

Abstract
Colorectal cancer (CRC) is the second leading cause of cancer-related mortality in the United States. As such, it assumes a significant role in both health policy decision-making and scientific research. CRC has been a model for investigating the molecular genetics of cancer development and progression; this is in part due to the easily detectable, sequential transition of cells from normal colonic epithelium to adenoma and then to adenocarcinoma. In addition, familial syndromes that predispose to CRC, such as familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC), have significantly contributed to our understanding of the genetic mechanisms underlying CRC formation. It is now well recognized that hereditary CRC syndromes are due to germline mutations of genes that function as tumor suppressors or, less frequently, oncogenes. Accumulation of subsequent mutations in other genes with related functions results in the stepwise progression to carcinoma. It is important to note that somatic changes in similar genes are involved in the formation of sporadic CRC. The identification of these important CRC-related genes may help facilitate the early diagnosis, prevention, and treatment of CRC. This article reviews the various familial CRC syndromes along with their genetic etiology, as well as discusses the principle of genetic testing for these conditions.
Introduction
CRC is one of the most common malignancies in the Western world. Although the mortality rate associated with CRC has been gradually declining in recent years, likely as a result of increasing screening and diet/lifestyle modifications, it is still estimated that this malignancy will lead to over 56,000 deaths in the United States in 2004, making it the second leading cause of cancer mortality.[1] Thus, CRC remains a major health concern and demands continuing efforts in developing strategies of screening and prevention.
Among the various causes of CRC, approximately 75% can be attributed to sporadic disease, in which there is no apparent predisposing etiology. The remaining cases of CRC are accounted for by familial incidences and inflammatory bowel disease (Figure 1). Although some familial cases consist of well-described hereditary CRC syndromes, including FAP, HNPCC, and the hereditary hamartomatous polyposis syndromes, the majority of familial disease has no clearly identifiable genetic etiology.
Figure 1.
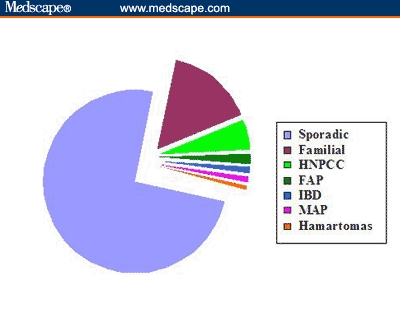
The causes of colorectal cancer. Sporadic CRC refers to those cases that occur in individuals over age 50 years without any identifiable predisposing factors. Familial cases are those with a family history of CRC but exclusive of FAP, HNPCC, and the hamartomatous polyposis syndromes. The approximate percentage of distribution for each condition is as follows: sporadic, 75%; familial, 15%; HNPCC, 5%; FAP, 1%; IBD, 1%; MAP, 1%; hamartomas, < 1%.
Key: IBD = inflammatory bowel disease; MAP = MYH-associated polyposis
In this report, we review the genetics of CRC, with a special emphasis on familial cases with defined genetic etiology. However, it is clear that additional research is needed to ultimately identify the genes involved in all of the hereditary forms of CRC. This latter determination will help improve the early detection of individuals at higher risk for developing this malignancy.
Classification of Cancer-Causing Genes
Genes whose mutations can cause cancer (cancer-causing genes) can be broadly divided into 2 classes: tumor suppressor genes (TSGs) and oncogenes. TSGs encode proteins that either inhibit cell proliferation or promote cell death (apoptosis). Loss of 1 of more of these "brakes" from the cell cycle contributes to the development of many cancers. A unique type of TSG belongs to a group of enzymes or proteins that are responsible for repairing DNA damage. Although these genes do not directly inhibit cell proliferation, their loss results in the accumulation of mutations in other genes that are essential in the control of cell proliferation. Because generally only 1 copy of a TSG is sufficient for function, both alleles of a TSG must be inactivated in order to promote tumor development. TSGs therefore act recessively.
The first TSG was identified in patients with hereditary retinoblastoma. Children with this syndrome inherit a single defective copy of the retinoblastoma susceptibility (Rb) gene. Inactivation of the second copy of Rb, usually occurring shortly after birth, results in the development of retinoblastoma (multiple tumors in the retinas of both eyes), often in both eyes. In contrast, individuals with sporadic retinoblastoma inherit 2 normal copies of Rb, each of which has to be inactivated before a tumor can form. Because the latter event is an unlikely occurrence, sporadic retinoblastoma develops much later in life and usually involves only 1 eye.
If TSGs are the "brakes" in cells, oncogenes represent the "gas pedal." Cells have many proto-oncogenes that are essential for the development of the organism. Mutations in these proto-oncogenes can sometimes lead to their activation, converting them into oncogenes. Once activated, oncogenes can greatly accelerate cell proliferation and often contribute to tumor formation. Sometimes, mutations that cause activation of cell surface receptors for various growth factors can also result in tumor formation. For example, many breast cancers contain excessive amounts of the Her2 receptor, which binds epidermal growth factor (EGF). Here, even very low levels of EGF, which normally is insufficient to stimulate cell proliferation, can significantly accelerate the growth of Her2-overexpressing breast cancer cells. On the basis of this finding, a monoclonal antibody specific for Her2 is now used clinically to treat certain breast cancers.
The Genetic Paradigm of Colorectal Cancer Formation
In 1990, Fearon and Vogelstein[2] presented evidence for a multistep genetic model for the formation of CRC. This model is based on the understanding that CRC is the result of mutations in key genes, including the inactivation of TSGs and the activation of oncogenes. Moreover, accumulation of mutations occurs in a sequential manner, with mutations of some genes preceding that of others. This genetic paradigm is shown in Figure 2. As seen, there are 2 different pathways that lead to the formation of CRC. One route is through inactivation of the TSG, adenomatous polyposis coli (APC), which accounts for approximately 85% of all CRC and is mutated in the germline of patients with FAP. The other pathway for development of CRC is through the mutational inactivation of a family of proteins involved in DNA mismatch repair (MMR), including MSH2, MLH1, and PMS2. MMR mutation is found in approximately 15% of all sporadic CRC and is responsible for the HNPCC syndrome.
Figure 2.
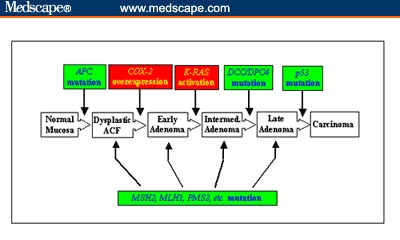
The genetic paradigm of colorectal cancer. The formation of CRC requires the sequential mutation of several genes. This includes the inactivation of TSGs (in green) and activation of oncogenes (in red). There are 2 independent pathways that can cause CRC, depending upon which TSG is first inactivated. Inactivation of the APC gene is found in about 85% of sporadic CRC, and inactivation of the mismatch repair genes, including MSH2, MLH1, and PMS2, is found in the remaining 15%.
Key: COX-2 = cyclo-oxygenase-2.
Mutations in the MMR genes cause genetic defects in other genes that are involved in growth control and behave like TSGs. The APC pathway of CRC formation often includes activation of oncogenes such as COX-2 and K-RAS and inactivation of additional TSGs such as DCC/DPC4 and p53 (Figure 2).
Hereditary Polyposis Syndromes
Familial Adenomatous Polyposis
FAP is an autosomal dominant disorder in which patients typically develop CRC in early adult life, secondary to extensive adenomatous polyp formation in the colon. Polyps also develop in the upper gastrointestinal tract, and malignancies may occur in other sites, including the brain and the thyroid. A phenotypic variant of FAP -- the Gardner syndrome -- is characterized by colonic polyposis with extraintestinal tumors, especially osteomas, and a rather characteristic retinal lesion known as congenital hypertrophy of the retinal pigment. The onset of polyposis in affected individuals often coincides with puberty, and untreated patients invariably develop CRC, with a median age at diagnosis of 40 years. Prophylactic colectomy is the treatment of choice once colonic polyposis is manifested.
The first clue to the location of the gene for FAP was provided in a study by Herrera and colleagues,[3] which demonstrated a constitutional deletion of chromosome 5q21 in a patient with Gardner syndrome. Subsequent linkage analysis demonstrated that 5q21 chromosome markers were linked to the development of FAP.[4,5] The gene responsible for FAP was identified in 1991 and was termed the APC gene.[6-8] Studies also demonstrated the presence of point mutations in the APC gene in the germline of patients with FAP.[7,9] In adenomas and carcinomas that develop in FAP patients, there is evidence for the inactivation of the second copy of the APC gene.[9-11] These findings strongly support Knudson's "2-hit" hypothesis for tumorigenesis[12] and indicate that APC is a TSG in CRC.
The APC gene contains 15 exons and a single open reading frame of 8538 nucleotides that encodes a protein of 2843 amino acids.[8] Mutations in the APC gene are present in 80% to 85% of patients with FAP[13] and in over 80% of cases of sporadic CRC.[14] It has been shown that a process termed loss of heterozygosity (LOH) is the main mechanism by which APC becomes inactivated. In this process, the first allele of the APC gene contains an inactivating point mutation in the germline or somatic DNA of patients with FAP or sporadic CRC, respectively. Approximately 90% of the point mutations in APC in CRC cause an inactive, truncated protein product. Most of these mutations accumulate in the central region of the APC gene, which is known as the mutation cluster region, and result in expression of carboxyl-terminally truncated proteins. APC mutations in the first or last third of the gene are associated with an attenuated polyposis with a late onset and a small number of polyps,[15] whereas mutations in the central region of the gene correlate with a severe phenotype characterized by thousands of polyps at a young age and with additional extracolonic manifestations. Nonneoplastic cells of patients with FAP are expected to retain normal APC function due to the presence of 1 wild-type allele, irrespective of the position of the mutation in the affected allele. Consistent with the Knudson 2-hit model, the wild-type APC allele is lost in a great majority of colorectal tumors in both sporadic and FAP disease.[14]
HNPCC
HNPCC is the most common form of inherited CRC to date. This syndrome is an autosomal dominant disease, with a population incidence of approximately 1:1000, and is responsible for up to 5% of all cases of CRC (Figure 2). Patients with HNPCC have up to an 80% lifetime risk of developing CRC, and a 60% lifetime risk of developing endometrial carcinoma. Affected individuals are also at an increased risk for other cancers, such as stomach, ovarian, small bowel, biliary, and kidney cancers. In contrast to FAP, patients with HNPCC develop adenomas at a normal rate but progress more rapidly through the stages of carcinogenesis.[16] Relative to sporadic CRC, adenomas and carcinomas in HNPCC occur predominantly in the proximal colon due to an increased rate of transformation.[17] Diagnosis of HNPCC is primarily based on clinical criteria, some of which are outlined in the Table. The Amsterdam Criteria were the first diagnostic guidelines to be developed, but they can only identify approximately 60% of the patients with HNPCC.[18] The latter led to the development of the revised criteria (Amsterdam II Criteria; Table), which take into consideration the presence of extracolonic cancer and have a sensitivity of detection of approximately 80%. New guidelines (The Bethesda Guidelines; Table) were also developed to aid in the decision process regarding whether patients who do not fulfill Amsterdam Criteria should undergo gene testing for HNPCC, and this further increased the sensitivity of detection to 94%.[18]
The pathogenesis of HNPCC is typified by the presence of widespread alterations in short, repeated DNA sequences in tumor cells -- a phenomenon called microsatellite instability (MSI).[13] This phenomenon indicates that numerous replication errors occur during the formation of tumors. Indeed, subsequent genetic analysis identified the culprits in HNPCC as a group of TSGs that encode proteins involved in repairing mismatched DNA sequences (MMR genes; Figure 2). The genes identified to date include MSH2, MLH1, PMS2, MSH6, and PMS1.[19] Among these, approximately 85% of the mutations involve MSH2 and MLH1. An important consequence of mutations in the MMR genes is the subsequent inactivation of other genes due to the presence of microsatellite sequences in their coding regions. Some of these targeted genes are involved in important aspects of regulation of cell proliferation. Examples include the type II receptor for transforming growth factor beta;[20,21] the type II receptor for the insulin-like growth factor 2;[22] and BAX, which is involved in the control of apoptosis.[23,24] Inactivation of these genes leads to derangement of cell proliferation and subsequent tumor formation.
The Hamartomatous Polyposis Syndromes
Hamartoma refers to an excessive but focal overgrowth of cells and tissues native to the organ in which it occurs. Although the cellular elements are mature and identical to those found in the remainder of the organ, they do not reproduce the normal architecture of the surrounding tissue. In the intestinal tract, several discrete familial syndromes characterized by multiple hamartomatous polyps have been described -- these include the Peutz-Jeghers syndrome, juvenile polyposis syndrome, and related syndromes such as Cowden's syndrome and Bannayan-Ruvalcaba-Riley syndrome. The hamartomatous polyps in these diseases are derived from the epithelial, stromal, or from both components, of the intestine. Affected patients are at risk for developing cancer of the intestinal tract.
Peutz-Jeghers Syndrome
This syndrome is an autosomal dominant disorder first described by Peutz in 1921, followed by Jeghers in 1949. Characteristics of this disease include the presence of pigmentation on the lips, buccal mucosa, hands, and feet; hamartomatous polyps throughout the gastrointestinal tract; and increased risk for gastrointestinal, breast, ovarian, and testicular cancers. The cumulative risk of colon cancer is 39%, with similar rates for gastric and pancreatic cancer.[25] Peutz-Jeghers syndrome is caused by germline mutations in STK11/LKB1, a serine-threonine kinase gene on chromosome 19p.[26,27] However, not all families with this syndrome are linked to defects in this gene locus, suggesting there is heterogeneity. Recent evidence for loss of STK11/LKB1 expression in Peutz-Jeghers polyps, even without dysplastic epithelium, raises the possibility that the STK11/LKB1 gene itself may be the gatekeeper to carcinogenesis in this syndrome, much as the APC gene is the gatekeeper in FAP.[28]
Juvenile Polyposis Syndrome
Juvenile polyps are distinctive hamartomas that have a smooth surface and are covered by normal colonic epithelium. Juvenile polyposis syndrome is defined by any 1 of the following criteria: 10 or more colonic juvenile polyps; juvenile polyps throughout the gastrointestinal tract; or any number of juvenile polyps, with a family history of juvenile polyposis. The risk of colon cancer is increased in familial juvenile polyposis, with cancer occurring at an average age of 34 years. Although the juvenile polyps per se are not considered neoplastic, the synchronous adenomatous polyps and mixed juvenile-adenomatous polyps of these patients may give rise to concern.
Most families with this syndrome have germline mutations of the DPC4/SMAD4 gene, a TSG that plays a role in signaling through the transforming growth factor-beta cascade.[29] Some families with juvenile polyposis syndrome carry mutations in the PTEN gene, a tumor suppressor with phosphatase activity.[30] It should be noted, however, that PTEN mutations have also been described in Cowden's syndrome and in Bannayan-Ruvalcaba-Riley syndrome.[31,32] Therefore, classifying a patient with a PTEN mutation can be problematic because, depending upon the syndrome (juvenile polyposis syndrome, Cowden's syndrome, and Bannayan-Ruvalcaba-Riley syndrome), the risks for various cancers and, hence, appropriate management, differ.
The APC I1307K Mutation
Most of the APC mutations found in hereditary and sporadic CRC are nonsense mutations (ie, they lead to the formation of a truncated protein). In contrast, the APC I1307K mutation is a missense point mutation in nucleotide position 3920, causing a T to A transversion and, consequently, the substitution of lysine (K) for isoleucine (I) in amino acid position 1307 of the APC protein.[33] This mutation is predominantly seen in Ashkenazi Jews and carries a predisposition to CRC.[34] The increased risk for CRC is about 2-fold, indicating that the mutation has a low penetrance despite a relatively high prevalence of about 6.1% in American Jews of European origin.[33] This polymorphism suggests a paradigm wherein subtle mutations in the APC gene, as compared with distinct well-characterized APC mutations found in FAP, may contribute to hereditary CRC.
MYH-Associated Polyposis
Although the majority of FAP is attributed to germline mutations in the APC gene, a small number of familial cases of polyposis have no identified APC mutations. Recent studies of these APC-negative probands identified germline mutations in the MYH gene, which functions in base excision repair. Somatic mutations in the APC gene can be found in tumors from carriers with biallelic mutations that are either missense or nonsense.[35] Hereditary polyposis due to MYH mutations has been termed MYH-associated polyposis and follows a distinct genetic pathway.[36] It has been estimated that MYH-associated polyposis contributes to approximately 1% of unselected CRC.[37]
Genetic Testing
The approach to genetic testing in individuals and family members at risk for CRC requires a well thought-out rationale, with a systematic identification of the proband, the at-risk family, appropriate pretest genetic counseling, laboratory investigations, and, finally, posttest counseling and clinical follow-up. If a mutation is detected in a proband, family members should be offered testing. If a family member tests negative, then he should be reassured as to the lack of need for further screening but be reminded of the fact that he still has the background population risk.
Genetic testing for FAP is performed using a commercially available in vitro synthesized protein assay to detect truncated APC protein.[13] The sensitivity of this test is approximately 65%, with false-negative results seen in about 20% of patients. Therefore, the disease is ruled out for an individual only if there is no mutation found in an affected family member.[38] Genetic testing for HNPCC is more complex because any 1 of a host of genes may be mutated, although defects in MLH1 and MSH2 are responsible for the disease in about 85% of cases. Furthermore, only 50% of patients who meet the Amsterdam criteria will have positive results on MLH1 and MSH2 testing, raising concerns about false negatives. Additional guidelines have been devised in an attempt to increase the yield for identification of HNPCC (Table).[39] Many of the newly published guidelines include examination of the tumors for MSI and level of MMR expression by immunohistochemistry.[40]
The issue of genetic testing has significant ethical connotations. The anxiety associated with testing, the implications of false-negative results, the financial aspects of testing, vis a vis insurance coverage, and the privacy issues associated with genetic testing should all be carefully and judiciously addressed. The process of testing should involve thorough genetic counseling of the patient and family members, both before and after the test. Testing should be offered after close consultation with the patient and family and in those cases where a reasonable correlation and benefit is evident.
Conclusion
Great progress has been made in the past 2 decades toward understanding the molecular genetics of CRC. Intense, ongoing research continues to shed light on new mechanisms of colorectal tumorigenesis. Despite this progress, there remains a relatively large population of individuals who have heritable CRC but an as yet unidentified genetic etiology. Advanced genetic testing hints at the vast potential of screening at-risk individuals, while, at the same time, the pitfalls of false-negative results remain to be addressed. An exciting future awaits us in the clinical application of new research and novel technologies that will ultimately lead to the universal goal -- better health for all.
Table 1.
Clinical Criteria for the Diagnosis of HNPCC
| The Amsterdam Criteria (1991) |
Three or more relatives with colorectal cancer, plus all of the following:
|
| Amsterdam II Criteria (1998) |
Three or more relatives with HNPCC-associated cancer (colorectal cancer or cancer of the endometrium, small bowel, ureter, or renal pelvis) plus all of the following:
|
| The Bethesda Guidelines (1997) |
Just 1 of these criteria needs to be met:
|
Footnotes
This work was in part supported by grants from the National Institutes of Health (DK52230 and CA84197).
Contributor Information
Irfan M Hisamuddin, Gastroenterology Fellow, Division of Digestive Diseases, Department of Medicine, Emory University School of Medicine, Atlanta, Georgia.
Vincent W Yang, R. Bruce Logue Professor of Medicine and Director, Division of Digestive Diseases, Department of Medicine; Professor, Department of Hematology and Oncology, Winship Cancer Institute, Emory University School of Medicine, Atlanta, Georgia.
References
- 1. American Cancer Society. Cancer Facts & Figures 2004. Available at: http://www.cancer.org/downloads/STT/CAFF_finalPWSecured.pdf. Accessed August 4, 2004.
- 2. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759-767. [DOI] [PubMed] [Google Scholar]
- 3. Herrera L, Kakati S, Gibas L, Pietrzak E, Sandberg AA. Gardner syndrome in a man with an interstitial deletion of 5q. Am J Med Genet. 1986;25:473-476. [DOI] [PubMed] [Google Scholar]
- 4. Bodmer WF, Bailey CJ, Bodmer J, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987;328:614-616. [DOI] [PubMed] [Google Scholar]
- 5. Leppert M, Dobbs M, Scambler P, et al. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science. 1987;238:1411-1413. [DOI] [PubMed] [Google Scholar]
- 6. Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661-665. [DOI] [PubMed] [Google Scholar]
- 7. Nishisho I, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665-669. [DOI] [PubMed] [Google Scholar]
- 8. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589-600. [DOI] [PubMed] [Google Scholar]
- 9. Miyoshi Y, Nagase H, Ando H, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229-233. [DOI] [PubMed] [Google Scholar]
- 10. Ichii S, Horii A, Nakatsuru S, et al. Inactivation of both APC alleles in an early stage of colon adenomas in a patient with familial adenomatous polyposis (FAP). Hum Mol Genet. 1992;1:387-390. [DOI] [PubMed] [Google Scholar]
- 11. Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54:5953-5958. [PubMed] [Google Scholar]
- 12. Knudson AG. Antioncogenes and human cancer. Proc Natl Acad Sci U S A. 1993;90:10914-10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aaltonen LA, Peltomaki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812-816. [DOI] [PubMed] [Google Scholar]
- 14. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-170. [DOI] [PubMed] [Google Scholar]
- 15. Spirio L, Olschwang S, Groden J, et al. Alleles of the APC gene: an attenuated form of familial polyposis. Cell. 1993;75:951-957. [DOI] [PubMed] [Google Scholar]
- 16. Ahlquist DA. Aggressive polyps in hereditary nonpolyposis colorectal cancer: targets for screening. Gastroenterology. 1995;108:1590-1592. [DOI] [PubMed] [Google Scholar]
- 17. Rijcken FE, Hollema H, Kleibeuker JH. Proximal adenomas in hereditary non-polyposis colorectal cancer are prone to rapid malignant transformation. Gut. 2002;50:382-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet. 2000;37:641-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peltomaki P. DNA mismatch repair and cancer. Mutat Res. 2001;488;77-85. [DOI] [PubMed] [Google Scholar]
- 20. Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336-1338. [DOI] [PubMed] [Google Scholar]
- 21. Parsons R, Myeroff LL, Liu B, et al. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995;55:5548-5550. [PubMed] [Google Scholar]
- 22. Souza RF, Appel R, Yin J, et al. Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet. 1996;14:255-257. [DOI] [PubMed] [Google Scholar]
- 23. Sakakibara T, Nakamura T, Yamamoto M, Matsuo M. Microsatellite instability in Japanese hereditary non-polyposis colorectal cancer does not induce mutation of a simple repeat sequence of the bax gene. Cancer Lett. 1998;124:193-197. [DOI] [PubMed] [Google Scholar]
- 24. Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967-969. [DOI] [PubMed] [Google Scholar]
- 25. Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447-1453. [DOI] [PubMed] [Google Scholar]
- 26. Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38-43. [DOI] [PubMed] [Google Scholar]
- 27. Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184-187. [DOI] [PubMed] [Google Scholar]
- 28. Gruber SB, Entius MM, Petersen GM, et al. Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res. 1998;58:5267-5270. [PubMed] [Google Scholar]
- 29. Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086-1088. [DOI] [PubMed] [Google Scholar]
- 30. Olschwang S, Serova-Sinilnikova OM, Lenoir GM, Thomas G. PTEN germ-line mutations in juvenile polyposis coli. Nat Genet. 1998;18:12-14. [DOI] [PubMed] [Google Scholar]
- 31. Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1998;16:64-67. [DOI] [PubMed] [Google Scholar]
- 32. Marsh DJ, Dahia PL, Zheng Z, et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet. 1997;16:333-334. [DOI] [PubMed] [Google Scholar]
- 33. Laken SJ, Petersen GM, Gruber SB, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet. 1997;17:79-83. [DOI] [PubMed] [Google Scholar]
- 34. Rozen P, Shomrat R, Strul H, et al. Prevalence of the I1307K APC gene variant in Israeli Jews of differing ethnic origin and risk for colorectal cancer. Gastroenterology. 1999;116:54-57. [DOI] [PubMed] [Google Scholar]
- 35. Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791-799. [DOI] [PubMed] [Google Scholar]
- 36. Lipton L, Halford SE, Johnson V, et al. Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 2003;63:7595-7599. [PubMed] [Google Scholar]
- 37. Halford SE, Rowan AJ, Lipton L, et al. Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol. 2003;162:1545-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giardiello FM, Brensinger JD, Petersen GM, et al. The use and interpretation of commercial APC gene testing for familial adenomatous polyposis. N Engl J Med. 1997;336:823-827. [DOI] [PubMed] [Google Scholar]
- 39. Umar A, Risinger JI, Hawk ET, Barrett JC. Testing guidelines for hereditary non-polyposis colorectal cancer. Nat Rev Cancer. 2004;4:153-158. [DOI] [PubMed] [Google Scholar]
- 40. Frazier ML, Su LK, Amos CI, Lynch PM. Current applications of genetic technology in predisposition testing and microsatellite instability assays. J Clin Oncol. 2000;18:70S-74S. [PubMed] [Google Scholar]