

The stereodivergent construction of cyclic ethers remains an important area of synthetic interest, particularly given the ubiquity of C-glycoside derivatives in natural and unnatural pharmacologically important agents.1 Bismuth(III) halides are inexpensive and environmentally benign reagents, which have been utilized as mild Lewis acid catalysts for an array of synthetic transformations.2,3 Herein, we describe a series of stereoselective intramolecular etherification reactions of δ-trialkylsilyloxy aldehydes and ketones 1, using catalytic bismuth tribromide and various trialkylsilyl nucleophiles for the construction of cis- and trans-2,6-di- and trisubstituted tetrahydropyrans 2 (eq 1).4
Equation 1.
The mechanistic hypothesis, outlined in Scheme 1, describes the basis of the two-component etherification reaction. Since bismuth-(III) trihalides are known to readily undergo hydrolysis to afford bismuth oxyhalides and the requisite Brønsted acid,2,5 we anticipated that the latter would promote the formation of ii, which should then undergo in situ desilylation to afford the lactol iii.6 Acidcatalyzed dehydration of iii could then lead to the formation of the oxocarbenium ion iv and facilitate axial nucleophilic attack to furnish the cyclic ether v.7 The potential advantage of this approach over that involving a Lewis acid is the unusual chemoselectivity, which will provide a powerful tool for the construction of cyclic ethers. This selectivity is presumably the result of the low reactivity of the protonated carbonyl, which requires addition of the pendant triorganosilyloxy group to afford the more reactive oxocarbenium ion.8 Moreover, the acid concentration is modulated using the hydrolysis of the triorganosilyl halide, making it an exceedingly mild and convenient method.
Scheme 1.

Preliminary studies demonstrated that the nature of the bismuth-(III) halide was inconsequential in terms of both efficiency and selectivity (Cl ∼ Br ∼ I). In light of this fact we elected to utilize the less expensive BiBr3. Interestingly, while the selectivity was unaffected by the nature of the triorganosilyl ether, the efficiency of the reaction was found to be directly related to the rate of protodesilylation (TMS ∼ TES > TBS >> TIPS).9 Additional studies using pendant triethylsilyl ethers then focused on providing evidence for the proposed hypothesis outlined in Scheme 1. Initial studies examined the proposal that BiBr3 or triethylsilyl bromide provided a source of HBr, which then functions as the catalyst (Table 1, entries 1, 5, and 7). Consistent with this hypothesis, the addition of water to BiBr3 did not prove detrimental to the reaction and further supports the notion that the BiBr3 is not acting as a Lewis acid (entry 2). Moreover, the addition of activated molecular sieves to each of these reagents, to sequester HBr and water, gave none of the desired product (entries 3, 6, and 8).10 This idea was further supported by the addition of 2,6-di-tert-butyl-4-methylpyridine (DTBMP), which neutralizes the hydrogen bromide and leads to no observable reaction (entry 4). The relatively poor catalytic activity of HBr and triethylsilyl bromide (entries 5 and 7) prompted additional studies to determine the potential role of bismuth in the reaction. Interestingly, the addition of an equimolar amount of BrBi=O appears to reconstitute the catalytic activity (entries 7 and 10 vs 9).11 Further investigations are underway to determine the precise role of the BrBi=O.
Table 1.
Elucidation of the Role of BiBr3 in the Two-Component Etherification Reaction (eq 1, 1a, R = Et; R1 = Bn; R2 = H; Nu = CH2CH=CH2)
| entry | catalysta | mol% | additive | ratio of 2a:3ae,f | (%) yieldg |
|---|---|---|---|---|---|
| 1 | BiBr3 | 10 | - | ≥99:1 | 99 |
| 2 | “ | H2Ob | ≥99:1 | 92 | |
| 3 | “ | “ | 4Å sievesc | NA | 0 |
| 4 | “ | “ | DTBMPd | NA | 0 |
| 5 | HBr | 20 | - | ≥99:1 | 34 |
| 6 | “ | “ | 4Å sievesc | NA | 0 |
| 7 | TESBr | “ | - | ≥99:1 | 32 |
| 8 | “ | “ | 4Å sievesc | NA | 0 |
| 9 | “ | “ | BrBi=Od | ≥99:1 | 91h |
| 10 | BrBi=O | “ | - | NA | 0 |
All reactions were carried out on a 0.1 mmol reaction scale in CH3CN at room temperature using 3 equiv of allyltrimethylsilane.
10 equiv based on BiBr3.
Molecular sieves were activated at 150 °C under high vacuum.
20 mol % based on 1a.
Ratios of diastereoisomers were determined by capillary GLC analysis.
The cis-diastereoisomer 3a was prepared from the ketone via a reductive etherification reaction.
GLC yields.
The addition of 4Å molecular sieves to this reaction also led to no observable reaction.
Table 2 outlines the scope of the two-component etherification reaction in terms of various nucleophiles. This study demonstrated that δ-triethylsilyloxy-substituted aldehydes and ketones serve as substrates for the etherification reaction. Interestingly, while the two-component coupling reactions with carbon nucleophiles furnished the trans-diastereoisomer (entries 1-6),12 the reductive coupling furnished the cis-diastereoisomer consistent with axial addition of the nucleophile (entries 7 and 8).13,14
Table 2.
| entry | 1; R2= | R3Si-Nub | cyclic ether 2/3Nu= | ds = 2:3c | yield (%)f | |
|---|---|---|---|---|---|---|
| 1 | H | A | -CH2CH=CH2 | a | ≥99:1d | 90 |
| 2 | Me | A | “ | b | ≥19:1 | 88 |
| 3 | H | B | -CH=C=CH2 | c | ≥19:1e | 80 |
| 4 | Me | B | “ | d | ≥19:1e | 72 |
| 5 | H | C | -CH2C(O)CH3 | e | ≥19:1 | 73 |
| 6 | Me | C | “ | f | ≥19:1 | 80 |
| 7 | 2H | D | -H | g | ≥19:1 | 85 |
| 8 | Me | D | “ | h | ≥99:1d | 95 |
All reactions were carried out on a 0.2-0.3 mmol reaction scale in CH3CN at room temperature using 5-10 mol % BiBr3 and 1.2-3.0 equiv. of R3Si-Nu.
A = Me3SiCH2CH=CH2; B = Me3SiCH2C≡CH; C = CH2=C(OSiMe3)CH3; D = Et3SiH.
Ratios of diastereoisomers were determined by 400 MHz 1H NMR on the crude reaction mixture unless otherwise indicated.
Determined by capillary GLC.
Contaminated with 5-10% of the propargylated derivative.
Isolated yields.
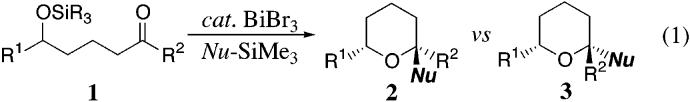
We envisioned that the stereoselective construction of adjacent tertiary ethers would prove challenging and thereby highlight the synthetic utility of the tandem two-component etherification reaction (eq 2). Treatment of the triethylsilyl ether 4 with BiBr3 and excess allyltrimethylsilane at room temperature furnished the bicyclic tetrahydropyran derivatives 5/6 in 77-81% yield, with excellent diastereoselectivity favoring 5.15,16 The ability to accomplish the selective formation of this bis-tertiary ether from the cis-ring fusion is particularly interesting given there are potentially two conformers of the oxocarbenium ion. Indeed, the stereochemical outcome is consistent with the Woerpel model,14 which predicts the 4-alkoxy substituent will adopt a pseudoaxial orientation in the transition state, thereby favoring the trans-addition of the allylsilane.
Equation 2.
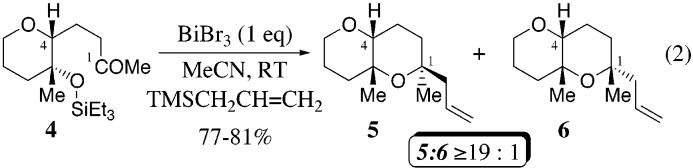
Encouraged by the preliminary results in Table 2, we also examined the feasibility of a sequential two-component reaction that involved an intermolecular addition followed by an intramolecular reductive etherification as outlined in eq 3.15,17 Treatment of the aldehyde 7 with excess trimethylsilyl enol ether 8 installs the trans-2,6-disubstituted tetrahydropyran, which upon addition of triethylsilane facilitates the reductive etherification to afford the bis-tetrahydropyran 9 in 73% yield with excellent diastereoselectivity (by GLC, eq 3). Hence, this cross-coupling reaction provides a one-step method for the installation of nonadjacent tetrahydropyran rings having complementary stereochemistry.
Equation 3.
In conclusion, we have developed a tandem two-component etherification reaction for the stereoselective construction of cis- and trans-2,6-di- and trisubstituted tetrahydropyran rings in which excellent selectivity could be obtained for either stereoisomer through the judicious choice of the nucleophile and substrate. This work also provides compelling evidence for hydrogen bromide and bismuth oxybromide to be responsible for the catalysis. The synthetic utility of this protocol is highlighted in the ability to construct adjacent tertiary ethers in a highly stereoselective manner and the sequential two-component cross-coupling followed by reductive etherification process for the expiditious synthesis of nonadjacent tetrahydropyrans. These methods will undoubtedly be widely applicable to target-directed synthesis.
Acknowledgment.
We sincerely thank the National Institutes of Health (GM58877) for generous financial support and the College of William and Mary for funding a Faculty Research Assignment (R.J.H.). We also thank Johnson and Johnson for a Focused Giving Award and Pfizer Pharmaceuticals for the Creativity in Organic Chemistry Award. The Camille and Henry Dreyfus Foundation is thanked for both a Camille Dreyfus Teacher-Scholar Award (P.A.E.) and a Henry Dreyfus Teacher-Scholar Award (R.J.H.).
Footnotes
Supporting Information Available: Experimental procedures, X-ray analysis of the p-nitrobenzoate derivative of 5, and spectral data for 1a—c, 2a—h, 4-5, and 7-9. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1). For a review on recent advances in the stereoselective construction of C-Glycosides, see:; Du Y, Linhardt RJ, Vlahov IR. Tetrahedron. 1998;54:9913. [Google Scholar]
- (2).Matano Y, Ikegami T. In: Organobismuth Chemistry. Suzuki H, Matano Y, editors. Elsevier; New York: 2001. pp. 21–245. [Google Scholar]
- (3). For a recent review on the uses of Bi(III) compounds in organic synthesis, see:; Leonard NM, Wieland LC, Mohan RS. Tetrahedron. 2002;58:8373. [Google Scholar]
- (4).Komatsu N, Uda M, Suzuki H, Takahashi T, Domae T, Wada M. Tetrahedron Lett. 1997;38:7215. [Google Scholar]; Komatsu N, Ishida J-Y, Suzuki H. Tetrahedron Lett. 1997;38:7219. [Google Scholar]
- (5). For an example of protodesilylation of alkyl triorganosilyl ethers using bismuth bromide, see:; Bajwa JS, Vivelo J, Slade J, Repic O, Blacklock T. Tetrahedron Lett. 2000;41:6021. [Google Scholar]
- (6). For an example of using silyl ethers as masked hydroxyl groups, see:; Angle SR, El-Said NA. J. Am. Chem. Soc. 1999;121:10211. [Google Scholar]
- (7). For approaches to the formation of C-glycosides involving nucleophilic addition to oxocarbenium ions, see:; Lewis MD, Cha JK, Kishi Y. J. Am. Chem. Soc. 1982;104:4976. [Google Scholar]; Sassaman MB, Prakash GKS, Olah GA. Tetrahedron. 1988;44:3771. [Google Scholar]; Homma K, Mukaiyama T. Chem. Lett. 1989. p. 259. [Google Scholar]
- (8). For the comparison of the kinetics of allylation of oxocarbenium ions and aldehyde Lewis acid complexes, see:; Mayr H, Gorath G. J. Am. Chem. Soc. 1995;117:7862. [Google Scholar]
- (9).Schelhaas M, Waldmann H. Angew. Chem., Int. Ed. Engl. 1996;35:2056. [Google Scholar]
- (10). For an example of using molecular sieves to scavenge hydrogen chloride, see:; Weinstock LM, Karady S, Roberts FE, Hoinowski AM, Brenner GS, Lee TBK, Lumma WC, Sletzinger M. Tetrahedron Lett. 1975;46:3979. [Google Scholar]
- (11).The structure of bismuth oxybromide, isolated from the hydrolysis of bismuth bromide, was confirmed via X-ray powder diffraction
- (12).Representative Experimental Procedure for the Two-Component Allylative Etherification: 6-Phenyl-5-(triethylsilyloxy)hexanal 1a (57.0 mg, 0.186 mmol) was dissolved in acetonitrile (2.0 mL) and stirred at room temperature. Bismuth tribromide (8.9 mg, 0.020 mmol) prepared as a solution in acetonitrile at 1 mg/10 μL was added via syringe directly followed by the rapid addition of allyltrimethylsilane (90 μL, 0.56 mmol). The reaction mixture was stirred at room temperature for ca. 16 h (tlc control). The solvent was removed in vacuo to afford the crude oil. Purification by flash chromatography (5% ethyl acetate/hexanes) furnished 2a (34.6 mg, 90%) as a colorless oil (ds ≥ 99:1 by GLC)
- (13). An alternative mechanistic proposal suggests that triethylsilyl bromide is formed from triethylsilane and bismuth tribromide and behaves as the Lewis acid catalyst, see:; Bajwa JS, Jiang X, Slade J, Prasad K, Repic O, Blacklock TJ. Tetrahedron Lett. 2002;43:6709. [Google Scholar]
- (14). For a discussion of substituent effects on the stereochemical outcome of additions to tetrahydropyran derived oxocarbenium ions, see:; Romero JAC, Tabacco SA, Woerpel KA. J. Am. Chem. Soc. 2000;122:168. [Google Scholar]
- (15).In the more demanding reactions outlined in eqs 2 and 3, use of stoichiometric BiBr3 improved the overall efficiency. This trend is presumably a function of the ease of desilylation of the tertiary alcohol (eq 2) and increased rate of the initial intermolecular reaction in the sequential sequence (eq 3)
- (16).The stereochemistry of 5 was established through X-ray crystallographic analysis of the p-nitrobenzoate derivative formed through reductive ozonolysis and esterification of the intermediary alcohol
- (17). For a related tandem Mukaiyama aldol—Prins cyclization cascade reaction, see:; Kopecky DJ, Rychnovsky SD. J. Am. Chem. Soc. 2001;123:8420. doi: 10.1021/ja011377n. [DOI] [PubMed] [Google Scholar]