

Abstract
The effects of inserting un-substituted ω-amino acids into the strand segments of model β-hairpin peptides was investigated by using four synthetic decapeptides, Boc-Leu-Val-Xxx-Val-d-Pro-Gly-Leu-Xxx-Val-Val-OMe: peptide 1 (Xxx=Gly), peptide 2 (Xxx = βGly = βhGly = homoglycine, β-glycine), peptide 3 (Xxx = γAbu = γ-aminobutyric acid), peptide 4 (Xxx = δAva = δ-aminovaleric acid). 1H NMR studies (500 MHz, methanol) reveal several critical cross-strand NOEs, providing evidence for β-hairpin conformations in peptides 2–4. In peptide 3, the NMR results support the formation of the nucleating turn, however, evidence for cross-strand registry is not detected. Single-crystal X-ray diffraction studies of peptide 3 reveal a β-hairpin conformation for both molecules in the crystallographic asymmetric unit, stabilized by four cross-strand hydrogen bonds, with the γAbu residues accommodated within the strands. The d-Pro-Gly segment in both molecules (A,B) adopts a type II′ β-turn conformation. The circular dichroism spectrum for peptide 3 is characterized by a negative CD band at 229 nm, whereas for peptides 2 and 4, the negative band is centered at 225 nm, suggesting a correlation between the orientation of the amide units in the strand segments and the observed CD pattern.
Keywords: amino acids, conformation analysis, crystal structure, peptides , protein folding
The higher homologues of the α-amino acids, in which additional carbon atoms are introduced into the residues, may be used to expand the range of folded polypeptide structures. Oligo-β-peptides have been shown to adopt variants of helical structures, with new patterns of backbone hydrogen bonds.[1] Polypeptide sheets with altered polarity can also be constructed.[2] Considerable recent work has reported the conformation of peptide oligomers of β-[1] and γ-[1m,3] residues. The incorporation of β-, γ-, and δ-residues into defined secondary structures formed by α-amino acids has also been reported. Earlier studies from this laboratory have demonstrated the incorporation of β-, γ-, and δ-amino acid residues into polypeptide helices.[4] Substituted β-residues have also been incorporated into the turn[2a,2,5a] and strand segments of model β-hairpins.[2] Homooligomers of (R,R)-2-aminocyclopentane carboxylic acid (trans-ACPC) adopt 12 helical structures[1h], whereas homooligomers of (1R,2S)-2-aminocyclopentane carboxylic acid ((1R,2S)-cis-ACPC) adopt nonpolar, sheetlike structures.[6a] The constrained γ-amino acid trans-3-ACPC has been incorporated into the strand segments of a parallel-sheet structure.[6b] Results of NMR studies established the accommodation of 3-amino-benzoic acid into the strand segments of a model hairpin structure.[6c] δAva has been shown to be accommodated at the i+2 position of a nucleatingd-Pro-Xxx turn in a model octapeptide hairpin, Boc-Leu-Val-Val- d-Pro-δAva-Leu-Val-Val-OMe.[5b] The incorporation of additional backbone atoms into the strand segments of β-hairpins permits alteration of the local polarity of β-sheets. In the case of unsubstituted ω-amino acids, lengthening of the spacer segment between the amino and the carboxyl ends of the residue results in a considerable increase in the range of accessible backbone conformations. Nevertheless, the earlier demonstrations of accommodation of β-, γ-, and δ-residues into folded structures suggest that further exploration of the conformational space available to these residues may be useful in defining parameters for their use in peptide design.
Here, we analyze the effects of introducing β, γ, and δ-residues into the strand segments of peptide hairpins. The following peptides were investigated: Boc-Leu-Val-Xxx-Val- d-Pro-Gly-Leu-Xxx-Val-Val-OMe, in which Xxx = Gly (1), Xxx = βGly (2) (βGly = βhGly, homoglycine, betaglycine; note that the previously used common name β-alanine is misleading in the context of the rapidly developing literature on β-peptides),[1a] Xxx = γAbu (3) (γAbu = γ-aminobutyric acid), and Xxx = δAva (4) (δAva = δ-aminovaleric acid).
The choice of sequences used was based on previous studies that established a β-hairpin conformation in solution and in crystals for the octapeptide Boc-Leu-Val-Val-d-Pro-Gly-Leu-Val-Val-OMe.[7] In addition, insertion of βPhe residues ((S)-β3-homophenylalanine) into strand segments at the non-hydrogen-bonding position in the model peptide (Boc-Leu-Val-βPhe-Val- d -Pro-Gly-Leu-βPhe-Val-Val-OMe results in a stable hairpin structure.[2c] Figure 1 shows a schematic view of an anticipated β-hairpin fold and defines the degrees of backbone torsional freedom in ω-amino acid residues. In the sequences of peptides 2–4, the unsubstituted ω-amino acid residues are inserted at two facing non-hydrogen-bonding positions. In the canonical β-hairpin that includes exclusively α-residues, peptide 1, the achiral α-amino acid residue (Gly) is incorporated at the facing non-hydrogen-bonding positions. The results presented in this paper provide strong evidence for a predominant population of β-hairpins in solution in the cases of peptides 2 (Xxx = βGly) and 4 (Xxx = δAva). X-ray diffraction studies of peptide 3 reveal the β-hairpin conformation for two independent molecules in the crystallographic asymmetric unit.
Figure 1.
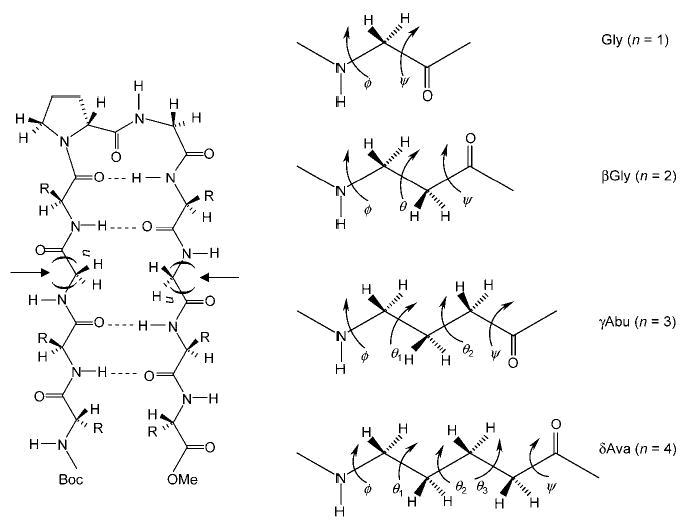
Schematic view of a hairpin (left) and definition of backbone torsional freedom in ω-amino acid residues (right). The arrows (left) represent the sites of insertion of ω-amino acid residues, and “n” refers to the number of methylene units in each strand segment.
Experimental Section
Peptide synthesis
Peptides 1–4 were synthesized by conventional solution-phase methods by using a fragment-condensation strategy.[2e] The tert-butyloxycarbonyl group was used for N-terminus protection, and the C-terminus was protected as a methyl ester. Deprotections were performed with 98% formic acid and saponification for the N- and C-termini, respectively. Couplings were mediated by dicyclohexylcarbodiimide (DCC)/1-hydroxybenzotriazole (HOBT). At the final step, the tetrapep-tide acid (Boc-Leu-Val-Xxx-Val-OH) was coupled to the N-terminus-de-protected hexapeptide (H-d-Pro-Gly-Leu-Xxx-Val-Val-OMe). The tetra-peptide Boc-Leu-Val-Xxx-Val-OMe was prepared by [1+3] condensation involving Boc-Leu-OH and H-Val-Xxx-Val-OMe. The tripeptide Boc-Val-Xxx-Val-OMe was prepared by [2+1] condensation involving an N-terminus dipeptide acid Boc-Val-Xxx-OH and H-Val-OMe, by using DCC/N-hydroxysuccinimide (HOSu). The hexapeptide Boc- d-Pro-Gly-Leu-Xxx-Val-Val-OMe was prepared by [2+4] coupling involving Boc- d-Pro-Gly-OH and H-Leu-Xxx-Val-Val-OMe. The tetrapeptide Boc-Leu-Xxx-Val-Val-OMe was prepared by [2+2] condensation involving an N-terminus dipeptide acid Boc-Leu-Xxx-OH and a C-terminus-deprotected dipeptide H-Val-Val-OMe, by using DCC/HOSu. Intermediates were characterized by 80 MHz NMR spectroscopy and/or by mass spectrometry (MALDI-TOF and ESI-MS). The target peptides were purified by reverse-phase medium-pressure liquid chromatography on a C18 (40–63 μm) column with methanol/water gradients. Peptides 1 and 3 were further purified by HPLC on a reverse-phase C18 (5–10 μm) column with methanol/water gradients.
Mass spectrometry
The purified peptides were analysed by mass spectrometry using a Kratos PC-Kompact MADLI-TOF mass spectrometer; m/z: calcd for 1: 1022 Da; found: 1046.7 Da [M+Na+], 1063.0 Da [M+K+]; m/z: calcd for 2: 1050 Da; found: 1074.0 Da [M+Na+], 1089.7 Da [M+K+]; m/z: calcd for 3: 1079 Da; found: 1102.3 Da [M+Na+], 1118.1 Da [M+K+]; m/z: calcd for 4: 1106 Da; found: 1129.4 Da [M+Na+], 1145.8 Da [M+K+]. Peptides 2–4 were fully characterized by 500 MHz 1H NMR spectroscopy.
NMR spectroscopy:
All NMR studies were carried out by using a Bruker DRX-500 MHz spectrometer at a probe temperature of 300 K. Resonance assignments were obtained by TOCSY and ROESY analysis. All two-dimensional data were collected in phase-sensitive mode, by using the time-proportional phase incrementation (TPPI) method. Sets of 1024 and 450 data points were used in the t2 and t1 dimensions, respectively. For TOCSY and ROESY analysis, 24 and 64 transients were collected, respectively. A spectral width of 6000 Hz was used in both dimensions. A spin-lock time of 250 ms was used to obtain ROESY spectra. Zero-filling was carried out to finally yield a data set of 2 K×1 K. A shifted square-sine-bell window was used before processing.
Circular dichroism (CD):
CD spectra were recorded by using a JASCO J-715 spectropolarimeter. The instrument was calibrated with (+)-10-camphor sulfonic acid. The path length used was 1 mm. The data were acquired in the wavelength scan mode, with a 2 nm band and a step size of 0.2 nm. Spectra were acquired at 300 K. Typically, four scans were acquired from 200–260 nm by using a scan speed of 50 nm min−1. The resulting data were baseline-corrected and smoothened.
X-ray diffraction
Crystals of peptide 3, in the form of thin, flat needles, were obtained from both methanol/water and methanol/dioxane/water solvent mixtures by slow evaporation. Numerous trials for X-ray data collection at several different temperatures and from different crystallization attempts resulted in a “best” data set, with single diffraction spots at ambient temperature. Decreasing the temperature resulted in fractured spots. The effective scattering resolution was 1.1 Å, in which the ratio of mean intensity over σ was ≥1.8. X-ray data were obtained from a colorless crystal, 0.50×0.30×0.10 mm in size, on a four-circle diffractometer (Bruker P4) by using CuKα radiation (λ = 1.54178 Å). The θ–2θ scan mode was used, with a scan width of 1.5°+2θ(α1−α2) and a scan speed of 13°min−1. There were no diffraction spots with measurable intensity beyond 2θ = 105°. Crystal data for C53H94N10O13·H2O: space group P1, a = 9.742(3) b = 10.842(3) c = 31.473(12) Å, α = 89.46(2) β = 83.28(4) γ = 78.85(3)°, V = 3238.8 Å3, Z = 2, and ρcalcd = 1.120 gcm −3. The structure was solved by using a 25-atom fragment from a known type II′ β-turn[7b] in a vector-search procedure,[8a] followed by the expansion of the partial structure by using the tangent formula[8b] and difference maps. Full-matrix least-squares refinement of F2 data of the non-hydrogen atoms, and of hydrogen atoms placed in idealized positions and riding on the carbon or nitrogen atoms to which they are bonded, resulted in a reliability factor of R1 = 10.2% for 4028 observed data [Fo > 4.0σ(Fo)] and 1380 parameters. CCDC 262350 contains the supplementary crystallographic data for this paper (atomic coordinates, bond lengths, and bond angles). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data request/cif.
Results and Discussion
The 500 MHz 1H NMR spectrum for peptide 1 in methanol solution did not yield well-dispersed amide resonances. In addition, the presence of minor conformers, presumably corresponding to cis–trans isomerization about the Val4- d-Pro5 bond, was also observed. The overlap of amide resonances precluded a detailed analysis. The introduction of Gly residues into the strand segments may contribute to the destabilization of registered, antiparallel strands. Subsequent NMR studies were, therefore, confined to peptides 2–4. All three peptides yielded sharp, well-resolved resonances, with adequate chemical-shift dispersion for the amide resonances. Resonances due to the presence of minor conformers, arising from cis–trans isomerization about the Val4- d–Pro5 bond, are observed. The subsequent analysis is restricted to the major trans conformer. Assignment of resonances was accomplished by using a combination of TOCSY and ROESY experiments.
Figure 2 shows a representative TOCSY spectrum of peptide 4 (Xxx = δAva). Resonance assignments are indicated. Peaks arising from a minor conformation are marked “m”. Partial ROESY spectra of peptide 2, indicating NOE connectivities for NH↔CαH (for α-residues) and NH↔CβH (for β-residues) (top panel) and for NH↔NH (bottom panel), are shown in Figure 3. Figure 4 illustrates NOEs for NH↔CαH (for α-residues) and NH↔CδH (for δ-residues) (top panel) and for NH↔NH (bottom panel) for peptide 4. The NMR parameters for peptides 2–4 are provided as Supporting Information.
Figure 2.
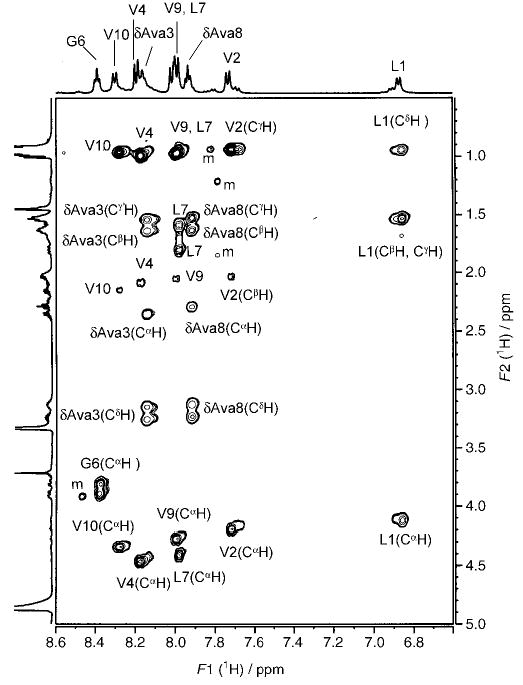
500 MHz partial TOCSY spectrum of peptide 4 in CD3OH at 300 K, illustrating the spin systems of individual amino acids.
Figure 3.
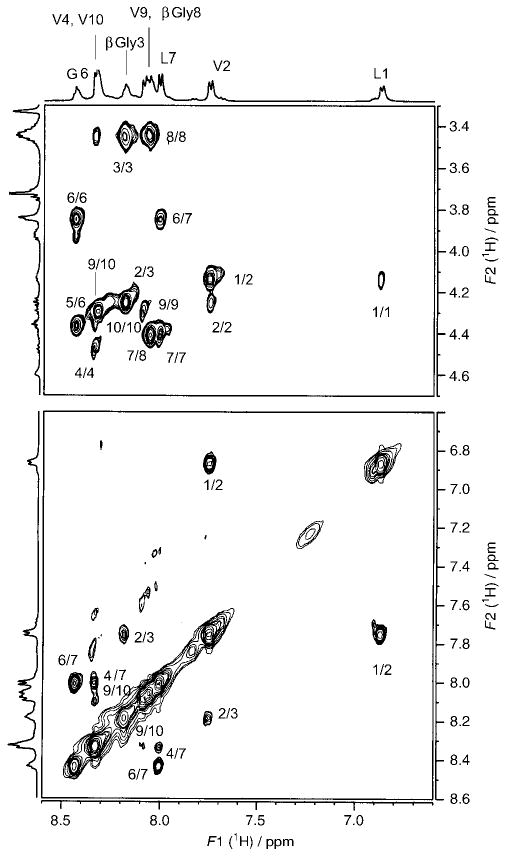
500 MHz partial ROESY spectrum of peptide 2 in CD3OH at 300 K, indicating NOEs for NH↔CαH (for α-residues) and NH↔CβH (for β-residues) (top) and for NH↔NH (bottom).
Figure 4.
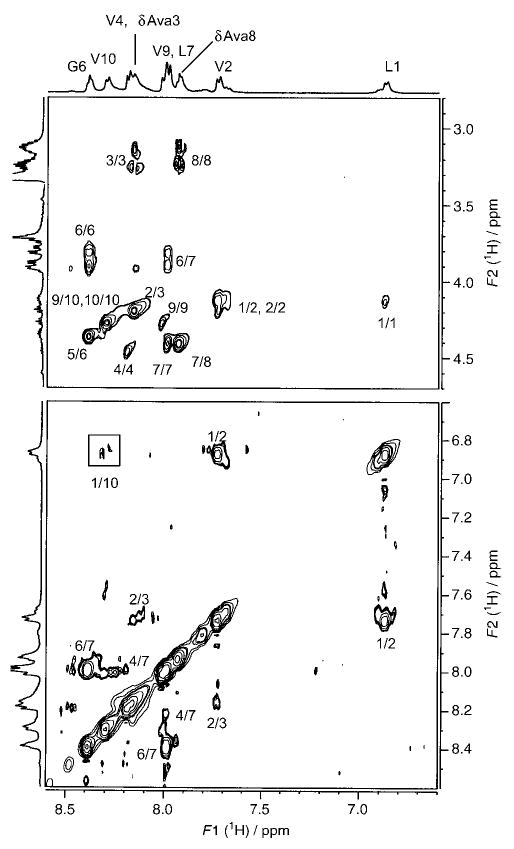
500 MHz partial ROESY spectrum of peptide 4 in CD3OH at 300 K, indicating NOEs for NH↔CαH (for α-residues) and NH↔CδH (for δ-residues) (top) and for NH↔NH (bottom).
The observed NOE connectivities for the three peptides are indicated schematically in Figure 5. The NOEs compatible with the hairpin conformation are represented as filled arrows, and open arrows identify NOEs that are incompatible with the β-hairpin structure. The presence of both types of NOEs is a reflection of conformational heterogeneity. Notably, the hairpin-incompatible NOEs are observed towards the N- and C-termini of the peptides. Fraying of hairpins at the termini has been established earlier in model peptides, both in solution and in the solid state.[2e] In all three peptides, strong d-Pro4(CαH)↔Gly5(NH), Gly5(NH)↔Leu6(NH), and Gly5(CαH)↔Leu6(NH) NOEs, consistent with the presence of type II′ β-turns, are observed. The results shown in Figures 3–5 provide reasonably strong NMR evidence for a predominant population of a β-hairpin conformation in peptide 2 (Xxx = βGly) and peptide 4 (Xxx = δAva). Interstrand NOEs in the case of peptide 4 are observed between Val2 (CαH) and Val9 (CαH) (data not shown) and Leu1 (NH) and Val10 (NH) (Figure 4, bottom panel), indicating spatial proximity of the termini. In addition, a strong NOE between δAva3 (CαH) and δAva8 (CδH) is also detected (data not shown), providing support for chain reversal and strand registry. In the case of peptide 2, NOEs between residues 1 and 10 and residues 2 and 9 were not detectable.
Figure 5.
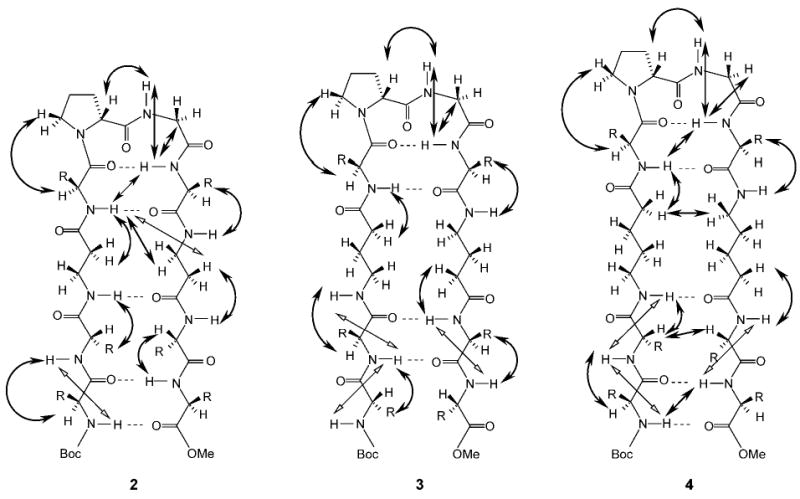
Schematic representation of hairpins 2–4, showing all observed NOEs. Filled arrows indicate NOEs supportive of hairpin conformations and open arrows refer to NOEs that are inconsistent with the anticipated structure.
Nevertheless, NOE evidence for a hairpin fold encompassing residues 3 and 8 is observed. In peptide 3 (Xxx = γAbu), NOE evidence supports β-turn formation at the d-Pro-Gly segment. However, the critical dNN NOE between Val4 (NH) and Leu7 (NH) was not observed. This short dNN distance, which is characteristic of the formation of the second hydrogen bond in a β-hairpin, is sometimes lengthened by solvation. In crystal structures of several model hairpins, solvent molecules often bridge the central peptide unit of the β-turn and one of the peptide units of the strand segments.[9] All the observed NOEs correspond to short inter-residue distances, suggestive of extended conformations in the strand segments of both the N- and C-termini. However, the absence of cross-strand NOEs precludes a firm conclusion regarding the population of β-hairpin conformations in peptide 3. A notable difference between the peptides is in the schematic β-hairpin structure, as illustrated in Figure 5. In peptides 2 and 4, five cross-strand hydrogen bonds may be anticipated in an ideal β-hairpin, whereas in peptide 3, only four cross-strand hydrogen bonds are expected, with residues 1 and 10 not forming a part of the registered antiparallel-strand structure. This difference arises because the Xxx residues contain an even number of backbone methylene groups in peptides 2 and 4, but an odd number in peptide 3.
Interestingly, single crystals of peptide 3 were obtained and X-ray diffraction analysis, described below, establishes a β-hairpin conformation in the solid state. Ironically, despite several attempts, peptides 2 and 4, which show strong evi dence for β-hairpin structures in solution, did not yield single crystals.
X-ray diffraction
Single crystals of peptide 3 grown from both methanol/water and methanol/water/dioxane solvent mixtures diffracted to a resolution of approximately 1.1–1.2 Å. The crystallographic asymmetric unit contains two independent molecules, A and B. Figure 6 shows a view of the molecular conformation of molecule A and the associated intra- and intermolecular hydrogen bonds. The relevant torsion angles and hydrogen-bond parameters are summarized in Tables 1 and 2, respectively. The individual molecules A and B within the crystal of peptide 3 have very similar conformations, as seen in the superimposition of molecules A and B in Figure 7. Each γ-inserted molecule is folded into a β-hairpin conformation and stabilized by four cross-strand hydrogen bonds with appropriate dimensions. The directions of the cross-strand NH···O = C hydrogen bonds alternate in the same manner as in hairpin turns containing only α-residues in the strands. The β-turns encompassing Pro5-Gly6 and Pro15-Gly16 are type II′ turns, such as those found in Boc-Leu-Val-Val- d-Pro-Gly-Leu-Val-Val-OMe.[7b] Type I′ turns have also been found in crystals of some of the β-hairpins containing β-amino acid residue inserts.[2c,e]
Figure 6.
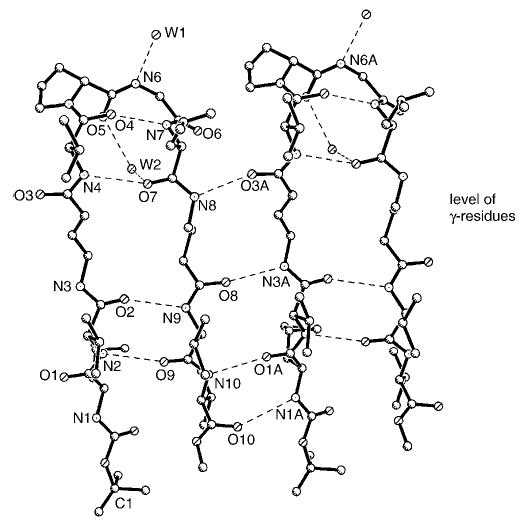
Hydrogen bonds in molecule A of peptide 3. Molecules with hairpin turns assemble by lateral translation into a continuous β-sheet. A similar, but separate, β-sheet is formed by molecule B (not shown). The γ-residues in molecule A are in positions 3 and 8 and face each other across the strand.
Table 1.
Torsion angles [°] in crystals of peptide 3.[a]
| Residue name | Torsion angles [°] | |||||
|---|---|---|---|---|---|---|
| ϕ | θ1 | θ2 | ψ | χ1 | χ2 | |
| Leu1 | −161 (−158) | 132 (122) | 176 (−176) | 169, 77 (−175, 60) | ||
| Val2 | −130 (−118) | 117 (125) | −173, −50 (−63, 173) | |||
| γAbu3 | −119 (−116) | +107 (−173) | 173 (176) | −158 (106) | ||
| Val4 | −131 (−128) | 100 (90) | −55, 180 (−62, −174) | |||
| d-Pro5[b] | 64 (64) | −127 (−134) | 7 (−29) | −21 (39) | ||
| Gly6 | −88 (−77) | −3 (−7) | ||||
| Leu7 | −76 (−75) | 138 (133) | −94 (−53) | 179, 1 (176, −57) | ||
| γAbu8 | 94 (179) | −175 (−171) | −65 (−135) | 143 (132) | ||
| Val9 | −130 (−134) | 127 (127) | 177, −57 (−45, 171) | |||
| Val10 | −114 (−112) | −117 (−59) | −55, 180 (−57, 180) | |||
Values are for two independent molecules, A and B, in the crystallographic asymmetric unit. Values in parentheses correspond to molecule B.
χ3: 25.2° (−33.2°); χ4: −20.3° (15.2°); χ5: 7.7° (8.6°)▪▪please define χ▪▪. The estimated standard deviations are ~3°.
Table 2.
Hydrogen bonds in peptide 3.
| Donor | Acceptor | D–A [Å] | H–A [Å] | C=O···N angle [°] |
|---|---|---|---|---|
| molecule A | ||||
| N1 | O10[a] | 3.276 | 2.49 | 140 |
| N2 | O9 | 2.973 | 2.12 | 160 |
| N3 | O8[a] | 2.894 | 2.03 | 168 |
| N4 | O7 | 3.078 | 2.20 | 143 |
| N5 (Pro) | ||||
| N6 | W1 | 2.815 | 1.95 | |
| N7 | O4 | 2.881 | 2.03 | 139 |
| N8 | O3[b] | 2.930 | 2.04 | 149 |
| N9 | O2 | 2.957 | 2.09 | 157 |
| N10 | O1[b] | 2.932 | 2.05 | 151 |
| molecule B | ||||
| N11 | O20′[b] | 3.290 | 2.40 | 144 |
| N12 | O19 | 2.887 | 2.02 | 155 |
| N13 | O18[b] | 2.813 | 1.96 | 174 |
| N14 | O17 | 3.054 | 2.17 | 147 |
| N15 (Pro) | ||||
| N16 | O6[c,d] | 2.990 | 2.11 | 123 |
| N17 | O14 | 2.933 | 2.08 | 133 |
| N18 | O13[a] | 2.891 | 2.01 | 148 |
| N19 | O12 | 2.937 | 2.08 | 154 |
| N20 | O11[a] | 2.865 | 1.98 | 166 |
| solvent | ||||
| W1 | O15 | 2.746 | ||
| W1 | O17 | 2.882 | ||
| W2[e] | O5 | 2.813 | ||
| W2 | O7 | 2.914 | ||
Symmetry of acceptor, 1+x, y, z.
Symmetry of acceptor, −1+x, y, z.
Symmetry of acceptor, x, 1+y, z.
Direct hydrogen bond between donor and acceptor▪▪ok?▪▪.
The N16 atom does not participate in a hydrogen bond with W2 (N16···W2=3.76 Å). Compare to N6···W1= 2.81 Å.
Figure 7.
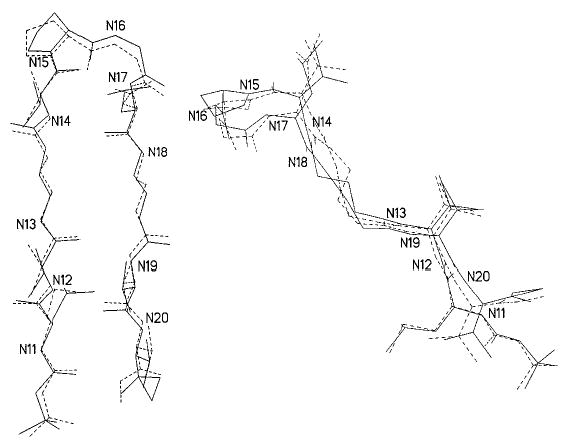
Face (left) and side (right) views of superimpositions, fitted by least-squares, of nitrogen atoms (1–10) in molecule A (dashed lines) to nitrogen atoms (11–20) in molecule B (solid lines).
The four – (CH2)3 – segments in the strands of molecules A and B (Figure 8) begin to show the type of conformational instability often observed for – (CH2)n – chains. A greater torsional flexibility about C–C bonds has been observed in crystal structures as n increases from four to twelve in the families of polymethylene-bridged cystine-based cyclobisamides and cyclobisureas, hybrid peptides that assemble into nanotubes.[10] In the present peptide 3, the –(CH2)3– groups are almost extended and do not disturb unduly the classic pleated-sheet conformation of β-hairpins (Figure 7). It is possible that peptide 4, with δ-amino acid residue inclusions, has sufficient disorder in the –(CH2)4– moieties to preclude neat β-sheet formation, and, therefore, cannot easily form crystals.
Figure 8.
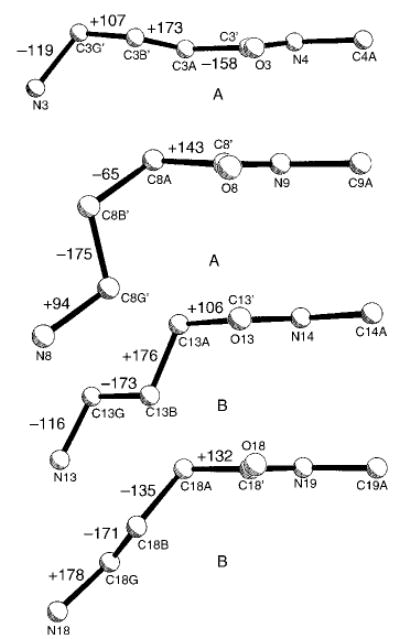
Comparison of four γAbu residues, indicating the variations in the backbone torsion angles.
The crystal packing in peptide 3, shown in Figure 9, has essentially the same features as the packing for an all-α-hairpin peptide,[7b] except that the α-sheet stacks in peptide 3 have a V-shaped tilt, in contrast to the relatively flat arrangement for the all-α-hairpin. In both crystals, the hydrogen bonds linking the heads of the left-side molecules to the heads of the right-side molecules are mediated by two water molecules, utilizing the carbonyl groups from d-Pro and Leu and the NH from Gly. The water molecules act as a scaffold that connects the β-sheets into a supramolecular assembly. Note, however, that in peptide 3, the W2–N16 distance is too large for a hydrogen bond, and instead, N16H is a donor to O6 directly. The packing of the stacks of β-sheets shown in Figure 9 is repeated along the horizontal direction (c axis), creating a hydrophobic boundary between the C-and N-termini of molecules A and B, as was also observed in the crystal of the all-α-hairpin.[7b]
Figure 9.
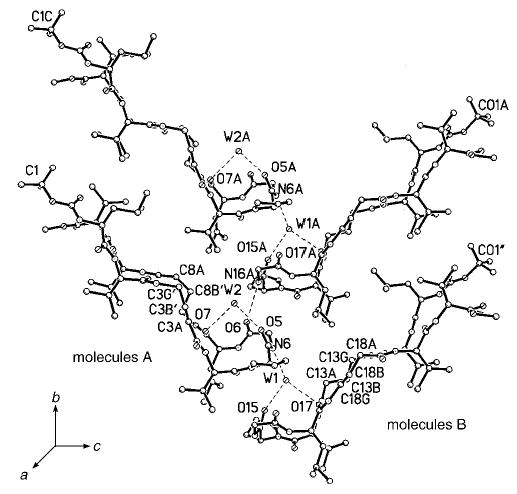
Crystal packing of the β-sheets (side view). The heads of the hairpin β-turns are connected in the vertical direction by one direct hydrogen bond, N16A···O6, and several hydrogen bonds mediated by water molecules, N6···W1, W1···015, and W1···017; and W2···05 and W2···07. If the assemblage is translated by one cell-length in the c direction, the tails of molecules A and B interdigitate with van der Waals distances between the hydrophobic tails and side chains.
Circular dichroism
Model β-hairpin peptides containing only aliphatic α-amino acid residues with d-Pro-Gly as the nucleating turn segment have generally yielded CD spectra characterized by a negative CD band at ~218 nm. The CD spectra of model β-hairpin peptides may arise as a consequence of contributions from both the nucleating turn segment and registered antiparallel strands. Peptides 2–4 provide an example in which backbone peptide units of the strand segments have been systematically separated by an increasing number of methylene groups, thereby altering the spatial relationships between the amide chromophores. Figure 10 shows the far UV-CD spectra of peptides 2–4. In each case, a negative CD band is observed. Notably, in peptide 3, the minimum is at 229 nm, whereas in peptides 2 and 4, the minimum is blue-shifted to 225 nm. A crossover point at 212 nm for peptide 3 is indicative of a positive CD band at a shorter wavelength. In constrast, in peptides 2 and 4, the crossover is not observed, suggestive of a short-wavelength, negative CD band. It is tempting to correlate the observed CD pattern with the insertion of methylene groups between the amide chromophores; peptide 3 corresponds to insertion of an odd number of methylene groups, whereas peptides 2 and 4 contain an even number of methylene groups. The effect of changing the local polarity of the sheet by inverting the orientation of amide units merits further investigation. It is not yet clear whether CD spectra are influenced by transition from registered hairpin structures to frayed structures that retain the turn and strand segments, but have lost antiparallel hydrogen bonds.
Figure 10.
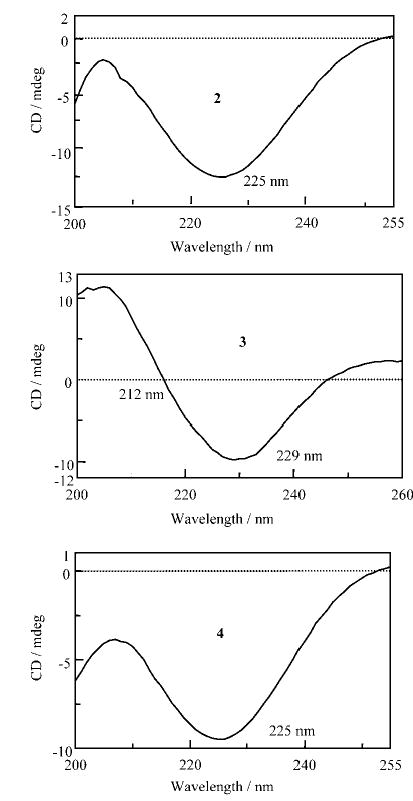
Far UV-CD spectra of peptides 2–4 in methanol at 300 K recorded at concentrations of 0.44–0.47 mm.
Conclusion
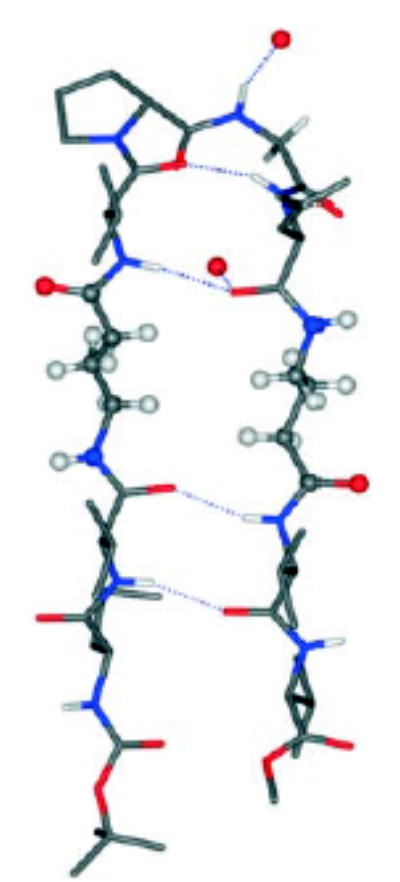
The insertion of ω-amino acid residues into the strand segments of a model peptide hairpin can be accomplished without disruption of the overall fold of the molecule. Unsubstituted β, γ, and δ-amino acid residues were successfully inserted into facing positions of antiparallel strands in model decapeptides. This study establishes that hybrid (α,ω) peptide hairpins can be obtained by rational design.
Hairpins that stay in place! The insertion of ω-amino acid residues into the strand segments of a model peptide hairpin can be accomplished without disruption of the overall fold of the molecule (see figure). Unsubstituted β, γ-, and δ-amino acid residues were successfully inserted into facing positions of antiparallel strands in model decapeptides.
Figure.
Acknowledgments
This work was supported in Bangalore by a program support grant in the area of Molecular Diversity and Design by the Department of Biotechnology, Government of India. R.S.R. is a recipient of a senior research fellowship of the Council of Scientific and Industrial Research, Government of India. The work at the Naval Research Laboratory was supported by National Institutes of Health Grant GM30902 and the Office of Naval Research.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/or from the author.
References
- 1.a Seebach D, Beck AK, Bierbaum DJ. Chem Biodiversity. 2004;1:1111–1239. doi: 10.1002/cbdv.200490087. [DOI] [PubMed] [Google Scholar]; b Seebach D, Overhand M, Kühnle FNM, Martinoni B, Oberer L, Hommel U, Widmer H. Helv Chim Acta. 1996;79:913–941. [Google Scholar]; c Appella DH, Christianson LA, Karle IL, Powell DR, Gellman SH. J Am Chem Soc. 1996;118:13071–13072. [Google Scholar]; d Seebach D, Gademann K, Schreiber JV, Matthews JL, Hintermann T, Jaun B, Oberer L, Hommel U, Widmer H. Helv Chim Acta. 1997;80:2033–2038. [Google Scholar]; e Seebach D, Matthews JL. Chem Commun. 1997:2015–2022. [Google Scholar]; f Appella DH, Christianson LA, Klein DA, Powell DR, Huang L, Barchi JJ, Gellman SH. Nature. 1997;387:381–384. doi: 10.1038/387381a0. [DOI] [PubMed] [Google Scholar]; g Appella DH, Christianson LA, Karle IL, Powell DR, Gellman SH. J Am Chem Soc. 1999;121:6206–6212. [Google Scholar]; h Appella DH, Christianson LA, Klein DA, Richards MR, Powell DR, Gellman SH. J Am Chem Soc. 1999;121:7574–7581. [Google Scholar]; i Abele S, Seiler P, Seebach D. Helv Chim Acta. 1999;82:1559–1571. [Google Scholar]; j Rueping M, Schreiber JV, Lelais G, Jaun B, Seebach D. Helv Chim Acta. 2002;85:2577–2593. [Google Scholar]; k Lelais G, Seebach D. Biopolymers. 2004;76:206–243. doi: 10.1002/bip.20088. [DOI] [PubMed] [Google Scholar]; l Cheng RP, Gellman SH, DeGrado WF. Chem Rev. 2001;101:3219–3232. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]; m Hill DJ, Mio JM, Prince RB, Hughes TS, Moore JS. Chem Rev. 2001;101:3893–4011. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 2.a Seebach D, Abele S, Gademann K, Jaun B. Angew Chem. 1999;111:1700–1703. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1595::AID-ANIE1595>3.0.CO;2-0. Angew. Chem. Int. Ed. 1999, 38, 1595–1597. [DOI] [PubMed] [Google Scholar]; b Krauthäuser S, Christianson LA, Powell DR, Gellman SH. J Am Chem Soc. 1997;119:11719–11720. [Google Scholar]; c Karle IL, Gopi HN, Balaram P. Proc Natl Acad Sci USA. 2001;98:3716–3719. doi: 10.1073/pnas.071050198. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Karle IL, Gopi HN, Balaram P. Proc Natl Acad Sci USA. 2002;99:5160–5164. doi: 10.1073/pnas.022616499. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gopi HN, Roy RS, Raghothama S, Karle IL, Balaram P. Helv Chim Acta. 2002;85:3313–3330. [Google Scholar]
- 3.a Seebach D, Brenner M, Rueping M, Schweizer B, Jaun B. Chem Commun. 2001:207–208. [Google Scholar]; b Seebach D, Brenner M, Rueping M, Jaun B. Chem Eur J. 2002;8:573–584. doi: 10.1002/1521-3765(20020201)8:3<573::AID-CHEM573>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]; c Hanessian S, Luo X, Schaum R, Michnick S. J Am Chem Soc. 1998;120:8569–8570. [Google Scholar]
- 4.a Karle IL, Pramanik A, Banerjee A, Bhattacharjya S, Balaram P. J Am Chem Soc. 1997;119:9087–9095. [Google Scholar]; b Banerjee A, Pramanik A, Bhattacharjya S, Balaram P. Biopolymers. 1996;39:769–777. doi: 10.1002/(SICI)1097-0282(199612)39:6%3C769::AID-BIP4%3E3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 5.a Chung YJ, Huck BR, Christianson LA, Stanger HE, Krauthäuser S, Powell DR, Gellman SH. J Am Chem Soc. 2000;122:3995–4004. [Google Scholar]; b Shankaramma SC, Singh SK, Sathyamurthy A, Balaram P. J Am Chem Soc. 1999;121:5360–5363. [Google Scholar]
- 6.a Martinek TA, Tóth GK, Vass E, Hollósi M, Fülöp F. Angew Chem. 2002;114:1794–1797. doi: 10.1002/1521-3773(20020517)41:10<1718::aid-anie1718>3.0.co;2-2. Angew. Chem. Int. Ed 2002, 41 1718–1721. [DOI] [PubMed] [Google Scholar]; b Woll MG, Lai JR, Guzei IA, Taylor SJC, Smith MEB, Gellman SH. J Am Chem Soc. 2001;123:11077–11078. doi: 10.1021/ja011719p. [DOI] [PubMed] [Google Scholar]; c Ramana Rao MHV, Kiran Kumar S, Kunwar AC. Tetrahedron Lett. 2003;44:7369–7372. [Google Scholar]
- 7.a Awasthi SK, Raghothama S, Balaram P. Biochem Biophys Res Commun. 1995;216:375–381. doi: 10.1006/bbrc.1995.2634. [DOI] [PubMed] [Google Scholar]; b Karle IL, Awasthi SK, Balaram P. Proc Natl Acad Sci USA. 1996;93:8189–8193. doi: 10.1073/pnas.93.16.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) G. M. Sheldrick, SHELXTL PLUS, Release 4.2 for Bruker R3m/V Crystal Search System, Bruker Analytical X-ray Instruments, Madison, WI, 1992; ; b Karle J. Acta Crystallogr Sect B. 1968;24:182–186. doi: 10.1107/s0567740868001731. [DOI] [PubMed] [Google Scholar]
- 9.a Das C, Naganagowda GA, Karle IL, Balaram P. Biopolymers. 2001;58:335–346. doi: 10.1002/1097-0282(200103)58:3<335::AID-BIP1010>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; b Aravinda S, Harini VV, Shamala N, Das C, Balaram P. Biochemistry. 2004;43:1832–1846. doi: 10.1021/bi035522g. [DOI] [PubMed] [Google Scholar]
- 10.a Ranganathan D, Lakshmi C, Karle IL. J Am Chem Soc. 1999;121:6103–6107. [Google Scholar]; b Ranganathan D, Haridas V, Sivakama Sundari C, Balasubramanian D, Madhusudanan KP, Roy R, Karle IL. J Org Chem. 1999;64:9320–9340. [Google Scholar]