products of first strand cDNA synthesis were directly amplified by PCR using AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA) using three separate sets of primers based on the human β-arrestins and G-protein-coupled receptor kinase cDNA sequence. PCR was performed with thermal cycling parameters of 94 °C for 3 min, 94 °C for 1 min, 60 °C for 1 min, and 72 °C for 2 min for 35 cycles followed by a final extension at 72 °C for 10 min. The primers for GPRC6A application were GPRC6A.F737 (5′-tgtgcattgccttcaaagag) and GPRC6A.R1812 (5′-gagagccaaggagtcatccc). Human glyceraldehyde-3-phosphate dehydrogenase was amplified as a control. Amplification products were resolved by electrophoresis on a 1.0% agarose gel and visualized by ethidium bromide staining.
Cell Culture
All culture reagents were from Invitrogen. Human embryonic kidney HEK-293 cells were obtained from American Type Culture Collection, Manassas, VA. HEK-293 cells stably expressing rat CASR were created as previously described (45, 46, 51).
HEK-293 cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal calf serum and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 95% air and 5% CO2. To evaluate cation responses, cells were induced to undergo growth arrest by overnight incubation in serum-free media (Dulbecco’s modified Eagle’s medium/F-12 containing 0.1% bovine serum albumin and 1% penicillin/streptomycin (Invitrogen). Cations or amino acids were added at the concentrations stated.
Transfection
HEK-293 cells were transiently co-transfected with SRE-luc and the following plasmids: β-arrestin 1, β-arrestin 2, Gαq-(305–355), C3 toxin, RGS2, RGS4, or RGS12 (46).
All plasmid DNAs were prepared using the EndoFree™ Plasmid Maxi Kit (Invitrogen). Transient transfections were performed as follows. 1 × 105 HEK-293 were plated in the 6-well plate and incubated overnight at 37 °C. A DNA-liposome complex was prepared by mixing DNA of the SRE-luciferase reporter plasmid, pCMV-β-gal, and other expression vectors as indicated with TransFast™ transfection reagent (Promega). The total plasmid DNA was equalized in each well by adjusting the total amount of DNA to 2 μg per well with the empty vector. Quiescence of transfected cells was achieved in subconfluent cultures by removing the media and washing with Hanks’ balanced salt solution (Invitrogen) to remove residual serum followed by incubation for an additional 24 h in serum-free quiescent media. Luciferase activity was assessed after 8 h of stimulation. The luciferase activity in cell extracts was measured using the luciferase assay system (Promega) following the manufacturer’s protocol using a BG-luminometer (Gem Biomedical, Inc., Hamden, CT).
To assess the impact of inhibition of Gαi, we used HEK-293 cells stably transfected with GPRC6A and SRE-luciferase plasmids plated in 6-well plates at 1 × 105 cells/well and incubated for 48 h at 37 °C. Quiescence of transfected cells was achieved in subconfluent cultures by removing the media and washing with Hanks’ balanced salt solution (Invitrogen) to remove residual serum followed by incubation for an additional 24 h in serum-free quiescent media. Then cells were pre-treated with 100 ng/ml pertussis toxin (Sigma) for 5 h. Luciferase activity was assessed after 8 h, and ERK activity was assessed after 10 min of stimulation with 10 mm calcium.
Confocal Microscopic Analysis
The 5 × 105 HEK-293 cells were plated in Dulbecco’s modified Eagle’s medium on the coverslips in 60-mm plates and grown overnight at 37 °C. The cells were transfected with Myc-tagged mouse GPRC6A cDNA using TransFast™ transfection reagent (Promega). At 48 h post-transfection the cells were washed with PBS and fixed in 4% paraformaldehyde for 30 min. The cells were then washed twice with PBS. The cells were subsequently incubated with the anti-Myc-fluorescein isothiocyanate antibody (Invitrogen) at a 1:500 dilution in PBS containing 1% bovine serum albumin for 1 h at room temperature in the dark. The cells were washed twice for 5 min each with PBS containing 1% bovine serum albumin, mounted onto microscope slides using the ProLong Antifade Kit (Molecular Probes), and examined under a Zeiss LSM 510 laser-scanning microscope.
Assay for ERK1/2 Mitogen-activated Protein Kinase
After 10 min of agonist treatment for the specified concentrations and duration, cells were washed twice with ice-cold PBS and scraped into 250 μl of lysis buffer (25 mm HEPES pH 7.2, 5 mm MgCl2, 5 mm EDTA, 1% Triton X-100, 0.02 tablet/ml of protease inhibitor mixture). Equal amounts of lysates were subjected to 10% SDS-PAGE, and phospho-ERK1/2 levels were determined by immunoblotting using anti-phospho-ERK1/2 mitogen-activated protein kinase antibody (Cell Signaling Technology, Beverly, MA). To confirm that variations in the amount of ERK did not contribute to stimulated ERK activity, in selected studies we used an anti-ERK1/2 mitogen-activated protein kinase antibody (Cell Signaling Technology) to measure ERK levels.
Intracellular Calcium Measurement
HEK-293 cells stably expressing the chimeric G protein Gqi5 were transfected with an expression vector encoding the full-length GPRC6A gene using FuGENE 6 (Roche Applied Science) according to the manufacturer’s recommended protocol. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 250 μg/ml hygromycin, and 3 μg/ml puromycin at 37 °C, 5% CO2. Clones were selected and analyzed for GPRC6A expression by quantitative PCR.
Cells were seeded at 2 × 104 cells/well in a collagen-coated, black-walled 384-well plate (Corning, Corning, NY) in growth media and placed in a 37 °C, 5%CO2 incubator overnight. Media was aspirated and replaced with 137 mm NaCl, 5.4 mm KCl, 0.5 mm CaCl2, 0.02 mm MgSO4, 3 mm NaHCO3, 0.64 mm KH2PO4, 5.5 mm glucose, 20 mm HEPES, pH 7.4, containing 1 × Calcium 3 dye (Molecular Probes) and 2.5 mm probenecid (Sigma). Cells were placed in a 37 °C, 5% CO2 incubator for 1 h. CaCl2 or SrCl2 was added to 5, 40, or 80 mm final concentration, and intracellular calcium measurements were made using a Fluorescence Imaging Plate Reader (Molecular Devices, Sunnyvale, CA).
Statistics
We evaluated differences between groups by one-way analysis of variance. Values sharing the same superscript are not significantly different at p < 0.05. All computations were performed using the Statgraphic statistical graphics system (STSC, Inc., Rockville, MD).
RESULTS
GPRC6A Has Conserved Putative Binding Residues for Calcium and Calcimimetics
We aligned the human and mouse GPRC6A amino acid sequences with those of CASR and mGluRs to determine whether the potential sites required for calcium and calcimimetics actions are conserved in GPRC6A (Fig. 1). We found that two of the amino acids in the extracellular domain and one in the 7-transmembrane loop region that are important in calcium sensing by CASR (Ser-147, Ser-169, Pro-823) correspond to Ser-149, Ser-171, and Pro-798 of human GPRC6A and Ser-149, Ser-171, and Pro-800 for mouse GPRC6A (Fig. 1, A and B). Interestingly, the calcium binding site Ser-170 in CASR is not conserved in GPRC6A, but instead, this residue is Thr-172 in GPRC6A, corresponding to Thr-188 in mGluR that is necessary for amino acid sensing (41). With regard to potential calcimimetic binding, Glu-814 in mouse GPRC6A and Glu-812 in human GPRC6A correspond to the consensus calcimimetic binding site Glu-837 of CASR (Fig. 1B) (36–39).
FIGURE 1. Comparison of conserved calcium and calcimimetic binding residues in members of the family C GPCRs.

A, extracellular region containing calcium binding sites. The amino acids required for calcium sensing in CASR, Ser-147, Ser-169, and Pro-823 (boxed residues) but not Ser-170 (*) are conserved between CASR and GPRC6A. m-, mouse; h-, human. B, a segment of the transmem-brane region containing putative calcimimetic binding sites. The Glu-837 binding site for calcimimetics in CASR is conserved in GPRC6A but not mGluRs. Regions of the extracellular domain and transmembrane domain from human and mouse GPRC6A (NM_148963 for human GPRC6A, and NM_153071 for mouse GPRC6A) were aligned with the human CASR (NM_000388) amino acid sequence and with members of the mGluRs (NM-000838 for human mGluR1, NM_000840 for human mGluR3, and NM_000842 for human mGluR5) with calcium-sensing properties.
GPRC6A Is an Extracellular Cation-sensing Receptor
We transfected an epitope-tagged GPRC6A cDNA into HEK-293 cells and assessed cell surface expression by confocal microscopy (Fig. 2). We found an immunofluorescence pattern consistent with cell surface expression, similar to previously published findings (40).
FIGURE 2. Cell surface expression of GRPC6A.
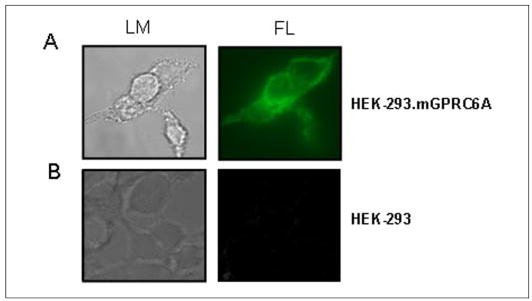
A, confocal microscopy images of HEK-293 cells transiently transfected with the Myc-tagged mouse GPRC6A. B, non-transfect HEK-293 controls. GPRC6A was detected with an anti-c-Myc fluorescein isothiocyanate antibody. Left panel, representative view using light microscopy (LM). Right panel, the same view under fluorescent microscopy (FL), magnification 100×. Immunofluorescence was observed in a peripheral pattern consistent with cell surface membrane expression.
We measured cation stimulation of CASR transfected into HEK-293 cells as assessed by ERK phosphorylation (52), SRE promoterluciferase reporter activity (46, 48, 51), and increments in intracellular calcium (53), which have previously been used to assess CASR activity. Initially, we compared GPRC6A and CASR activation in transfected HEK-293 cells using ERK activity (Fig. 3A). We observed a dose-dependent activation by extracellular calcium of GPRC6A, with demonstrable activation occurring at 3 mm and continued activation at concentrations up to 60 mm calcium (Fig. 3A, upper panel). In contrast, CASR transfected into HEK-293 cells also was activated by extracellular calcium in a dose-dependent fashion, with a maximal response occurring at a calcium concentration of 5 mm (Fig. 3A, lower panel). In addition the CASR agonists, strontium, gadolinium, and magnesium were able to activate ERK through GPRC6A (Fig. 3B). In addition, the putative Ob. CASR agonist aluminum, which activates Ob. CASR but not CASR (45), also activated ERK in HEK-293 cells transfected with GPRC6A (Fig. 3B). None of the cations stimulated ERK activity in HEK-293 cells not transfected with GPRC6A (Fig. 3 and data not shown).
FIGURE 3. Effect of various extracellular cations on GPRC6A-mediated EKR activation.
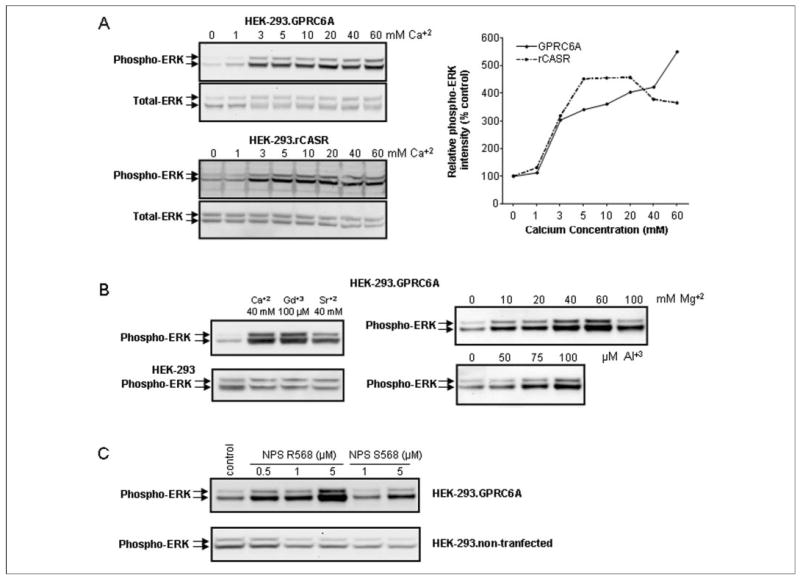
A, comparison of dose-dependent effects of Ca2+on GPRC6A- and CASR-mediated ERK activation. Extracellular calcium activated both receptors, but GPRC6A responded to higher concentrations of extracellular calcium. B, cation specificity of GPRC6A activation. In addition to Ca2+, Gd3+ (100 μm) and Sr2+ (40 mm) as well as Mg2+ and Al3+ stimulated GPRC6A-mediated ERK activation. Magnesium and aluminum, similar to calcium (A) also resulted in a dose-dependent stimulation of ERK activity in HEK-293 cells transfected with GPRC6A. ERK was not activated by cations in controls HEK-293 cells without GPRC6A. C, the calcimimetic NPS R568 stimulates GPRC6A-mediated EKR activity. Application of the active isomer NPS R568 resulted in a dose-dependent stimulation of GPRC6A-mediated ERK activation (at concentrations of 0.5 μm) in the presence of 1 m m extracellular Ca2+, but the less active isomer NPS S568 was effective only at concentrations 5 μm. The HEK-293 cells transfected with GRPC6A or CASR were incubated in Dulbecco’s modified Eagle’s medium/F-12 containing 0.1% bovine serum albumin quiescence media and exposed to the various cations or calcimimetics for 10 min, and ERK activation was determined as described under “Experimental Procedures.”
We also evaluated if GPRC6A is activated by calcimimetics. In the presence of 1 mm [Ca2+]e in the media, we found that the phenylalkylamine NPS R568 stimulated SRE-luciferase and ERK activity in HEK-293 cells overexpressing GPRC6A (Fig. 3D). GPRC6A is very sensitive to NPS R568, responding to concentrations of 0.5 μm. The R enantiomer of NPS 568 is ~10-fold more potent than its corresponding S enantiomer in activating CASR (8). Similarly, NPS S568 is ~10-fold less potent in activation of GPRC6A (Fig. 3C).
Next, we evaluated the extracellular calcium-sensing properties of GPRC6A by testing the ability of a panel of divalent and trivalent cations to stimulate GPRC6A-dependent SRE-luciferase activity in HEK-293 cells co-transfected with GPRC6A and the SRE-luciferase promoter/reporter construct (Fig. 4). We found that the divalent cations calcium, magnesium, and strontium dose-dependently stimulated SRE-luciferase activity, achieving a maximum stimulation at ~40–60 mm (Fig. 4A). The trivalent cation gadolinium, which activates CASR, stimulated GPRC6A-dependent SRE-luciferase activity, achieving maximum stimulation at 100 μm (4A).
FIGURE 4. Effects of polyvalent cations on GPRC6A-induced SRE-reporter gene activity and intercellular calcium in HEK-293 cells.
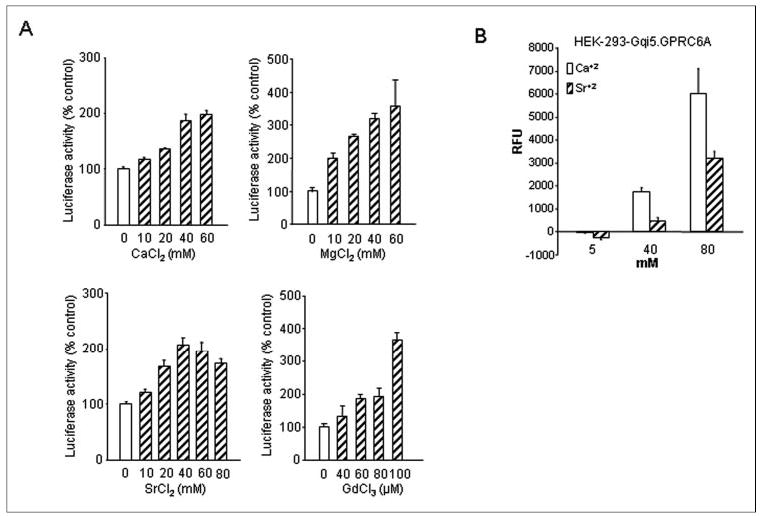
A, dose-dependent effects of various cations, Ca2+, Mg2+, Sr2+, and Gd3+, on SRE-luciferase activity in HEK-293 cells stably transfected with human GPRC6A cDNA and the SRE-luciferase reporter construct. Growth arrest was induced in 3-day-old subconfluent cultures by preincubation for 24 h under serum-free conditions before the addition of cations at the indicated concentrations. Luciferase activities were measured as described under “Experimental Procedures.” The values depicted represent the mean ± S.E. B, GPRC6A-mediated intracellular calcium mobilization in response to Ca2+ and Sr2+. HEK-293/Gqi5 cells stably expressing GPRC6A were seeded at 2 × 104 cells/well and loaded with Calcium 3 dye (Molecular Probes). Cells were treated with 5, 40, and 80 mm Ca2+ or Sr2+, and fluorescence was measured using Fluorescence Imaging Plate Reader as described under “Experimental Procedures.” The graph shows the magnitude of the response due to GPRC6A. RFU, relative fluorescence units.
We also evaluated the ability of GPRC6A to activate intracellular calcium signaling. Intracellular calcium was assessed in HEK-293 cells transfected with GPRC6A and a chimeric Gαq/i construct (54, 55) to enhance signaling sensitivity (Fig. 4B). Both calcium and strontium resulted in dose-dependent stimulation of GPRC6A as assessed by in vitro fluorescence-based measurement of [Ca2+]i mobilization. Whereas the addition of 5 mm Ca2+ or Sr2+ to Fluo 3-loaded HEK-CASR cells had no effect, the addition of 40 and 60 mm Ca2+ or Sr2+ induced a significant increase in intracellular fluorescence. The response to extracellular calcium was rapid and remained steady after reaching a maximal response within 20–30 s, whereas the response to strontium is characterized by a rapid increase in intracellular calcium and the resolution of the response to base line. HEK-293 cells that were not transfected with GPRC6A did not demonstrate this response to either Ca2+ or Sr2+. These data suggest that the rapid extracellular calcium-induced rise in fluorescence in HEK-GPRC6A cells is a receptor-mediated intracellular calcium response that is specific for GPRC6A.
GPRC6A Is Coupled to Gαi, Gαq, and Rho A
To confirm that GPRC6A has characteristics of other GPCRs, we examined the effects of the ubiquitously expressed β-arrestin1 and β-arrestin 2 on GPRC6A activation (56). To accomplish this, we tested agonist-stimulated SRE-luciferase activity in HEK-293 cells transfected with GPRC6A in the presence and absence of coexpressed β-arrestin1 and 2 (Fig. 5A). β-Arrestin 1 and 2 each resulted in ~20–30% reduction in strontium-stimulated GPRC6A activity.
FIGURE 5. G-protein coupling of GPRC6A.
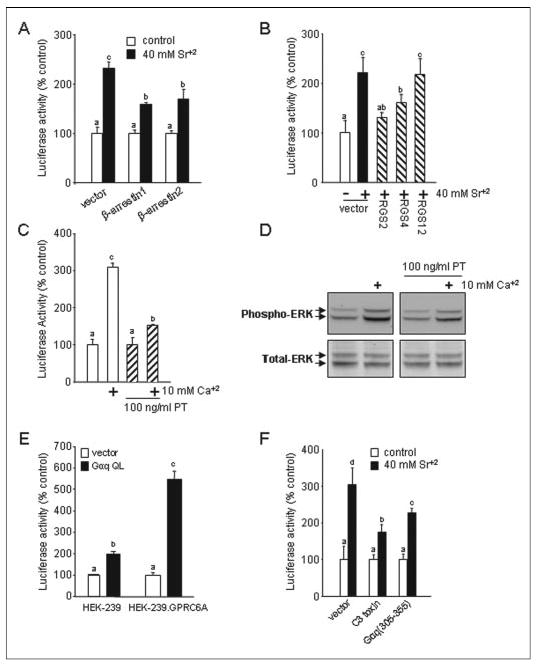
A, overexpression of β-arrestins block GPRC6A-mediated activation of the SRE. HEK-293 cells were cotransfected SRE-luciferase and pCMV-β-gal (0.015 μg) along with the construct (1.0 μg) directing the expression of β-arrestin 1 or β-arrestin 2. B, inhibition of GPRC6A activity by expression of RGS2, -4, -12. HEK-293 cells were cotransfected with expression vectors for RGS2, RGS4, or RGS12 (1 μg) along with GPRC6A (0.5 μg), the SRE-luciferase reporter gene (0.01 μg), and pCMV-β-gal (0.015 μg). C and D, pertussis toxin (PT) inhibits GPRC6A activation of the SRE (C) and ERK (D). HEK-293 cells, which stably cotransfected GPRC6A and SRE-luciferase plasmid DNAs, were cultured in serum-free media for 24 h. After pretreatment with 100 ng/ml pertussis toxin for 5 h, the cells were stimulated by 10 mm calcium. E, effect of activated mutants of Gαq subunits on the activity of the SRE. HEK-293 cells were cotransfected with 0.5 μg of expression vectors for the constitutively activated mutant of Gαq (Gαq QL) along with expression vectors for GPRC6A (0.5 μg) or empty vector, the SRE-luciferase reporter gene (0.01 μg), and p-cytomegalovirus-β-galactosidase (pCMV-β-gal) (0.015 μg). F, GPRC6A activity is inhibited by the expression of Gαq minigene construct, Gαq-(305–359), and Rho A-specific inhibitor C3 toxin. HEK-293 cells were cotransfected with the constructs directing the expression of Gαq-(305–359) (1.0 μg) or C3 toxin construct (1.0 μg) with the GPRC6A (0.5 μg), the SRE-luciferase reporter gene (0.01 μg), and pCMV-β-gal (0.015 μg). Data are shown as relative luciferase activity reported as the percent induction compared with the activity under non-stimulated conditions and normalized for β-galactosidase. Values represent the mean ± S.E. of at least three experiments. Values sharing the same superscript are not significantly different at p < 0.05.
To determine the G-protein subunits that mediate GPRC6A stimulation of SREs, we tested RGS2, which lacks in vitro GAP activity for Gαi but is a potent inhibitor of Gαq, RGS4, which has GTPase-activating effects for the Gαi and/or Gαq subunits, and RGS2, which inhibits G12-and G13-mediated signaling (50, 57). Coexpression of either RGS2 or -4 with GPRC6A significantly inhibited cation-induced SRE activation, whereas RGS2 had no effect (Fig. 4B). To further establish the role of Gαi, we pretreated HEK-293 cells expressing GPRC6A with pertussis toxin, a selective irreversible inactivator of Gαi/Gαo (58). Pretreatments with pertussis toxin partially inhibited the ability of extracellular calcium to stimulate GPRC6A-dependent activation of SRE-luciferase (Fig. 5C) and ERK (Fig. 5D) in HEK-293 cells. We cotransfected GPRC6A-expressing HEK-293 cells with the SRE reporter gene plasmid and a cDNA encoding the constitutively active Gαq subunit (Gαq QL). We found that cells expressing the activated α-subunit of Gαq produced the highest induction of luciferase activity in the presence of GPRC6A (Fig. 5E). The activation of SRE by Gαq QL was significantly less in the absence of GPRC6A (Fig. 5E), suggesting that Gαi and Gαq might act in concert.
To further examine the potential involvement of Gαq, we attempted inhibition of Gαq-mediated signaling in the HEK-293 cells with a Gαq minigene, Gαq-(305–359). This minigene consists of the COOH-terminal peptide residues 305–359 and has previously been shown to uncouple Gαq-coupled receptors without affecting coupling to other classes of Gαsubunits (47). Coexpression of Gαq-(305–359) with GPRC6A resulted in significant inhibition of cation-stimulated luciferase activity (Fig. 5F). Collectively these data indicate that GRPC6A is coupled to both Gαi and Gαq.
Finally, we investigated whether Rho is a downstream effector of GPRC6A activation of SRE, as has been reported for CASR (46) using a DNA construct that expresses the C3 exoenzyme (Fig. 5F). This toxin ADP ribosylates Rho A at asparagine 41, thereby preventing the exchange of GDP by GTP and retaining Rho A in its GDP-bound inactive form (59). Cotransfection with a plasmid expressing the C3 toxin inhibits GPRC6A-stimulated SRE activity (Fig. 5F).
GPRC6A Is Expressed in Bone and Cultured Osteoblasts
Prior studies indicate that the mRNA for GPRC6A is widely expressed in many tissues and organs, including lung, liver, spleen, heart, kidney, skeletal muscle, testis, and brain (40–42), but no studies to date have investigated GPRC6A expression in bone and osteoblasts. We examined the expression of GPRC6A in a commercial mouse cDNA tissue panel (Fig. 6A) and in mouse tissues and cell lines (Fig. 6, B and C). Transcripts for GPRC6A were detected in all tissues analyzed, including bone, calvaria, and fat, which have not been previously tested. The sequence of the RT-PCR product obtained from kidney (Fig. 6B) was confirmed to be GPRC6A by direct sequencing. In addition we were able to amplify the predicted size band from cultured osteoblasts (Fig. 6C). In contrast, we have previously reported the inability to amply CASR in these osteoblasts (60).
FIGURE 6. Expression of GPRC6A in mouse tissues and osteoblasts.
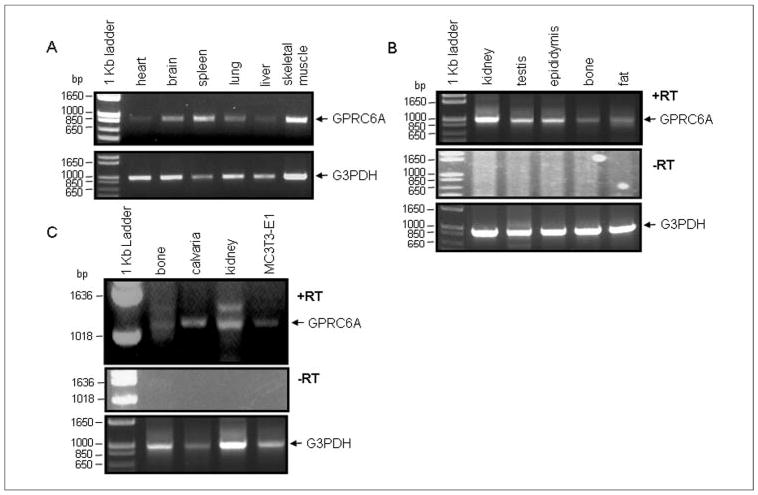
A, expression pattern of GPRC6A from the mouse cDNA panel. PCR products were amplified from multiple tissue cDNA panels that contain normalized adult mouse cDNA preparations. kb, kilobase. B, GPRC6A expression in mouse tissues by RT-PCR. The primers for GPRC6A application are GPRC6A.F737 (tgtgcattgccttcaaagag) and GPRC6A.R1812 (gagagccaaggagtcatccc). C, GPRC6A expression in mouse bone tissues and osteoblastic cell line MC3T3-E1 by RT-PCR. The primers for GPRC6A application are GPRC6A.F1321 (gctcgagactgcaagaaacc) and GPRC6A.R2320 (tgaaggccagaactgtgatg). −RT indicates negative control from omitting the reverse transcription step. We used the housekeeping control gene glyceraldehyde-3-phosphate dehydrogenase (G3PDH) for a positive control of RNA integrity.
Osteocalcin Acts in Concert with Calcium to Stimulate GPRC6A but Not CASR
Given evidence for GPRC6A expression in bone and osteoblasts, the reported presence of high concentrations of calcium in the bone microenvironment and the potential of extracellular matrix proteins in bone to act as calcium binding proteins, we investigated the ability of osteocalcin, the most abundant calcium-binding extracellular matrix protein in bone, to modify the effects of calcium and other cations on GPRC6A activation (61). Consistent with the observation that millimolar levels of free calcium promote osteocalcin to adopt the α-helical conformation (62), we found that osteocalcin in the presence of extracellular calcium significantly enhanced SRE-luciferase activity in HEK-293 cells that stably expressed GPRC6A (Fig. 7A). These effects depended on the prevailing level of extracellular Ca2+. In this regard, at sub-threshold extracellular Ca2+ (≤1.0 mm), osteocalcin (up to 40 ng/ml) had no effect (Fig. 7A). However, at 1 and 5 mm Ca2+, osteocalcin had a dose-dependent effect to stimulate GPRC6A-dependent increments in SRE-luciferase activity. The addition of both strontium and magnesium to the extracellular media also enhanced osteocalcin stimulation of GPRC6A, but the addition of the trivalent cation aluminum did not (Fig. 7B).
FIGURE 7. Osteocalcin is a co-factor for GPRC6A.
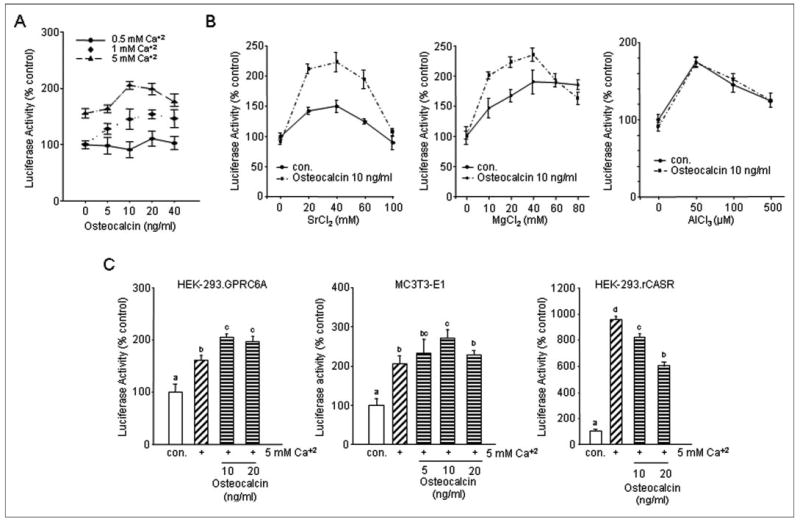
A, dose-dependent enhancement by osteocalcin of calcium-mediated activation of GPRC6A. B, osteocalcin augments strontium-and magnesium-stimulated SRE-luciferase activation by GPRC6A transfected into HEK-293 cells but not the effect of aluminum. C, comparison of osteocalcin activation of CASR. Ob. CASR and GPRC6A. HEK-293 cells transfected with GPRC6A respond to osteocalcin in the presence of extracellular calcium similar to MC3T3-E1 osteoblasts. Osteocalcin inhibits calcium stimulation of CASR transfected into HEK-293 cells. Values represent the mean ± S.E. of at least three experiments. Values sharing the same superscript are not significantly different at p < 0.05. con, control.
Finally, we compared the ability of osteocalcin to stimulate SRE-luciferase activity in the MC3T3-E1 cell line (Fig. 7C), which lacks CASR but has a functional response to extracellular cations (19, 63), with that of HEK-293 cells transfected with GPRC6A (Fig. 7C). In the presence of 5 mm calcium, the addition of osteocalcin to the media resulted in a dose-dependent increase in SRE-luciferase activity, achieving a maximum response at osteocalcin concentrations of 10 ng/ml in both MC3T3-E1 osteoblasts and HEK-293 cells transfected with GPRC6A. In contrast, osteocalcin had the opposite effect to inhibit calcium-stimulated SRE-luciferase activity in HEK-293 cells expressing CASR (Fig. 7C).
DISCUSSION
Several members of the C family of GPCRs have dual amino acid and calcium-sensing properties due to conserved binding sites in their large extracellular domains. In the current study we show that GPRC6A, an orphan receptor recently shown to sense amino acids (40, 41), has conserved binding sites for calcium (i.e. Ser-149 and Ser-171) and an amino acid (Thr-172) in its large extracellular domain and a conserved consensus calcimimetic binding site (Fig. 1), suggesting that this receptor may also dually sense both extracellular calcium and amino acids as well as calcimimetics. Indeed, we found that GPRC6A is capable of responding to extracellular cations, including calcium as well as the calcimimetic NPS 568, which was previously thought to be specific for CASR (Figs. 3 and 4). These findings suggest that GPRC6A is a unique receptor with similarities to both CASR and amino acid sensing mGluRs and is the second member of this family shown to be a target for calcimimetics.
There are important differences, however, between the extracellular cation-sensing properties of GPRC6A and CASR. For example, the apparent affinity for extracellular calcium appears to be lower for GPRC6A compared with CASR. In this regard, calcium in doses of up to 40 mm was necessary to maximally activate GPRC6A, whereas the CASR is maximally activated by lower calcium concentrations (Fig. 3A). In addition, using intracellular calcium to detect GPRC6A, we consistently failed to see evidence for activation by calcium at concentrations of 5 mm, which we have shown will activate CASR (Fig. 4B). This suggests that the affinity of GPRC6A for calcium might be lower than for CASR. In contrast, using ERK or SRE-luciferase to detect GPRC6A activation, we observed activation by 5 mm extracellular calcium but not 1 mm calcium unless osteocalcin was present (Figs.3and4). Cautionisneeded, however, in estimating the affinity of receptors in vitro, since this might be influenced by the level of receptor expression, the cell context, and the sensitivity of the method used to detect receptor activation. Indeed, millimolar levels of calcium are required to activate CASR transfected into heterologous cells in vitro, whereas this receptor is sensitive to 0.1 mm changes in ionized calcium in vivo. The ligand specificity of GPRC6A is also different from CASR. Whereas GPRC6A shares with CASR ligands such as magnesium, strontium, and gadolinium, the trivalent cation aluminum, which activates GPRC6A at μm concentrations, is only a weak agonist for CASR (45). Finally, osteocalcin in the presence of a threshold concentration of calcium can stimulate GPRC6A, but these conditions inhibit calcium-mediated CASR activation (Fig. 7). These concentrations of osteocalcin necessary to activate GPRC6A are well with the physiological concentrations found in normal serum. Thus, GPRC6A is evolutionarily linked to CASR but has distinct apparent affinities for extracellular cations and differences in ligand specificity.
The expression of GPRC6A in bone and osteoblasts (Fig. 6) raises the possibility that GPRC6A may account for the novel calcium-sensing response reported in bone and osteoblasts derived from CASR knock-out mice (41). The ability of GPR6A to respond to aluminum, which is capable of stimulating de novo bone formation under certain experimental conditions (64), and the ability of osteocalcin to stimulate GPRC6A in the presence of calcium qualify this receptor as a candidate for Ob. CASR, which is also activated by aluminum (65) and osteocalcin in combination with calcium (Fig. 7C). Moreover, calcimimetics, which activate the putative osteoblastic cation-sensing receptor (66), also activate GPRC6A (Fig. 3C). Other characteristics of GPRC6A, however, differ from the putative Ob. CASR. In this regard, magnesium, which activates GPRC6A, is not a ligand for Ob. CASR. In addition, Ob. CASR appears to be coupled to SRE-luciferase activity via pathways not linked to RhoA (10), whereas GPRC6A is coupled to RhoA-dependent pathways in HEK-293 cells. Whether some of these discrepancies can be explained by the differences between evaluating a transfected receptor in HEK-293 cells and endogenous receptor osteoblasts remains to be established. Regardless, our data raise the novel possibility that calcium and osteocalcin released from bone resorption may act in concert to stimulate osteoblast-mediated bone formation through the activation of GPRC6A.
At present we have insufficient data to determine the physiologically relevant ligand for GPRC6A or to establish whether it is a primary amino acid-sensing, calcium-sensing, or osteocalcin-sensing receptor. Other members of the C family of GPCRs, such as mGluRs and GABA receptors, also are stimulated by extracellular calcium in vitro, but their physiological ligands are not calcium (29, 31). Conversely, l-amino acids are capable of stimulating CASR, but calcium, rather than amino acids, is the most important physiologically relevant ligand for CASR. Additional information is needed to determine whether calcium and/or other cations are the physiological ligands for GPRC6A or whether its ability to sense calcium represents a secondary property common to all members of the family C GPCRs. At present, mutations of GPRC6A, which is located on human chromosome 6 and mouse chromosome 10, have not been implicated in any human diseases. Therefore, mouse genetic approaches will be necessary to elucidate the physiological function of this receptor and to establish whether it has a role in regulating calcium homeostasis. Efforts are currently under way to characterize a GPRC6A null mouse model.
Our findings differ from other recent studies of GPRC6A, which failed to find an effect of extracellular calcium to activate the receptor transfected into Xenopus oocytes (40). These discrepancies might be explained by difference in cell type, the sensitivity of the method used to assess receptor activation, or the use of insufficient concentrations of extracellular calcium. These studies also did not evaluate the effects of other cations, which we have shown to activate GRPC6A transfected into HEK-293 cells. It is also important to note that we used a full-length GPRC6A cDNA construct used in our studies, whereas others examined GPRC6A function using a using a chimeric receptor generated using the extracellular domain of GPRC6A and transmembrane domain and COOH terminus of the homologous goldfish 5.24 receptor (67). Also, prior studies of the human GPRC6A activation were confounded by the inability to achieve cell surface expression in mammalian cell lines (41). On the other hand, others have demonstrated trafficking of mouse GPRC6A to the cell surface membrane (40). We have confirmed cell surface expression of GPRC6A (Fig. 2), and the functional responses to the cell-impermeable aluminum and gadolinium of our transfected human GPRC6A cDNA are consistent with its cell surface expression. In addition, β-arrestins, which uncouple the receptors from their cognate G-protein-mediated signaling (56), also was effective in inhibiting GPRC6A activation (Fig. 5A). Additional data indicate that GPRC6A is coupled to Gαi and Gαq. Evidence for a role Gαi include the effects of pertussis toxin (PT) and RGS4 to inhibit GPRC6A activation (Fig. 5, B–D). There is also evidence of involvement of Gαq. In this regard RGS2, which has GTPase-activating effects for Gαq subunits (Fig. 5B) dominant negative Gαq-(305–355) (Fig. 5F), all inhibited GPRC6A signaling. Thus, GPRC6A appears to be a cell surface-expressed GPCR that is coupled to Gαi and Gαq. It is not clear, however, why we were unable to demonstrate an effect of GPRC6A activation to increase intracellular calcium unless HEK-293 cells were transfected with the chimeric Gαq/i construct, which allows Gαi-coupled receptors to activate phosphatidylinositol-phospholipase C (55). A similar paradox exists for Ob. CASR, which appears to be both Gαi and Gαq, but does not lead to increments in intracellular calcium (10). Also, the activation of the rabbit PGF2αreceptor, which is coupled to Gαi and Gαq, failed to increase intracellular calcium in HEK-293 cells (68), suggesting that activation of Gαq is not always sufficient for stimulating signal transduction pathway required for increasing intracellular calcium.
Regardless, the similar response to amino acids, extracellular calcium, and calcimimetics by the related calcium-sensing receptor homolog, CASR, along with the conserved residues between GPRC6A and CASR that are responsible for cation and calcimimetic responses suggest that CASR and GPRC6A have overlapping functions. Whether GPRC6A is the functionally important extracellular cation-sensing receptor in bone or other tissues and explains the GPCR response to extracellular cations found in CASR deficient tissues is speculative. However, the bone microenvironment, which consists of high extracellular calcium concentrations and bone-specific extracellular matrix calcium binding proteins such as osteocalcin, likely imposes unique requirements for osteoblast sensing of extracellular calcium, possibly through a novel, low calcium affinity receptor. Additional studies are warranted to investigate the functional role of GPRC6A in osteoblasts and the role of GPRC6A in regulating bone formation as well as its function in other tissues, such as skeletal muscle, adipose tissue, and kidney, where it is also expressed. Moreover, the response of GPRC6A to calcimimetics indicates that it may be possible to develop specific ligands or allosteric modulators to target GPRC6A and to develop therapeutic agents that regulate receptor function in tissues where it is of physiological importance.
Footnotes
The work was supported by NIAMS, National Institutes of Health Grant 2R01AR037308.
The abbreviations used are: GPCR, G-protein coupled receptors; GPRC6A, G-protein-coupled receptor family C, group 6, subtype A; CASR, calcium-sensing receptor; mGluR, metabotropic glutamate receptor; Ob., osteoblasts; RGS, regulators of G protein signaling; SRE, serum response element; HEK, human embryonic kidney cells; GABA, γ-aminobutyric acid receptor; RT, reverse transcription; p-CMV, p-cytomegalovirus; ERK, extracellular signal-regulated kinase; PBS, phosphate-buffered saline.
References
- 1.Hofer AM, Brown EM. Nat Rev Mol Cell Biol. 2003;4:530–538. doi: 10.1038/nrm1154. [DOI] [PubMed] [Google Scholar]
- 2.Chattopadhyay N, Brown EM. Cell Signal. 2000;12:361–366. doi: 10.1016/s0898-6568(00)00082-6. [DOI] [PubMed] [Google Scholar]
- 3.VanHouten J, Dann P, McGeoch G, Brown EM, Krapcho K, Neville M, Wysolmerski JJ. J Clin Investig. 2004;113:598–608. doi: 10.1172/JCI18776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng SX, Okuda M, Hall AE, Geibel JP, Hebert SC. Am J Physiol Gastrointest Liver Physiol. 2002;283:240–250. doi: 10.1152/ajpgi.00500.2001. [DOI] [PubMed] [Google Scholar]
- 5.Conigrave AD, Quinn SJ, Brown EM. Proc Natl Acad Sci U S A. 2000;97:4814–4819. doi: 10.1073/pnas.97.9.4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown EM, Katz C, Butters R, Kifor O. J Bone Miner Res. 1991;6:1217–1225. doi: 10.1002/jbmr.5650061112. [DOI] [PubMed] [Google Scholar]
- 7.Quinn SJ, Ye CP, Diaz R, Kifor O, Bai M, Vassilev P, Brown E. Am J Physiol. 1997;273:C1315–C1323. doi: 10.1152/ajpcell.1997.273.4.C1315. [DOI] [PubMed] [Google Scholar]
- 8.Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG, Balandrin MF. Proc Natl Acad Sci U S A. 1998;95:4040–4045. doi: 10.1073/pnas.95.7.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen A, Silverberg SJ. Curr Opin Pharmacol. 2002;2:734–739. doi: 10.1016/s1471-4892(02)00210-2. [DOI] [PubMed] [Google Scholar]
- 10.Pi M, Quarles LD. J Cell Biochem. 2005;95:1081–1092. doi: 10.1002/jcb.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi T, Chattopadhyay N, Kifor O, Ye C, Vassilev PM, Sanders JL, Brown EM. Am J Physiol Cell Physiol. 2001;280:C382–C393. doi: 10.1152/ajpcell.2001.280.2.C382. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi T, Chattopadhyay N, Kifor O, Butters RR, Jr, Sugimoto T, Brown EM. J Bone Miner Res. 1998;13:1530–1538. doi: 10.1359/jbmr.1998.13.10.1530. [DOI] [PubMed] [Google Scholar]
- 13.Kameda T, Mano H, Yamada Y, Takai H, Amizuka N, Kobori M, Izumi N, Kawashima H, Ozawa H, Ikeda K, Kameda A, Hakeda Y, Kumegawa M. Biochem Biophys Res Commun. 1998;245:419–422. doi: 10.1006/bbrc.1998.8448. [DOI] [PubMed] [Google Scholar]
- 14.Shalhoub V, Grisanti M, Padagas J, Scully S, Rattan A, Qi M, Varnum B, Vezina C, Lacey D, Martin D. Crit Rev Eukaryotic Gene Expression. 2003;13:89–106. doi: 10.1615/critreveukaryotgeneexpr.v13.i24.30. [DOI] [PubMed] [Google Scholar]
- 15.Kim YH, Kim JM, Kim SN, Kim GS, Baek JH. Biochem Biophys Res Commun. 2003;304:729–735. doi: 10.1016/s0006-291x(03)00661-2. [DOI] [PubMed] [Google Scholar]
- 16.Bapty BW, Dai LJ, Ritchie G, Jirik F, Canaff L, Hendy GN, Quamme GA. Kidney Int. 1998;53:583–592. doi: 10.1046/j.1523-1755.1998.00790.x. [DOI] [PubMed] [Google Scholar]
- 17.Choudhary S, Wadhwa S, Raisz LG, Alander C, Pilbeam CC. J Bone Miner Res. 2003;18:1813–1824. doi: 10.1359/jbmr.2003.18.10.1813. [DOI] [PubMed] [Google Scholar]
- 18.Tu Q, Pi M, Karsenty G, Simpson L, Liu S, Quarles LD. J ClinInvestig. 2003;111:1029–1037. doi: 10.1172/JCI17054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pi M, Garner SC, Flannery P, Spurney RF, Quarles LD. J Biol Chem. 2000;275:3256–3263. doi: 10.1074/jbc.275.5.3256. [DOI] [PubMed] [Google Scholar]
- 20.Pin JP, Duvoisin R. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 21.Mohler H, Fritschy JM. Trends Pharmacol Sci. 1999;20:87–89. doi: 10.1016/s0165-6147(99)01323-1. [DOI] [PubMed] [Google Scholar]
- 22.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 23.Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJ, Zuker CS. Cell. 2001;106:381–390. doi: 10.1016/s0092-8674(01)00451-2. [DOI] [PubMed] [Google Scholar]
- 24.Nelson G, Chandrashekar J, Hoon MA, Feng L, Zhao G, Ryba NJ, Zuker CS. Nature. 2002;416:199–202. doi: 10.1038/nature726. [DOI] [PubMed] [Google Scholar]
- 25.Robbins MJ, Michalovich D, Hill J, Calver AR, Medhurst AD, Gloger I, Sims M, Middlemiss DN, Pangalos MN. Genomics. 2000;67:8–18. doi: 10.1006/geno.2000.6226. [DOI] [PubMed] [Google Scholar]
- 26.Brauner-Osborne H, Krogsgaard-Larsen P. Genomics. 2000;65:121–128. doi: 10.1006/geno.2000.6164. [DOI] [PubMed] [Google Scholar]
- 27.Calver AR, Michalovich D, Testa TT, Robbins MJ, Jaillard C, Hill J, Szekeres PG, Charles KJ, Jourdain S, Holbrook JD, Boyfield I, Patel N, Medhurst AD, Pangalos MN. Brain Res Mol Brain Res. 2003;110:305–317. doi: 10.1016/s0169-328x(02)00662-9. [DOI] [PubMed] [Google Scholar]
- 28.Mun HC, Franks AH, Culverston EL, Krapcho K, Nemeth EF, Conigrave AD. J Biol Chem. 2004;279:51739–51744. doi: 10.1074/jbc.M406164/200. [DOI] [PubMed] [Google Scholar]
- 29.Wise A, Green A, Main MJ, Wilson R, Fraser N, Marshall FH. Neuropharmacology. 1999;38:1647–1656. doi: 10.1016/s0028-3908(99)00119-7. [DOI] [PubMed] [Google Scholar]
- 30.Tabata T, Araishi K, Hashimoto K, Hashimotodani Y, van der Putten H, Bettler B, Kano M. Proc Natl Acad Sci U S A. 2004;101:16952–16957. doi: 10.1073/pnas.0405387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubo Y, Miyashita T, Murata Y. Science. 1998;279:1722–1725. doi: 10.1126/science.279.5357.1722. [DOI] [PubMed] [Google Scholar]
- 32.Saunders R, Nahorski SR, Challiss RA. Neuropharmacology. 1998;37:273–276. doi: 10.1016/s0028-3908(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z, Qiu W, Quinn SJ, Conigrave AD, Brown EM, Bai M. J Biol Chem. 2002;277:33727–33735. doi: 10.1074/jbc.M200976200. [DOI] [PubMed] [Google Scholar]
- 34.Hu J, McLarnon SJ, Mora S, Jiang J, Thomas C, Jacobson KA, Spiegel AM. J Biol Chem. 2005;280:5113–5120. doi: 10.1074/jbc.M413403200. [DOI] [PubMed] [Google Scholar]
- 35.Galvez T, Urwyler S, Prezeau L, Mosbacher J, Joly C, Malitschek B, Heid J, Brabet I, Froestl W, Bettler B, Kaupmann K, Pin JP. Mol Pharmacol. 2000;57:419–426. doi: 10.1124/mol.57.3.419. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Jiang Y, Quinn SJ, Krapcho K, Nemeth EF, Bai M. J Biol Chem. 2002;277:33736–33741. doi: 10.1074/jbc.M200978200. [DOI] [PubMed] [Google Scholar]
- 37.Hu J, Reyes-Cruz G, Chen W, Jacobson KA, Spiegel AM. J Biol Chem. 2002;277:46622–46631. doi: 10.1074/jbc.M207100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrel C, Kessler A, Dauban P, Dodd RH, Rognan D, Ruat M. J Biol Chem. 2004;279:18990–18997. doi: 10.1074/jbc.M400724200. [DOI] [PubMed] [Google Scholar]
- 39.Miedlich SU, Gama L, Seuwen K, Wolf RM, Breitwieser GE. J Biol Chem. 2004;279:7254–7263. doi: 10.1074/jbc.M307191200. [DOI] [PubMed] [Google Scholar]
- 40.Kuang D, Yao Y, Lam J, Tsushima RG, Hampson DR. J Neurochem. 2005;93:383–391. doi: 10.1111/j.1471-4159.2005.03025.x. [DOI] [PubMed] [Google Scholar]
- 41.Wellendorph P, Hansen KB, Balsgaard A, Greenwood JR, Egebjerg J, Brauner-Osborne H. Mol Pharmacol. 2005;67:589–597. doi: 10.1124/mol.104.007559. [DOI] [PubMed] [Google Scholar]
- 42.Wellendorph P, Brauner-Osborne H. Gene (Amst) 2004;335:37–46. doi: 10.1016/j.gene.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Conigrave AD, Mun HC, Delbridge L, Quinn SJ, Wilkinson M, Brown EM. J Biol Chem. 2004;279:38151–38159. doi: 10.1074/jbc.M406373200. [DOI] [PubMed] [Google Scholar]
- 44.Yamauchi K, Holt K, Pessin JE. J Biol Chem. 1993;268:14597–14600. [PubMed] [Google Scholar]
- 45.Spurney RF, Pi M, Flannery P, Quarles LD. Kidney Int. 1999;55:1750–1758. doi: 10.1046/j.1523-1755.1999.00432.x. [DOI] [PubMed] [Google Scholar]
- 46.Pi M, Spurney RF, Tu Q, Hinson T, Quarles LD. Endocrinology. 2002;143:3830–3838. doi: 10.1210/en.2002-220240. [DOI] [PubMed] [Google Scholar]
- 47.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Science. 1998;280:574–577. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 48.Pi M, Oakley RH, Gesty-Palmer D, Cruickshank RD, Spurney RF, Luttrell LM, Quarles LD. Mol Endocrinol. 2005;19:1078–1087. doi: 10.1210/me.2004-0450. [DOI] [PubMed] [Google Scholar]
- 49.Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 50.Mao J, Yuan H, Xie W, Simon MI, Wu D. J Biol Chem. 1998;273:27118–27123. doi: 10.1074/jbc.273.42.27118. [DOI] [PubMed] [Google Scholar]
- 51.Pi M, Quarles LD. J Bone Miner Res. 2004;19:862–869. doi: 10.1359/JBMR.040114. [DOI] [PubMed] [Google Scholar]
- 52.Hobson SA, Wright J, Lee F, McNeil SE, Bilderback T, Rodland KD. Mol Cell Endocrinol. 2003;200:189–198. doi: 10.1016/s0303-7207(01)00749-3. [DOI] [PubMed] [Google Scholar]
- 53.Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, Gowen M, Gleason JG, Bhatnagar PK, Fox J. J Pharmacol Exp Ther. 2001;299:323–331. [PubMed] [Google Scholar]
- 54.Joshi SA, Fan KP, Ho VW, Wong YH. FEBS Lett. 1998;441:67–70. doi: 10.1016/s0014-5793(98)01527-0. [DOI] [PubMed] [Google Scholar]
- 55.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 56.Kohout TA, Lefkowitz RJ. Mol Pharmacol. 2003;63:9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- 57.Kehrl JH, Sinnarajah S. Int J Biochem Cell Biol. 2002;34:432–438. doi: 10.1016/s1357-2725(01)00141-8. [DOI] [PubMed] [Google Scholar]
- 58.West RE, Jr, Moss J, Vaughan M, Liu T, Liu TY. J Biol Chem. 1985;260:14428–14430. [PubMed] [Google Scholar]
- 59.Aktories K, Hall A. Trends Pharmacol Sci. 1989;10:415–418. doi: 10.1016/0165-6147(89)90191-0. [DOI] [PubMed] [Google Scholar]
- 60.Pi M, Hinson TK, Quarles L. J Bone Miner Res. 1999;14:1310–1319. doi: 10.1359/jbmr.1999.14.8.1310. [DOI] [PubMed] [Google Scholar]
- 61.Hauschka PV, Carr SA. Biochemistry. 1982;21:2538–2547. doi: 10.1021/bi00539a038. [DOI] [PubMed] [Google Scholar]
- 62.Nishimoto SK, Waite JH, Nishimoto M, Kriwacki RW. J Biol Chem. 2003;278:11843–11848. doi: 10.1074/jbc.M211449200. [DOI] [PubMed] [Google Scholar]
- 63.Quarles LD, Hartle JE, Jr, Siddhanti SR, Guo R, Hinson TK. J Bone Miner Res. 1997;12:393–402. doi: 10.1359/jbmr.1997.12.3.393. [DOI] [PubMed] [Google Scholar]
- 64.Quarles LD. J Clin Investig. 2003;112:642–646. doi: 10.1172/JCI19687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quarles LD, Wenstrup RJ, Castillo SA, Drezner MK. Endocrinology. 1991;128:3144–3151. doi: 10.1210/endo-128-6-3144. [DOI] [PubMed] [Google Scholar]
- 66.Dvorak MM, Siddiqua A, Ward DT, Carter DH, Dallas SL, Nemeth EF, Riccardi D. Proc Natl Acad Sci U S A. 2004;101:5140–5145. doi: 10.1073/pnas.0306141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuang D, Yao Y, Wang M, Pattabiraman N, Kotra LP, Hampson DR. J Biol Chem. 2003;278:42551–42559. doi: 10.1074/jbc.M307120200. [DOI] [PubMed] [Google Scholar]
- 68.Hebert RL, Carmosino M, Saito O, Yang G, Jackson CM, Qi Z, Breyer RM, Natarajan C, Hata AN, Zhang Y, Guan Y, Breyer MD. J Biol Chem. 2005;280:35028–35037. doi: 10.1074/jbc.M505852200. [DOI] [PubMed] [Google Scholar]